A GFP-based assay for monitoring post-transcriptional regulation of ARE-mRNA turnover
Don
Benjamin
,
Marco
Colombi
,
Georg
Stoecklin
and
Christoph
Moroni
*
Institute for Medical Microbiology, Petersplatz 10, 4003 Basel, Switzerland. E-mail: christoph.moroni@unibas.ch; Fax: +41 61 2673283; Tel: +41 61 2673264
First published on 27th September 2006
Abstract
Interleukin-3 (IL3) mRNA is intrinsically labile due to the presence of a destabilizing AU-rich element (ARE) that targets the transcript for rapid degradation. We review our experience with a sensitive reporter system where changes in IL3 mRNA stability are translated into increased/decreased green fluorescent protein (GFP) signals. A GFP reporter gene was fused to the full-length IL3 3′UTR containing the ARE motif that responds to regulatory signals that control transcript stability. The reporter system was tested against known IL3 mRNA stabilizing/destabilizing agents either through pharmacological treatment, siRNA knock-down of components of the decay machinery, mutation of the ARE motif, or in tumour lines harbouring stable IL3 mRNA. In all cases, the reporter transcript responds in an identical fashion to the endogenous IL3 message thereby verifying the fidelity of the system. This reporter system allows screening and identification of novel ARE-mRNA stabilizing compounds, or the isolation of mutants defective in ARE-mRNA turnover. We also report preliminary attempts to modify the system for high-throughput screening of an extensive compound library. The simplicity and effectiveness of this screen makes it ideal for screening of modulators of clinically important ARE-bearing transcripts such as TNFα, VEGF, the interferons and other cytokines.
Introduction
Post-transcriptional regulation is an extra layer of control that determines the ultimate levels of gene expression under given conditions in a cell. A major post-transcriptional mechanism is mRNA turnover where the balance between the rate of transcription and mRNA degradation sets the actual gene expression level.1 Transcripts under post-transcriptional regulation are intrinsically unstable, in most cases this is due to the presence in the 3′UTR of a destabilizing AU-rich element (ARE). An estimated 5–8% of genes contain putative AREs, many of these are of clinical significance such as cytokines, interferons, cell-cycle regulators and proto-oncogenes.2 Under conditions where the activity of these genes are urgently required, the mRNA can be stabilized causing rapid accumulation in mRNA and protein levels.3 Therefore it is important to be able to measure the rate of mRNA decay for a gene of interest.The classical method of measuring mRNA decay rates are by decay chase assays. This involves a transcription block (usually with the RNA polymerase inhibitor actinomycin D), and RNA extraction at appropriate intervals. Northern hybridization with a specific probe allows measurement of the decay rate for a transcript of interest. A drawback to this method is that it is labour-intensive and time consuming. In order to quickly and simply measure changes in mRNA stability for our gene of interest, interleukin-3 (IL3), we have devised a green fluorescent protein (GFP)-based assay that is rapid, non-invasive, and responds to pharmacologically induced stabilization, siRNA targeting of the decay apparatus, as well as reflecting oncogenic or other mutations that affect IL3 mRNA stability. The system allows adaptation to high-throughput screening for compounds or mutations that affect ARE-mRNA turnover.
Results
Establishment of a GFP-ARE mRNA reporter system in PB3c mast cells
mRNA transcripts are destabilized by the presence of an ARE that targets the transcript for rapid degradation. Transcripts of a GFP reporter gene fused to the IL3-3′UTR that contains a well-defined and functional ARE4 (Fig. 1A) should undergo similar post-transcriptional regulation as the IL3 mRNA and hence be unstable, but nevertheless, become stabilized under conditions where the endogenous IL3 transcript is stable. This is reflected as an increase in GFP fluorescence as visualized by fluorescence activated cell sorting (FACS) analysis (Fig. 1B), or vice versa in the event of further destabilization of the already labile mRNA.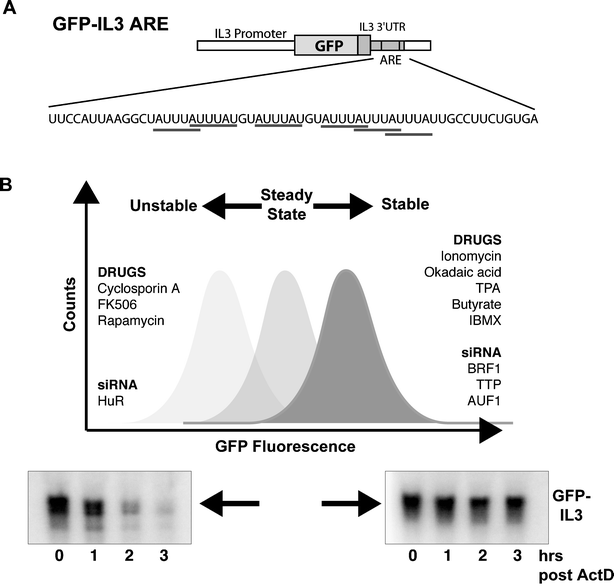 | ||
| Fig. 1 (A) The GFP-reporter construct consisting of the EGFP coding sequence fused to the full-length IL3 3′UTR. The 59 nt ARE sequence contains 6 AUUUA motifs. (B) GFP fluorescence of the GFP-ARE reporter cell-line increases under conditions where the GFP-ARE mRNA is stabilized, resulting in increased GFP protein synthesis and vice versa. A list of treatments (discussed in the text) that modulate IL3 mRNA stability is shown. mRNA decay chase assay of cells undergoing various treatments followed by northern hybridisation with an IL3 specific probe confirms if the reporter transcript is stabilized/destabilized. | ||
PB3c murine mastocytes are an ideal background to study and manipulate IL3 ARE-mRNA stability as they rapidly degrade IL3 mRNA, yet respond to stabilizing agents such as ionomycin.5 In addition, PB3c derived tumors are available with an intrinsic defect in ARE-mRNA decay and correspondingly stable IL3 mRNA.6 The GFP-IL3 3′UTR reporter construct was transfected into a PB3c subclone, PB3c-15, and GFP expressing cells were screened by FACS to identify clones having a sharp, well-formed peak for GFP fluorescence. In our experience, great variation in the level and signal-to-noise ratio of GFP fluorescence is obtained after the initial transfection, most probably due to the site of integration of the construct. Therefore it is necessary to first select for GFP expressing clones by fluorescent microscopy followed by FACS screening to identify clones having smooth, well-defined peaks. Several suitable clones were obtained and one, designated 15-GFP-IL3ARE, was selected for further use (Fig. 2A).
 | ||
| Fig. 2 (A) Untransfected PB3c-15 cells have low auto-fluorescence, after transfection with the GFP-IL3 ARE construct, the mass culture shows considerable variation in fluorescence intensity. Clone 15-GFP-IL3ARE was selected after fluorescence activated cell sorting (FACS), sub-cloning, expansion and FACS analysis. (B) A control cell line, 15-GFP-ΔARE, is deleted for the ARE and produces more GFP due to increased mRNA stability. (C) The reporter cell-line shows a shift towards increased GFP expression after ionomycin treatment (1 μM, 4 h). (D) Similar stimulation as in (C) was performed on the ΔARE control cell-line. (E) The GFP-reporter was stably expressed in PB3c-clone 20 and a tumorigenic derivative, V2D1, which is defective in IL3 mRNA decay. The reporter transcript behaves as expected in the precursor and tumor cell backgrounds as confirmed by FACS analysis and mRNA decay assays. (F) Destabilization of ARE-mRNA by FK506 (1 μg ml−1) can be visualized in this reporter system as a decrease in GFP fluorescence. | ||
In parallel, a construct deleted for the ARE was also transfected and selected as described. GFP levels from clones expressing the ΔARE reporter are substantially higher due to higher reporter transcript stability and confirm the ARE-dependence of the assay system (Fig. 2B).
To test whether the reporter cell line faithfully reflects known conditions for IL3 mRNA stabilization, cells were stimulated with ionomycin for 4 hours before FACS analysis. The fluorescence peak displayed a large shift to the right consistent with ionomycin-induced stabilization of the reporter mRNA resulting in increased GFP levels (Fig. 2C). Independent confirmation was obtained by actinomycin D RNA decay chase experiments that showed reporter mRNA stabilization.7 As a specificity control for transcriptional effects on reporter gene expression, the 15-GFP-ΔARE cell line was also stimulated with ionomycin followed by FACS analysis (Fig. 2D). Only a minor shift is observed compared to that previously obtained by actual mRNA stabilization.
Oncogenic transformation of PB3c can also constitutively stabilize IL3 mRNA.6 Therefore the system was tested in V2D1 cells, a PB3c-derived tumor cell line with stable IL3 mRNA. Gratifyingly, the GFP-IL3 transcript in this background was stable as expected but labile in the non-transformed precursor cells (Fig. 2E). IL3 mRNA can be destabilized in V2D1 by FK506. Therefore we treated V2D1-GFP-IL3 cells with FK506 for 72 hours before proceeding with FACS analysis. The longer treatment was required due to stability of pre-existing GFP protein which has to be cleared before the actual steady-state protein levels can be measured. The predicted backshift in GFP fluorescence was observed (Fig. 2F), thereby confirming that this system faithfully recapitulates known events in IL3 mRNA stabilization and can therefore be used to monitor post-transcriptional regulation.
Validation of the GFP-ARE reporter in other contexts
The GFP-IL3 reporter in human HT1080 fibrosarcoma cells responds to stabilization by okadaic acid (Fig. 3A).7 A heterologous GFP-reporter system using the TNFα-ARE in murine RAW264.7 macrophages could also be stabilized upon exposure to bacterial lipopolysaccharide (LPS, a known stabilizer of TNFα mRNA, Fig. 3B).8 In both cases, reporter RNA stabilization was verified by actinomycin D decay assays (data not shown).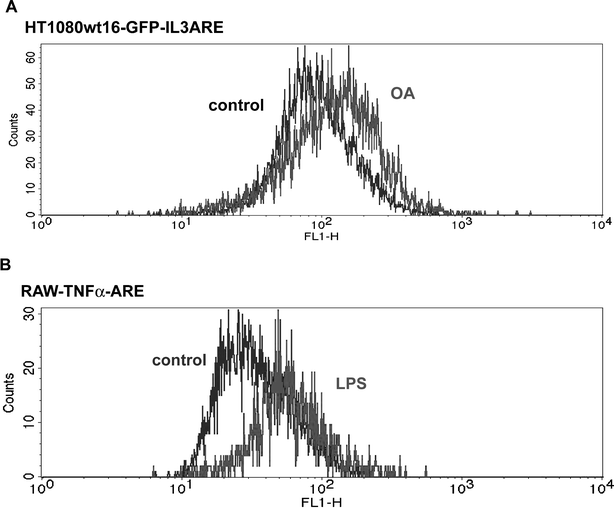 | ||
| Fig. 3 (A) The GFP-IL3 reporter was inserted in HT1080 cells and a clone, wt16-GFP-IL3ARE, was selected for further study. Reporter cells stimulated with okadaic acid (OA, 100 nM, 4 h) showed increased GFP fluorescence due to mRNA stabilization. (B) A heterologous system using the TNFα-ARE in RAW264.7 macrophages. Stimulation with LPS (2 μg ml−1, 8 h) gave the expected GFP shift due to increased TNFα-ARE mRNA stability. | ||
Application of the GFP-ARE assay
We have used the reporter system to identify agents that modulate IL3 mRNA stability. A screen for IL3 mRNA stabilizing compounds in PB3c cells identified okadaic acid.7 As shown in Fig. 3A, it also stabilizes the reporter in HT1080 cells suggesting conservation of the regulatory mechanism. siRNA mediated knockdown of genes implicated in the mRNA turnover pathway was performed in the HT1080-based reporter system and allowed demonstration of genes having a stabilizing or destabilizing effect.9A more innovative application of the system is to obtain mutants defective in ARE-mRNA decay. This exploits the non-invasive nature of the assay, and the ability to sort and enrich for cells having a desired property (Fig. 4A). We previously described mutagenesis of HT1080wt16-GFP-IL3 which yielded three mRNA turnover mutants.10 Retroviral cDNA library transfection revealed the lesion in one clone as BRF1, an ARE-binding protein with mRNA decay promoting activity.11 A similar procedure was applied to PB3c-15 cells in the hope of obtaining additional mutants. After several rounds of mutagenesis with the frame-shift mutagen ICR 191, cells were sorted by flow cytometry and a small fraction of cells (0.02 to 0.05%) displaying fluorescence emission above an arbitrarily chosen threshold were directly sub-cloned (Fig. 4B). After expansion, clones were inspected by fluorescence microscopy to select candidates for further confirmation by FACS. Clones expressing GFP at least two-fold more than basal levels were retained and tested by mRNA decay chase assays to confirm that the increase is due to the ablation of some component of the ARE-mRNA machinery by mutation, resulting in reporter transcript stabilization. Work is presently underway on further characterizing these high-GFP expressing clones.
 | ||
| Fig. 4 (A) Strategy for obtaining ARE-mRNA decay mutants.10,11 Mutagenized cells with increased GFP are sorted by FACS and genuine mutants confirmed by mRNA decay chase assays. (B) Representative example of sorting procedures. The pool of mutagenized PB3c-15 cells shows variable GFP expression. High GFP-expressing cells are collected by FACS-sorting (M, highest 0.05%) and sub-cloned for further characterisation of clones containing a defect in ARE-mRNA turnover. | ||
Adaptation of the GFP-assay for high-throughput screening
While the screening approach could be successfully applied on a small-scale, a large-scale high-throughput screen (HTS) of even a modest compound library (around 104 compounds) would be onerous as FACS analysis does not lend itself easily to automation.We therefore adapted the system to a plate-reading format that is compatible with HTS. Cell-culture medium gives high background fluorescence at the excitation/emission spectra of GFP, hence it is necessary to resuspend the cells in a fluorometrically neutral medium such as PBS prior to measurement. Fluorescence of the reporter cells is proportional to cell numbers over a wide range for plate reading (Fig. 5A). PB3c grows in suspension, and unfortunately, washing and resuspension of the cells results in unequal cell numbers per well with unacceptable variability in GFP measurement. Therefore we switched to the adherent reporter cell lines: GFP-IL3ARE in HT1080 and GFP-TNFα in RAW macrophages. Readings were acquired with a FLIPRtetra (Molecular Devices) and Safire2 (Tecan) fluorometric plate readers, two platforms that are commonly integrated into a HTS. As seen in Fig. 5B and C, both methods give robust measurements with good signal-to-noise ratios that are consistent with expectations, indicating that the GFP–ARE assays are reliable in a plate-reading format and hence can feasibly be used for HTS.
 | ||
Fig. 5 (A) Low and high GFP expressing cells, (15-GFP-IL3ARE vs. VG59) were pelleted, resuspended in PBS+2% FCS and counted with a Coulter counter. Increasing numbers of cells were plated in a 96-well plate for fluorescence measurement (Safire2, Tecan). (B) RAW-GFP-TNFα ARE cells were stimulated with LPS (2 μg ml−1, 6 h). Medium was aspirated and replaced with 40 μl PBS before fluorescence reading (Safire2). Empty wells contain PBS. (C) HT1080 wt16-GFP-IL3ARE cells were stimulated with OA (4 h) before fluorescence measurement with a FLIPRtetra plate reader. Optimum signal-to-background ratios were obtained at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells/well. Values shown are means for arbitrary fluorescence units (FU) ± SD. 000 cells/well. Values shown are means for arbitrary fluorescence units (FU) ± SD. | ||
Discussion
Post-transcriptional regulation is a rapid and dynamic process allowing cells to set the level of gene expression independently of transcription. This mechanism is of biomedical interest and represents an as yet poorly understood mechanism for pharmacological intervention. We have used GFP levels to track changes in reporter mRNA stability as it is non-invasive and therefore ideal for monitoring on-going progress throughout the course of an experiment. In addition, mRNA stabilization is not an all-or-nothing process and different levels of stabilization are attained as post-transcriptional regulation only modulates the level of gene expression without switching it completely on or off. Changing GFP levels, that can be measured in real-time, provides a simple and effective method to detect such changes in mRNA stability.Nevertheless, there are several technical innovations that can be implemented to improve the reporter assay. For high-throughput screening, it would be desirable to construct a three-colour reporter cell-line for data normalization and to minimize the number of false positive hits. Such a cell-line would express the unmodified GFP vector construct as a fluorescence marker for determination of cell numbers. A second fluorescent protein would be linked to the ARE-bearing 3′UTR of the gene of interest and serve as the actual reporter construct. The third colour fluorescence marker would be linked to an ARE-deleted version of the same 3′UTR and serve as an internal control against possible transcriptional activation by the test compounds. Of course, judicious selection of the colours to be used must be exercised and it may be necessary to screen for clones expressing the various markers at just the right ratio in order to establish the reporter system.
Another consideration, is that in terms of clinical significance, it is often more important to be able to detect destabilization of an abnormally stable ARE-mRNA. This is true for inflammation/rheumatoid arthritis and angiogenesis where pathologically stable TNFα and VEGF mRNA is responsible for disease progression.12–14 The current assay requires at least 48–72 hours for clearance of pre-existing GFP protein before any reduction in mRNA levels can be accurately reflected (Fig. 1F). In a screening situation, this could conceivably allow for the development of non-specific secondary effects. However the commercial availability of unstable GFP variants or addition of destabilizing domains such as the PEST sequence could provide a solution.15
Another advantage of the system is its simplicity, with a bare minimum of steps required prior to measurement, which not only ensures a rapid and less labour-intensive means of obtaining data, but also renders the system amenable to automation for HTS.
Experimental
Cell culture
Cells were cultured in Iscove's medium supplemented with 10%FCS, 2 mM L-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 50 μM 2-mercaptoethanol. Mast cell medium was supplemented with 1% conditioned medium from X63-mIL3 cells as an IL3 source.5 Stable mast cell transfectants were obtained by electroporation with a GenePulser (BioRad). All other cells were transfected with Lipofectamine 2000 (Invitrogen).Plasmid constructs
All constructs used pEGFP-N1 (Clontech) as the backbone. Detailed construction of the IL3 ARE, ΔARE and TNFα ARE constructs have been described.8,10,11Flow cytometry and fluorescence measurement
Cells were resuspended in 200 ml PBS containing propidium iodide (5 μg ml−1). 104 cells were counted using a FACScalibur (Becton Dickinson) running Cellquest software. GFP fluorescence was excited at 488 nm and emission measured with a 510 nm filter. Propidium iodide staining was detected with a 580 nm filter and allowed exclusion of dead cells and cellular debris from data analysis. Cells were sorted using a FACSorter (Becton Dickinson), during sorting cells were kept in FCS-free medium at 4 °C and were either expanded for further sorting or directly sub-cloned in 60-well plates containing full medium. After recovery and expansion, clones were inspected by fluorescence microscopy to select candidates for further analysis.Two fluorescent plate readers were evaluated: Safire2 (Tecan) and a FLIPRTetra (Molecular Devices). A full-spectrum scan of the excitation/emission spectra identified Ex 488/Em 515 as optimal and was subsequently employed. Cells were cultured in 96-well plates (Costar, black, transparent bottom, poly-lysine coated). Prior to measurement, the plates were spun in a cytospin, medium aspirated and replaced with 40 μl PBS. Readings were taken from the bottom with multiple reads (12 flashes) per well.
Acknowledgements
We thank Hugo Albrecht and Urs Regenass (DPI, Allschwil) for discussions and access to plate reading instrumentation. This work was partly supported by a Swiss National Foundation grant to C.M.References
- J. Guhayinogi and G. Brewer, Gene, 2001, 265, 11–23 CrossRef.
- T. Bakheet, B. R. Williams and K. S. Khabar, Nucleic Acids Res., 2003, 31, 421–423 CrossRef CAS.
- J. Shim and M. Karin, Mol. Cells, 2002, 14, 323–331 Search PubMed.
- G. Stoecklin, S. Hahn and C. Moroni, J. Biol. Chem., 1994, 269, 28591–28597 CAS.
- A. Wodnar-Filipowicz and C. Moroni, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 777–781 CrossRef CAS.
- H. Hirsch, A. Nair, V. Backenstoss and C. Moroni, J. Biol. Chem., 1995, 270, 20629–20635 CrossRef CAS.
- D. Benjamin, M. Colombi and C. Moroni, Nucleic Acids Res., 2004, 32, e89 CrossRef.
- G. Stoecklin, M. Lu, B. Rattenbacher and C. Moroni, Mol. Cell. Biol., 2003, 23, 3506–3515 CrossRef CAS.
- I. Raineri, D. Wegmueller, B. Gross, U. Certa and C. Moroni, Nucleic Acids Res., 2004, 32, 1279–1288 CrossRef CAS.
- G. Stoecklin, X. F. Ming, R. Looser and C. Moroni, Mol. Cell. Biol., 2000, 20, 3753–3763 CrossRef CAS.
- G. Stoecklin, M. Colombi, I. Raineri, S. Leuenberger, M. Mallaun, M. Schmidlin, B. Gross, M. Lu, T. Kitamura and C. Moroni, EMBO J., 2002, 21, 4709–4718 CrossRef CAS.
- F. Lejbkowicz, I. Goldberg-Cohen and A. P. Levy, Acta Histochem., 2005, 106, 405–411 Search PubMed.
- L. B. Nabors, E. Suswam, Y. Huang, X. Yang, M. J. Johnson and P. H. King, Cancer Res., 2003, 63, 4181–4187 CAS.
- J. A. Baugh and R. Bucala, Curr. Opin. Drug Discovery Dev., 2001, 4, 635–650 CAS.
- X. Li, X. Zhao, Y. Fang, X. Jiang, T. Duong, C. Fan, C. C. Huang and S. R. Kain, J. Biol. Chem., 1998, 273, 34970–34975 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |