Direct detection of double-stranded DNA: molecular methods and applications for DNA diagnostics†
Indraneel Ghosh*a, Cliff I. Stainsa, Aik T. Ooib and David J. Segal*b
aDepartment of Chemistry, University of Arizona, Tucson, Arizona 85721. E-mail: ghosh@email.arizona.edu; Tel: 520-621-6331
bGenome Center and Department of Pharmacology, University of California, Davis, CA 95616. E-mail: djsegal@ucdavis.edu; Fax: 530-754-9658; Tel: 530-754-9134
First published on 28th September 2006
Abstract
Methodologies to detect DNA sequences with high sensitivity and specificity have tremendous potential as molecular diagnostic agents. Most current methods exploit the ability of single-stranded DNA (ssDNA) to base pair with high specificity to a complementary molecule. However, recent advances in robust techniques for recognition of DNA in the major and minor groove have made possible the direct detection of double-stranded DNA (dsDNA), without the need for denaturation, renaturation, or hybridization. This review will describe the progress in adapting polyamides, triplex DNA, and engineered zinc finger DNA-binding proteins as dsDNA diagnostic systems. In particular, the sequence-enabled reassembly (SEER) method, involving the use of custom zinc finger proteins, offers the potential for direct detection of dsDNA in cells, with implications for cell-based diagnostics and therapeutics.
 Indraneel Ghosh | Indraneel Ghosh was born on 30th December, 1968 in India. He obtained his BS degree in chemistry at Hobart College, Geneva, NY. He received his PhD in Chemistry at Purdue University, IN in 1998 with Professor Jean Chmielewski working on designing peptide inhibitors of protein–protein interactions and self-replicating peptides. Following his doctoral studies, Neel was a joint postdoctoral fellow at Yale University, New Haven, CT with Professor Andrew Hamilton in the Department of Chemistry and Professor Lynne Regan in Molecular Biophysics and Biochemistry. At Yale, he developed the split-Green Fluorescent Protein reporter system for the in vivo detection of protein–protein interactions. He started as an assistant professor at the University of Arizona, Tucson, AZ in 2001 and has been engaged in interdisciplinary research that spans the chemistry and biology interface. His current research interests are focused upon the design and selection of new proteins, peptides and small molecules that can be useful in the construction of therapeutics, biosensors, and biomaterials. He has been a recipient of a Research Innovation Award from the Research Corporation and a Career Award from the National Science Foundation. |
 Cliff Stains | Cliff Stains was born in Lewistown, PA in 1979. He attended Millersville University, Millersville, PA where he majored in Chemistry with an emphasis in Biochemistry. Under the direction of Dr Sandra Turchi he developed methods for the separation of methylated nucleotides, leading to an undergraduate honors thesis. Cliff joined the research group of Dr Indraneel Ghosh at the University of Arizona in 2003 where he is currently pursuing a PhD in Chemistry with a focus on Biological Chemistry. His research interests include designing protein–protein and protein–DNA assemblies for use in biological and materials applications. He has been the recipient of an institutional pre-doctoral Ruth L. Kirschstein National Research Service Award and mid-career awards at the University of Arizona. |
 Aik Ooi | Aik Ooi was born in Penang, Malaysia in 1980. He obtained his BS degree in Chemistry at Mount Union College, Alliance, OH, with a minor in Biology. For his undergraduate thesis, he worked under the supervision of Dr Laura Beal on the synthesis and application of substituted flavonoids as multi-drug resistant pump inhibitors in S. aureus. In 2002, Aik enrolled in the University of Arizona for his graduate studies in Medicinal and Natural Products Chemistry. He joined the research group of Dr David Segal, where he is working on the design and development of sequence-specific double-stranded DNA detection utilizing custom-designed zinc fingers and split-enzyme complementation. He has been the recipient of Yuma young investigator award and Caldwell research award at the University of Arizona. |
 David Segal | David Segal was born in Yonkers, NY in 1966. He obtained a BS with honors in Biology from Cornell University, Ithaca, NY, in 1989. He obtained a PhD in Biochemistry from the University of Utah, Salt Lake City, UT, in 1996 with Professor Dana Carroll working on the stimulation of homologous recombination by targeting double-strand breaks in DNA. Seeking better targeting methods, he joined the laboratory of Professor Carlos Barbas at The Scripps Research Institute, La Jolla, CA as a postdoctoral fellow, where he helped develop the most widely used methods for engineering custom zinc finger DNA-binding proteins. As an assistant professor at the University of Arizona, Tucson, AZ, from 2002–2005, and now as an assistant professor at the University of California, Davis, CA, his research continues to focus on the design of engineered zinc fingers and their application as diagnostic agents and therapeutics |
An introduction to DNA diagnostics
Molecular diagnostics that report on DNA sequence information are making increasingly important contributions to medicine and research. Pathogen identification based on a DNA sequence is more accurate, less subjective, and often much faster than culture-based methods.1,2 In addition to improving existing services, DNA diagnostics allow access to genomic information previously unavailable to clinicians. Genotyping methodologies, which reveal the status of the single nucleotide polymorphisms (SNP) that distinguish one individual's genome from another's, can provide insights into the most effective drug regimen for a particular patient, or provide clues to resistance or susceptibility for particular diseases. Such diagnostic information can usually be obtained by determining the presence, absence, or abundance of a particular known sequence. This feature distinguishes such methods from general sequencing methodologies, which aim to determine large stretches of unknown sequence.Many types of DNA diagnostic methodologies have been described. Some are in the very early stages of development while others are commercially available. One approach to categorize the myriad of techniques is to define how they address their common goals. Like all diagnostic technology, DNA diagnostics require both a detection method and a signaltransducer. Most current detection methods for the sequence-specific recognition of DNA make use of the special property of single-stranded DNA (ssDNA) to base pair with high specificity to a complementary molecule (Fig. 1A). The other molecule may be another ssDNA, ssRNA, peptide-nucleic acid (PNA),3 or other base-pairing molecular analog. Such specific annealing or hybridization forms the basis for such common technologies as PCR amplification with specific primer sets, Southern blot, Northern blot, DNA microarray, and fluorescent in situ hybridization (FISH).4 However, there are other ways to read the sequence information besides Watson–Crick base-pairing (Fig. 1B). For example, polyamides are small chemical compounds that can be designed to bind with high sequence specificity in the minor groove of double-stranded DNA (dsDNA).5 Similarly, triplex-forming DNA,6 and zinc finger DNA-binding proteins7 can all be engineered to achieve specific base-pair recognition of dsDNA in the major groove.
 | ||
| Fig. 1 An overview of DNA diagnostic methods. Detection methods can read the sequence information by either (A) Watson–Crick base pairing with one strand (orange), which requires denaturation of the duplex and subsequent hybridization with a complementary probe (purple), or (B) direct detection of dsDNA by specific interaction with base edges in the major or minor groove. A signal transducer converts the detection event into a quantitative signal, such as fluorescence intensity (green). | ||
The second component required is a signaltransducer, which converts the sequence-specific recognition event into a signal that can be quantitatively measured (Fig. 1). Typically the signal is optical (colorimetric, fluorescent, luminescent, turbidic, etc) or electrical (voltage, resistance, or current change). Transducers with fluorescent readouts are used most commonly, such as dyes that intercalate into dsDNA (ethidium bromide and SYBR green) or are attached to the base-pairing partner of ssDNA (labeled probes used in DNA microarrays,8 Taqman real-time PCR chemistry,9 or FISH). While the specificity of a DNA diagnostic will depend on the fidelity of the detection method, the sensitivity will largely be a function of the signal transducer. For example, PNA probes can be engineered to bind with extremely high specificity and affinity to their denatured chromosomal targets in a FISH assay.10 However, detection of unique genomic sequences is limited by the difficulty in detecting the weak signal of one fluorescent molecule over background. To improve sensitivity, several ingenious methods have recently been developed to sensitively detect the recognition event (such as hybridization-dependent current fluctuations across an α-hemolysin nanopore11,12), or amplify the transduced signal (such as hybridization-dependent release of barcode DNA from captured nanoparticles,13 or aggregation-enhanced fluorescence14). Some of these methods have proven to be extremely sensitive, able to detect molecules in the zeptomolar range (1–500 molecules per ml sample). Other strategies rely again on the special ability of nucleic acids to form specific base pairs and enzymatically amplify the DNA, either before or as part of the detection method (such as PCR, Strand Displacement Amplification (SDA),15 or Rolling Circle Amplification (RCA)16).
For an overview of recent advances in hybridization-based DNA diagnostics, the reader is directed to several outstanding reviews on this topic.17–19 The scope of this review will be restricted to methods for the direct detection of dsDNA, meaning methods that do not require dsDNA denaturation and subsequent hybridization. Progress in this area has been slower because of the difficulty in engineering highly specific detection methods for the major or minor groove of DNA. However, the emergence of such technologies in the past decade has now enabled their application as dsDNA diagnostics. In some cases, the ability to use dsDNA as a substrate enables capabilities beyond what would be possible for hybridization-based methods.
Beyond annealing: direct detection of dsDNA
Methods for direct detection of dsDNA in the minor groove
 | ||
| Fig. 2 Polyamide minor groove binders. Left: structural representation of a polyamide (green) bound in the minor groove of dsDNA (black and orange). Middle: a schematic illustration of the binding interactions. The abbreviations are pyrrole (Py), hydroxypyrrole (Hp), and imidazole (Im). Right: chemical structure of thiazole orange, used as a signal transducer. | ||
Laemmli and coworkers have recently utilized fluorescein-labeled polyamides as “chromosome paints” with the goal to visualize AT-rich satellite regions and scaffold-associated regions in the genome of Drosophila melanogaster.23 In a second study, designed polyamides conjugated to Texas-Red were used to target and visualize telomeric repeats in insects (TTAGG) and vertebrates (TTAGGG) with high specificity.24 These results demonstrated that telomere-specific polyamide-dye conjugates might allow for the rapid estimation of the telomere length. These elegant studies utilized fluorescence microscopy of fixed cells or of isolated nuclei, where excess labeled-polyamides can be removed. However, the detection of dsDNA in live cells or whole animals would require a method for removing unbound labeled-polyamides, as the background fluorescence would likely decrease the contrast.
With the goal of lowering background (signal from unbound labeled-polyamides), Dervan and coworkers have recently designed and tested polyamides conjugated to intercalating dyes tetramethyl rhodamine and thiazole orange (Fig. 2, right).25,26 In these studies, several fluorescent conjugates were synthesized and tested against dsDNA targets, 5′-WGGGWW-3′, 5′-WGGCCW-3′, and 5′-WGWWCW-3′ (W = A or T). It was found that the designed conjugates with thiazole orange exhibit >1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold fluorescence enhancement only in the presence of specific target dsDNA, where the dye likely intercalates at an adjacent site. The lowest concentration of oligonucleotide detected in this study was 1 nM, although lower concentrations were not examined. Mismatched targets reduced the signal by >90%. As polyamide binding site sizes and sequence specificities are being further refined, these new dsDNA-sensitive dyes attached to appropriate targeting molecules will likely find use in probing dsDNA in a cellular setting.27
000-fold fluorescence enhancement only in the presence of specific target dsDNA, where the dye likely intercalates at an adjacent site. The lowest concentration of oligonucleotide detected in this study was 1 nM, although lower concentrations were not examined. Mismatched targets reduced the signal by >90%. As polyamide binding site sizes and sequence specificities are being further refined, these new dsDNA-sensitive dyes attached to appropriate targeting molecules will likely find use in probing dsDNA in a cellular setting.27
Methods for direct detection of dsDNA in the major groove
 | ||
| Fig. 3 Triplex-DNA major groove binders. Left: structural representation of a TFO (green) bound in the major groove of dsDNA (black and orange). Middle: a schematic illustration of the binding interactions. Right: two types of “padlock” TFO strategies, (top) linear TFO with ends joined using a “splint” oligonucleotide (blue), and (bottom) stem-loop TFO with ligated fluorescently labeled DNA (purple). | ||
An early application of triplex-DNA as a diagnostic agent was to stain an alpha-satellite repeat in chromosomal spreads using an assay analogous to FISH (appropriately termed TISH).31 A 16 nt polypyrimidine TFO was designed to bind the 500–1000 repeats of the target sequence at pH 6 without denaturation. TFO binding was stabilized by crosslinking to the duplex via a tethered psoralen moiety. Signal transduction was accomplished by tagging the TFO with fluorescein isothiocyanate (FITC). The study found TISH comparable to FISH in both sensitivity and specificity. The dsDNA-based TISH was suggested to be more quantitative than FISH, since there was no competition between probe hybridization and duplex reannealing. Additionally, non-denatured DNA allowed TISH, but not FISH, to be compatible with G-banding chromosomal reference techniques.
Another application of triplex DNA is the so called “padlock” TFO.32 The detection method is based on the central part of a linear TFO, which forms a triplex with the target duplex DNA. The ends of the linear molecule can be joined covalently by base-pairing with an additional “splint” oligonuclotide, followed by DNA ligase (Fig. 3, top right). The result is a circular ssDNA molecule that is topologically linked to the target duplex. Signal transduction can be accomplished by RCA,16 or other amplification method. The ends of the TFO can alternatively be stabilized non-covalently by a stem-loop structure (Fig. 3, bottom right).33,34 Signal transduction in this case can be accomplished by designing the stem loop to have a short terminal overhang, to which a fluorescently labeled DNA can be ligated. Because the TFO is physically wrapped around the duplex molecule (hence the name “padlock”), the affinity of this complex is far greater than that of the triplex alone. In one study, a polypurine/polypyrimidine tract in individually spread molecules of lambda phage DNA was visualized with a 59 nt stem-loop oligonucleotide probe (containing a 15 nt central triplex forming region) and a 500 bp stem-loop-binding labeled duplex.33 For increased sensitivity, the 500 bp DNA was labeled with at least 20 molecules of AlexaFluor 546. The purified lambda DNA was stretched on glass slides using molecular combing methods. The sample had to be heated to unwind the stem-loop on the probe oligonucleotide, then slowly cooled to allow rewinding after triplex formation. The precise position of the target site along individual DNAs was easily observable by fluorescence microscopy. In another study, radiolabeled padlock TFO were able to detect subfemtomolar concentrations of target dsDNA using a signal transduction method of gel electrophoresis followed by autoradiography of dried gels.34
A wide variety of technological improvements have been made to expand recognition beyond strictly polypurine tracts, improve affinity, reduce pH dependence, and reduce degradation in cells. For example, artificial base analogs can extend recognition to all possible base pairs.35 Chemical compounds such as BQQ can act as triplex stabilizing agents.36 Triplex formation involving any sequence of bases was suggested to occur at physiological pH in the presence of YOYO-1, an oxazole yellow homodimer the fluorescent intensity of which increases over 1000-fold in the presence of dsDNA and 100000-fold in the presence of triplex DNA.37 This approach was reported to distinguish SNPs and single base pair deletions in PCR amplified fragments of cystic fibrosis gene, the DNA repair gene hMSH2, and the tumor suppressor genes BRCA1 and P16. The assay was extremely rapid and simple; however, the postulated triplexes were not unequivocally demonstrated. Such modified triplex paradigms and their general applicability for direct dsDNA recognition deserve further study.
Peptide nucleic acid (PNA) are synthetic nucleic acid homologs containing standard DNA bases but a polyamide backbone composed of N-(2-aminoethyl) glycine units. The nucleobases are attached with methylenecarbonyl linkers.40 The neutral backbone eliminates the charge repulsion in standard nucleic acid hybridization, therefore PNA is able to bind DNA and RNA with extremely high affinity and specificity. In the most common application, a homopyrimidine PNA forms a structure in which one PNA molecule forms Watson–Crick base pair interactions with a polypurine DNA strand, while another PNA forms Hoogsteen interactions. The other DNA strand is displaced, forming a “P loop”. However, a variety of PNA:DNA2, PNA2:DNA, PNA2:DNA2 structures have been observed under various experimental conditions.3
DNA detection applications have been developed using both RecA41 and PNA.42–44 Indeed, applications of PNA are so numerous that the reader is directed to a dedicated review on this subject.45 However, as both these technologies involve some aspect of local duplex disruption and are not strictly major or minor groove detection methods, they will not be discussed further here.
Beyond in vitro assays, the E. colilac repressor was fused to green fluorescent protein (GFP) to visualize inserted repeats of the lac operator in living yeast and mammalian cells.49 Single chromosomal integrations of a vector containing 256 repeats of the lac operator could be observed by fluorescence microscopy in cells expressing the GFP–lac fusion protein, with a signal-to-noise (background nuclear fluorescence) ratio of 12 : 1. In the absence of DNA binding, the expression of GFP–lac produced only diffuse fluorescence. The sensitivity of this live cell imaging method was found to be comparable to immunostaining and FISH. The study went on to demonstrate the utility of this method to examine chromonema fibers in interphase nuclei. The authors noted that previous failures to appreciate large-scale chromatin substructure within chromosome domains were consistent with structural perturbations in chromatin structure resulting from standard in situ hybridization procedures. More recently, the GFP–lac system was used to perform mosaic analysis in living C. elegans.50
The GFP–lac studies demonstrate successful detection in an environment in which the direct detection of dsDNA is a clear advantage, the living cell. However, these studies also illustrate the technological challenges to advancing beyond the proof-of-concept stage. First, the use of natural DNA-binding proteins as the detection method severely restricts the spectrum of DNA sequences that can be recognized. Second, signal transduction was successful because 256 GFP–lac molecules were spatially restricted to one locus. Detection of spatially distributed or unique target sequences, as is more typically desired, would be impossible because the individual bound GFP–lac molecules would have the same signal intensity as unbound molecules. We have attempted to address both of these challenges in the recently described sequence-enabled reassembly (SEER) detection methodology.51–53 As will be described below, the detection method is based on engineered zinc finger proteins, the specificity of which can be programmed by the investigator. The signal transducer is based on the binding-dependent reassembly of a reporter protein, such that no signal should be present unless DNA-binding occurs.
The SEER method for direct detection of dsDNA
 | ||
| Fig. 4 Zinc finger protein major groove binders. Top: structural representation of a three zinc finger protein (blue) bound in the major groove of dsDNA (black and orange). Bottom: recognition modules incorporated into the alpha helix of a zinc finger that will enable it to specifically bind the indicated 5′-ANN-3′, 5′-CNN-3′ or 5′-GNN-3′ DNA sequence. | ||
Using phage display technology, DNA contacting amino acids in the zinc finger domain were randomized for the selection of new variants that recognize desired DNA sequences.55,56 This allowed the selection of modules to construct multi-domain zinc fingers to bind to specific DNA sequences. Currently, domains have been identified that facilitate binding to all 5′-GNN-3′, most 5′-ANN-3′ and 5′-CNN-3′, and some 5′-TNN-3′ type sequences, enabling targeting to an extremely wide spectrum of target sites.57–59 Multi-finger proteins based on these custom DNA binding domains can be assembled by PCR using overlapping oligonucleotides or commercial synthesis.60,61
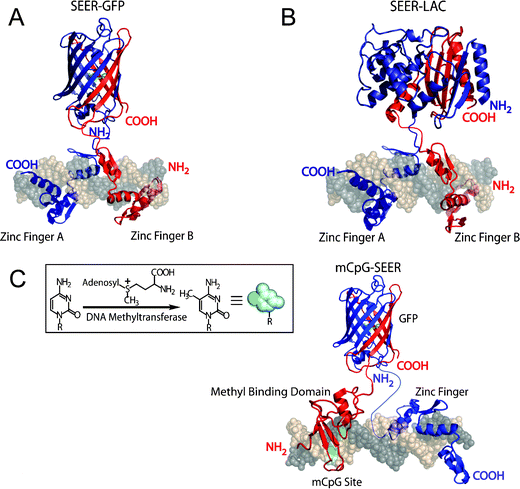 | ||
| Fig. 5 The SEER method for the direct detection of dsDNA. (A) SEER-GFP, (B) SEER-LAC, (C) mCpG-SEER with GFP. | ||
000-fold improvement in detection time over SEER-GFP, as it could differentiate target from non-target DNA sequences in 5 min. The colorimetric assay format had sufficient sensitivity to easily detect 20 nM of purified target DNA. The specificity of the system was high enough to distinguish a single base-pair mutation in the 18 bp binding site. The intensity of the signal remained the same in the presence of equal mass herring sperm DNA to target oligonucleotide.Towards the goal of a rapid method for the detection of known sites of hypermethylation at CpG islands, we have recently developed a new approach called mCpG-SEER (Fig. 5C).52 The mCpG-SEER system was designed based upon our existing SEER-GFP system, while incorporating a means for targeting methylated CpG sites. We chose the well-characterized MBD2 protein from humans, that has a binding affinity of 2.7 nM for mCpG sites while it has a 50–100 fold reduced binding affinity for unmethylated CpG sites. We hypothesized that this difference in binding affinities would allow us to selectively target mCpG sites versus unmethylated CpG sites. Since numerous sites on a genome are methylated, we needed to introduce sequence selectivity, which can be readily achieved by utilizing natural and designed zinc fingers as discussed. As proof of concept, we employed the Zif268 zinc finger to recognize a site next to the mCpG site. We found that the specificity of mCpG-SEER was >40-fold between a methylated versus a non-methylated CpG target site. We also found that the fluorescent signal was linear to 5 pmol of methylated target DNA in a 100 µL sample volume. Thus, mCpG-SEER represents a new and potentially useful method for the direct detection of CpG methylation, which may find numerous applications in delineating the epigenome and in cancer research.
Current status and future of direct detection methods for dsDNA
Why is direct detection of dsDNA important?
DNA is rarely present in single-stranded form, either naturally or after PCR amplification. Improvements in the detection of either type of dsDNA should lead to more robust and flexible DNA diagnostics. The dsDNA-based TISH was suggested to be more quantitative than FISH, since there was no competition between probe hybridization and duplex reannealing.31 Also, perturbations in chromatin structure may result from standard in situ hybridization procedures.49 Several of the assays described were extremely fast and simple, requiring no duplex denaturation or careful control of temperature. Conceivably, such methods could reduce assay time as well as the costs associated with sophisticated instrumentation and highly-trained technicians.Into the cell
Direct detection of dsDNA would also have an obvious advantage for the visualization of genomic information in living cells. Methods for determining genotype, chromatin status, and target copy number in individual living cells have been largely inaccessible using currently available hybridization-based techniques.78 Triplex DNA, polyamides and engineered zinc finger proteins have all been used in cells with some efficacy as gene regulators79 and inducers of DNA damage or homologous recombination.80 Therefore, they are good candidates for cellular diagnostic assays, although none have yet been evaluated in this role. Table 1 compares several features of the methods described in this review. Methods such as fluorescein-conjugated polyamides or triplex (TISH) could be applied to visualize repeated regions in living cells. Background fluorescence would be expected to be high, but the GFP–lac study suggests binding to spatially restricted loci might generate sufficient signal. Methods such as Thiazole orange-polyamides or SEER should have less background, and therefore should be even more sensitive. However, the sensitivity of all these methods will likely need to be improved in order to detect unique sequences in cells, such as SNPs, translocations, or mutations.| Method | Assay | Sensitivity | Sequence restrictions | Likely to be useful in cells | Reference |
|---|---|---|---|---|---|
| a Lowest concentration used in study, but lower concentrations were not examined. | |||||
| Polyamide: | |||||
| Fluorescein conjugate | FISH-like | Highly repeated sequences | None | Yes | 23,24 |
| Thiazole orange | Oligo targets | 1 nMa | None | Yes | 25 |
| Triplex: | |||||
| TISH | FISH-like | Highly repeated sequences | Polypurine tracts | Yes | 31 |
| Padlock-FITC | Spread molecules | Single molecule | Polypurine tracts | No, heat requirement | 33 |
| Padlock-radiolabeled | Southern | <1 fM | Polypurine tracts | No, heat requirement | 34 |
| YOYO-1 | Amplified DNA | 4 nMa | None | No, intercalator | 37 |
| Protein: | |||||
| Hin-oxazole yellow | Oligo targets | 50 nMa | Hin sites | Delivery? | 47 |
| EcoRI–nano | Spread molecules | Single molecule | EcoRI sites | No, cleavage | 48 |
| GFP–lac | Live cell | 256 tandem repeats | lac sites | Yes | 49 |
| SEER-GFP | Oligo targets | 2.5 µMa | Few | Yes | 53 |
| SEER-LAC | Oligo targets | 20 nM | Few | Yes | 51 |
| mCpG SEER | Oligo targets | 50 nM | Few | Yes | 52 |
Beyond detection
While any of these methods might eventually be suitable for dsDNA detection, SEER offers several additional capabilities. The ability to use other types of DNA-binding domains enables the recognition of other types of information, such as DNA methylation (mCpG-SEER), adducts and damage. In cells, the ability to reassemble enzymatic functions in response to genotype could have applications for gene therapy. Also, the use of cytotoxic substrates81 or enzymes in cells could enable sequence-dependent cell killing, with applications for therapeutics.Conclusions
If the field of DNA diagnostics is in its infancy, methods for the direct detection of dsDNA are embryonic. Most of the recent work is still proof-of-concept experiments. This is partially technological. TFO have been used since the 1980s, polyamides were developed in the mid 1990s and the first generally accessible methods for engineering zinc finger proteins were described in 1999. Polyamides have the least sequence recognition restrictions, but are generally limited to short (i.e. multiple) target sites. TFO can recognize long (i.e. unique) target sites, but are generally limited to polypurine tracts. Engineered zinc finger proteins can be designed to long target sites with less sequence restrictions, but are still currently unable to target all possible sequences. However, the pace and scope of new investigations continued to increase, and some promising candidate methods (such as triplex with YOYO-1) are already entering commercial development. The field is benefiting from intensifying research in other applications of dsDNA recognition technology, primarily in the areas of gene regulation and gene disruption/correction. There is also a growing consensus to bridge the gap between the basic scientists who develop these methods but are unfamiliar with their applications, and the clinicians who would use these methods but are unfamiliar with the technological challenges. The greatest progress will certainly be made by the collaboration between these two camps.Acknowledgements
C.I.S. was supported by an NIH training grant. D.J.S. and I.G. were supported by GM077403 and CA122630. We thank Bogdan Olenyuk for helpful comments on this manuscript.References
- S. D. Atkins and I. M. Clark, J. Appl. Genet., 2004, 45, 3–15 Search PubMed.
- M. M. Lopez, E. Bertolini, A. Olmos, P. Caruso, M. T. Gorris, P. Llop, R. Penyalver and M. Cambra, Int. Microbiol., 2003, 6, 233–243 Search PubMed.
- P. E. Nielsen, Curr. Med. Chem., 2001, 8, 545–550 CAS.
- J. M. Levsky and R. H. Singer, J. Cell Sci., 2003, 116, 2833–2838 Search PubMed.
- P. B. Dervan, Bioorg. Med. Chem., 2001, 9, 2215–2235 CrossRef CAS.
- P. P. Chan and P. M. Glazer, J. Mol. Med., 1997, 75, 267–282 CrossRef CAS.
- D. Segal and C. F. Barbas III, Curr. Opin. Biotechnol., 2001, 12, 632–637 CrossRef CAS.
- J. D. Hoheisel, Nat. Rev. Genet., 2006, 7, 200–210 CrossRef CAS.
- F. M. De la Vega, K. D. Lazaruk, M. D. Rhodes and M. H. Wenz, Mutat. Res., 2005, 573, 111–135 CrossRef.
- H. Stender, Expert. Rev. Mol. Diagn., 2003, 3, 649–655 Search PubMed.
- S. Howorka, S. Cheley and H. Bayley, Nat. Biotechnol., 2001, 19, 636–639 CrossRef CAS.
- W. Vercoutere, S. Winters-Hilt, H. Olsen, D. Deamer, D. Haussler and M. Akeson, Nat. Biotechnol., 2001, 19, 248–252 CrossRef CAS.
- J. M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932–5933 CrossRef CAS.
- K. Dore, M. Leclerc and D. Boudreau, J. Fluoresc., 2006, 16, 259–265 CrossRef CAS.
- T. J. Hellyer and J. G. Nadeau, Expert. Rev. Mol. Diagn., 2004, 4, 251–261 Search PubMed.
- V. V. Demidov, Expert. Rev. Mol. Diagn., 2005, 5, 477–478 Search PubMed.
- L. Bissonnette and M. G. Bergeron, Expert. Rev. Mol. Diagn., 2006, 6, 433–450 Search PubMed.
- K. K. Jain, Expert. Rev. Mol. Diagn., 2003, 3, 153–161 Search PubMed.
- R. Zhao, Clin. Lab. Sci., 2005, 18, 254–262 Search PubMed.
- M. L. Kopka, C. Yoon, D. Goodsell, P. Pjura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 1376–1380 CAS.
- M. Mrksich and P. B. Dervan, J. Am. Chem. Soc., 1993, 115, 9892–9899 CrossRef CAS.
- S. White, J. W. Szewczyk, J. M. Turner, E. E. Baird and P. B. Dervan, Nature, 1998, 391, 468–471 CrossRef CAS.
- S. Janssen, T. Durussel and U. K. Laemmli, Mol. Cell, 2000, 6, 999–1011 CrossRef CAS.
- K. Maeshima, S. Janssen and U. K. Laemmli, EMBO J., 2001, 20, 3218–3228 CrossRef CAS.
- E. J. Fechter, B. Olenyuk and P. B. Dervan, J. Am. Chem. Soc., 2005, 127, 16685–16691 CrossRef CAS.
- V. C. Rucker, S. Foister, C. Melander and P. B. Dervan, J. Am. Chem. Soc., 2003, 125, 1195–1202 CrossRef CAS.
- C. L. Warren, N. C. Kratochvil, K. E. Hauschild, S. Foister, M. L. Brezinski, P. B. Dervan, G. N. Phillips, Jr. and A. Z. Ansari, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 867–872 CrossRef CAS.
- T. Le Doan, L. Perrouault, D. Praseuth, N. Habhoub, J. L. Decout, N. T. Thuong, J. Lhomme and C. Helene, Nucleic Acids Res., 1987, 15, 7749–7760 CrossRef CAS.
- H. E. Moser and P. B. Dervan, Science, 1987, 238, 645–650 CrossRef CAS.
- A. J. Cheng, J. C. Wang and M. W. Van Dyke, Antisense Nucleic Acid Drug Dev., 1998, 8, 215–225 CAS.
- M. D. Johnson, Chromosoma, 1999, 108, 181–189 CrossRef CAS.
- C. Escude, T. Garestier and C. Helene, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10603–10607 CrossRef CAS.
- B. Geron-Landre, T. Roulon, P. Desbiolles and C. Escude, Nucleic Acids Res., 2003, 31, e125 CrossRef.
- B. Geron-Landre, T. Roulon and C. Escude, FEBS J., 2005, 272, 5343–5352 Search PubMed.
- J. S. Li, F. X. Chen, R. Shikiya, L. A. Marky and B. Gold, J. Am. Chem. Soc., 2005, 127, 12657–12665 CrossRef CAS.
- T. Roulon, C. Helene and C. Escude, Angew. Chem., Int. Ed., 2001, 40, 1523–1526 CrossRef CAS.
- J. I. Daksis and G. H. Erikson, Genet. Test., 2005, 9, 111–120 CrossRef CAS.
- E. Cassuto, S. C. West, J. Podell and P. Howard-Flanders, Nucleic Acids Res., 1981, 9, 4201–4210 CrossRef CAS.
- B. J. Rao, S. K. Chiu and C. M. Radding, J. Mol. Biol., 1993, 229, 328–343 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS.
- Y. Shigemori, H. Haruta, T. Okada and M. Oishi, Genome Res., 2004, 14, 2478–2485 CrossRef CAS.
- E. S. Baker, J. W. Hong, B. S. Gaylord, G. C. Bazan and M. T. Bowers, J. Am. Chem. Soc., 2006, 128, 8484–8492 CrossRef CAS.
- P. Schatz, J. Distler, K. Berlin and M. Schuster, Nucleic Acids Res., 2006, 34, e59 CrossRef.
- E. Y. Chan, N. M. Goncalves, R. A. Haeusler, A. J. Hatch, J. W. Larson, A. M. Maletta, G. R. Yantz, E. D. Carstea, M. Fuchs, G. G. Wong, S. R. Gullans and R. Gilmanshin, Genome Res., 2004, 14, 1137–1146 CrossRef CAS.
- K. E. Lundin, L. Good, R. Stromberg, A. Graslund and C. I. Smith, Adv. Genet., 2006, 56, 1–51 Search PubMed.
- J. C. Venter, Science, 2001, 291, 1304–1351 CrossRef CAS.
- M. Thompson, Bioconjugate Chem., 2006, 17, 507–513 CrossRef CAS.
- J. R. Taylor, M. M. Fang and S. Nie, Anal. Chem., 2000, 72, 1979–1986 CrossRef CAS.
- C. C. Robinett, A. Straight, G. Li, C. Willhelm, G. Sudlow, A. Murray and A. S. Belmont, J. Cell Biol., 1996, 135, 1685–1700 CrossRef CAS.
- A. S. Gonzalez-Serricchio and P. W. Sternberg, BMC Genet., 2006, 7, 36 Search PubMed.
- A. T. Ooi, C. I. Stains, I. Ghosh and D. J. Segal, Biochemistry, 2006, 45, 3620–3625 CrossRef CAS.
- C. I. Stains, J. L. Furman, D. J. Segal and I. Ghosh, J. Am. Chem. Soc., 2006, 128, 9761–9765 CrossRef CAS.
- C. I. Stains, J. R. Porter, A. T. Ooi, D. J. Segal and I. Ghosh, J. Am. Chem. Soc., 2005, 127, 10782–10783 CrossRef CAS.
- S. A. Wolfe, L. Nekludova and C. O. Pabo, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 183–212 CrossRef CAS.
- R. R. Beerli and C. F. Barbas, Nat. Biotechnol., 2002, 20, 135–141 CrossRef CAS.
- C. O. Pabo, E. Peisach and R. A. Grant, Annu. Rev. Biochem., 2001, 70, 313–340 CrossRef CAS.
- B. Dreier, R. R. Beerli, D. J. Segal, J. D. Flippin and C. F. Barbas III, J. Biol. Chem., 2001, 276, 29466–29478 CrossRef CAS.
- B. Dreier, R. P. Fuller, D. J. Segal, C. V. Lund, P. Blancafort, A. Huber, B. Koksch and C. F. Barbas, J. Biol. Chem., 2005, 280, 35588–35597 CrossRef CAS.
- D. J. Segal, B. Dreier, R. R. Beerli and C. F. Barbas III, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2758–2763 CrossRef CAS.
- M. Mani, K. Kandavelou, F. J. Dy, S. Durai and S. Chandrasegaran, Biochem. Biophys. Res. Commun., 2005, 335, 447–457 CrossRef CAS.
- D. J. Segal, Methods, 2002, 26, 76–83 Search PubMed.
- I. Remy, A. Galarneau and S. W. Michnick, Methods Mol. Biol., 2002, 185, 447–459 Search PubMed.
- I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2000, 122, 5658–5659 CrossRef CAS.
- W. Gao, B. Xing, R. Y. Tsien and J. Rao, J. Am. Chem. Soc., 2003, 125, 11146–11147 CrossRef CAS.
- A. Galarneau, M. Primeau, L. E. Trudeau and S. W. Michnick, Nat. Biotechnol., 2002, 20, 619–622 CrossRef CAS.
- A. Bird, Cell, 1992, 70, 5–8 CrossRef CAS.
- A. P. Bird, Nucleic Acids Res., 1980, 8, 1499–1504 CrossRef CAS.
- R. Jaenisch and A. Bird, Nat. Genet., 2003, 33 Suppl, 245–254 CrossRef.
- S. J. Clark, J. Harrison, C. L. Paul and M. Frommer, Nucleic Acids Res., 1994, 22, 2990–2997 CrossRef CAS.
- Y. Furuichi, Y. Wataya, H. Hayatsu and T. Ukita, Biochem. Biophys. Res. Commun., 1970, 41, 1185–1191 CrossRef CAS.
- J. G. Herman, J. R. Graff, S. Myohanen, B. D. Nelkin and S. B. Baylin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- Z. Xiong and P. W. Laird, Nucleic Acids Res., 1997, 25, 2532–2534 CrossRef CAS.
- A. M. Raizis, F. Schmitt and J. P. Jost, Anal. Biochem., 1995, 226, 161–166 CrossRef CAS.
- H. Thomassin, E. J. Oakeley and T. Grange, Methods, 1999, 19, 465–475 Search PubMed.
- T. Rein, D. A. Natale, U. Gartner, M. Niggemann, M. L. DePamphilis and H. Zorbas, J. Biol. Chem., 1997, 272, 10021–10029 CrossRef CAS.
- M. F. Fraga and M. Esteller, Biotechniques, 2002, 33, 632, 634, 636–649.
- P. M. Warnecke, C. Stirzaker, J. Song, C. Grunau, J. R. Melki and S. J. Clark, Methods, 2002, 27, 101–107 Search PubMed.
- H. J. Tanke, R. W. Dirks and T. Raap, Curr. Opin. Biotechnol., 2005, 16, 49–54 CrossRef CAS.
- T. G. Uil, H. J. Haisma and M. G. Rots, Nucleic Acids Res., 2003, 31, 6064–6078 CrossRef CAS.
- A. F. Kolb, C. J. Coates, J. M. Kaminski, J. B. Summers, A. D. Miller and D. J. Segal, Trends Biotechnol., 2005, 23, 399–406 CrossRef CAS.
- V. M. Vrudhula, D. E. Kerr, N. O. Siemers, G. M. Dubowchik and P. D. Senter, Bioorg. Med. Chem. Lett., 2003, 13, 539–542 CrossRef CAS.
Footnote |
| † Abbreviations: ZF, zinc finger; GFP, green fluorescent protein; PNA, peptide nucleic acid; dsDNA, double-stranded DNA; TFO, triplex-forming oligonucleotide; SNP, single nucleotide polymorphism; SEER, sequence-enabled reassembly. |
| This journal is © The Royal Society of Chemistry 2006 |