Proteome-on-a-chip: Mirage, or on the horizon?
Sergio L. S.
Freire
a and
Aaron R.
Wheeler
*abc
aDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5S 3H6, Canada. E-mail: awheeler@chem.utoronto.ca; Fax: (416) 946-3865; Tel: (416) 946-3864
bInstitute for Biomaterials and Biomedical Engineering, University of Toronto, 164 College St., Toronto, ON M5S 3G9, Canada
cBanting and Best Department of Medical Research, University of Toronto, 112 College St., Toronto, ON M5G 1L6, Canada
First published on 5th September 2006
Abstract
Proteomics has emerged as the next great scientific challenge in the post-genome era. But even the most basic form of proteomics, proteome profiling, i.e., identifying all of the proteins expressed in a given sample, has proven to be a demanding task. The proteome presents unique analytical challenges, including significant molecular diversity, an extremely wide concentration range, and a tendency to adsorb to solid surfaces. Microfluidics has been touted as being a useful tool for developing new methods to solve complex analytical challenges, and, as such, seems a natural fit for application to proteome profiling. In this review, we summarize the recent progress in the field of microfluidics in four key areas related to this application: chemical processing, sample preconcentration and cleanup, chemical separations, and interfaces with mass spectrometry. We identify the bright spots and challenges for the marriage of microfluidics and proteomics, and speculate on the outlook for progress.
Introduction
The push to sequence the human genome1 brought an unprecedented level of attention to the field of genomics, which, in-turn, has spawned an ever-increasing assortment of “–omics” disciplines (e.g., proteomics, metabolomics, transcriptomics, etc.). While the merits of this exercise in jargon generation are not clear, one effect will likely resonate for many years to come, namely, the emergence of the proteome, defined as the complement of all proteins expressed in a system, as one of the most important and pervasive research topics in all of science. From its first conceptualization in the mid 1990s,2 the field of proteomics has undergone a meteoric rise in popularity, leading to the publication of more than 6000 experimental papers in the field in the past five years.3 One reason for this prominence is the intimate relationship between disease states and protein constitution and dynamics.4 Another reason is the paradigm shift in thinking about the relationship between genome and proteome; we now know that the proteome owes as much to the wide range of post-translational modifications of proteins (i.e., phosphorylation, acetylation, sulfation, glycosylation, myristoylation, conjugation with lipids, and proteolysis), as it does to gene transcription. Indeed, as succinctly summarized in a recent editorial in Nature, “proteins, not genes, are the business end of biology.”5Unfortunately, the increasing importance of proteomics has not been accompanied by a corresponding improvement in analytical tools, especially when compared to genomics. For example, while DNA microarrays6 and multiplexed sequencing7 have ushered in an era of high-throughput genomics, proteomics still relies primarily on the low-throughput technique of two-dimensional (slab) gel electrophoresis (2DGE) combined with mass spectrometry. There are many reasons for the dearth of useful proteomics tools, including: (a) mammalian samples contain over 20,000 different proteins, which requires that proteomics methods be capable of collecting, storing, cataloguing, and analyzing vast amounts of information; (b) many structurally similar (but functionally different) isoforms of proteins may be expressed, complicating absolute identification; (c) a concentration range of six decades or more can separate the lowest abundance (and often the most interesting) from the highest abundance proteins; (d) proteins span a wide range of pH, polarity, and solubility, making global experimental protocols virtually impossible; (e) many proteins adsorb non-specifically to a wide range of surfaces; and (f) proteins are not endowed with an analogue to the nucleic acid property of base-complimentarity. These challenges and others combine to form the major bottleneck in proteomics research: a lack of robust tools capable of collecting and analyzing data on a proteome-wide scale.8 Here, we review the challenges inherent to the most basic form of “profiling” proteomics, viewed through a lens of how microfluidics and related techniques may be able to contribute to the development of the next generation of proteomics tools.
Proteome profiling is the identification of all proteins expressed in a sample. Conventionally, profiles are generated through a series of steps that may require several days of labor-intensive laboratory work.9–11 A typical protocol includes: (1) a sample of proteins is separated by 2DGE, (2) the relevant protein bands are excised, (3) the proteins are chemically processed (which may involve several sequential reactions, including reduction of disulfides, alkylation of thiols, enzymatic digestion, etc.), (4) the sample is purified and concentrated, (5) mass spectra are generated using Electrospray Ionization (ESI) or Matrix Assisted Laser Desorption/Ionization (MALDI), and (6) the protein is identified by its unique peptide mass fingerprint (PMF) using database searching software such as MASCOT.12 Steps 2–6 must be repeated hundreds of times to analyze each of the relevant 2DGE spots.
While 2DGE-based methods are most commonly used, an alternative, called “shotgun proteomics,” has recently become popular.13–15 In this technique, protein samples are (1) chemically processed, (2) purified and concentrated, (3) separated by two-dimensional liquid chromatography (2DLC, typically strong cation exchange, SCX, followed by reversed phase, RP) and (4) analyzed by mass spectrometry. The key advantage of shotgun proteomics is that analytes are eluted directly from the separation column into the mass spectrometer, eliminating the need for band selection and excision (a particularly time-consuming bottleneck for 2DGE-based methods). This advantage comes at a cost, however, as chemical processing prior to separations requires that peptides be fully sequenced by tandem mass spectrometry and identified using very complex algorithms (e.g., SEQUEST14). In addition, the chemical processing step, common to both shotgun proteomics and 2DGE-based methods, prevents the implementation of anything approaching a high-throughput analysis. For example, in a seminal study by the Yates group,13 1,484 proteins from yeast lysate were identified in a single “shot”; however, prior to taking this shot, samples were extensively processed, requiring more than three days of work.†
In the following, we review the state-of-the art in the field of microfluidics as it relates to the potential for the development of new proteomics tools. These new tools stand to benefit from the advantages of microfluidics, including favorable reaction kinetics, capacity to integrate multiple processes, reduced reagent consumption, and decreased analysis time. There are several good, comprehensive reviews of proteomic applications in microfluidics;16–19 there are also reviews of related lab-on-a-chip technologies, such as protein microarrays.20,21 Rather than reproduce these reviews, we focus here on a critical evaluation of selected microfluidics technologies that will be most useful for the development of high-throughput proteome profiling tools. These include approaches for implementing (a) chemical processing of proteins, (b) sample preconcentration and cleanup, (c) separations of peptides and proteins, and (d) interfaces with mass spectrometry.
Chemical processing
The identification of unknown proteins often requires that they be digested (or divided) into a group of constitutive peptides, which are interrogated by mass spectrometry. Comparing the masses of the peptides to genomic databases enables absolute identification by means of peptide mass fingerprinting (PMF). Proteins are digested by exposure to proteolytic enzymes (e.g., trypsin), or other lytic reagents (e.g., cyanogen bromide, CNBr); this is often part of a multistep process requiring 12–18 hours, including (1) mixing the sample with buffer and denaturant, (2) mixing and incubating the sample with reducing agent to reduce disulfide bonds, (3) mixing and incubating the sample with alkylating agent to prevent disulfide bonds from reforming, and finally, (4) mixing and incubating with lytic reagent. This process is extremely tedious and time-consuming, making it an attractive application for microfluidics.22‡Several device configurations have been used for proteomic processing in microfluidic devices, including open channels,23–25 immobilized beads,26–29 and other solid phase media.30–33 In the first report of on-chip proteolytic digestion, Gottschlich et al.23 used open-channel devices for tryptic digestion and reduction of disulfides in proteins. As shown in Fig. 1, this method enabled complete tryptic digestion in 15 minutes. More recently, Huang et al.24 and Liu et al.25 used surface adsorbed trypsin in channels to enable complete digestion in less than 5 seconds. To our knowledge, these are the fastest tryptic digestions that have been reported. To increase the surface-to-volume ratio further, Yue et al.28 used a bed of trypsin-modified agarose beads immobilized in a weir in a glass microfluidic device to digest β-casein. Slentz et al.29 developed a similar method for digestion of bovine serum albumin in a poly(dimethylsiloxane) (PDMS) device. An alternative method for increasing the surface area-to-volume ratio in microfluidic devices is to use trypsin-modified monoliths, formed from polymer plugs cured in-situ, or from membranes sandwiched between channels. Peterson et al.31 developed a methacrylate-based monolith for tryptic digestion of myoglobin in ∼11 seconds. Sakai-Kato et al.32 developed similar methods using trypsin immobilized in a sol-gel matrix. Gao et al.33 reported using a trypsin-modified polyvinylidiene fluoride (PVDF) membrane, in a PDMS microfluidic chip, enabling protein digestion in 3–10 minutes.
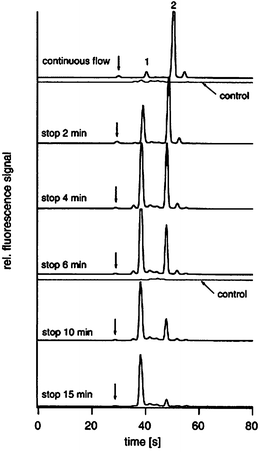 | ||
| Fig. 1 Electropherograms representing the digestion of a sample of insulin B. Peak 2 originates from the undigested peptide, and peak 1 originates from the digested products. As shown, the digestion is complete in ∼15 minutes. Reprinted from Gottschlich et al.,23 copyright 2000, with permission from Elsevier. | ||
Microfluidics is promising for developing tools with integrated chemical processing of proteomic analytes, by virtue of fast reaction kinetics. However, to our knowledge, there have been no reports of implementing a fully integrated process, including stepwise reduction, alkylation, and digestion. Microfluidic tools for chemical processing will likely not be widely adopted until this critical benchmark is achieved.
Sample preconcentration and cleanup
Sample preconcentration and cleanup (i.e., removal of unwanted constituents, such as non-protein biomolecules, surfactants, and buffers) is critical for the detection of low-abundance proteins. Sample preconcentration is particularly important for microfluidics; while reduced volumes are a tremendous advantage for analysis of precious samples, these constraints can pose a problem for detection and analysis of dilute analytes. In recognition of this drawback, several approaches have been developed to implement this critical application in microfluidic devices.One technique that is commonly used for sample concentration and cleanup in microfluidic devices, called sample stacking or isotachophoresis,34–37 is implemented by applying electrical fields to channels containing plugs of buffers with different conductivity. For example, Jung et al.36 report using sample stacking in microchannels to concentrate fluorescent analytes by 1000-fold. In more recent work, the same authors developed isotachophoresis devices capable of detecting the dye, Alexa Fluor 488, present at an initial concentration of 100 fM.37
Another method for sample preconcentration and cleanup is dialysis. In dialysis, samples are driven across a selectively permeable membrane; non-permeating analytes become concentrated at the interface, and can be subsequently analyzed. The drawback of this method (compared to sample stacking) is the necessity of fabricating these kinds of membranes in microfluidic devices. Foote et al.38 recently developed a creative method to implement dialysis in microchannels; by using a silicate adhesive to bond the cover-plate to the channel, a membrane was formed which was permeable to buffer ions but restrictive to larger molecules. Devices constructed in this manner were used to concentrate β-galactosidase and ovalbumin 600-fold. Zhang et al.39 sandwiched a polycarbonate membrane with track-etched pores between microchannels to selectively concentrate proteomic analytes. Finally, Song et al.40 fabricated a membrane at an intersection of microchannels by laser-induced polymerization. As shown in Fig. 2, this device was capable of concentrating proteins with molecular weight larger than the threshold (determined by the pore size in the membrane) by up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold.
000-fold.
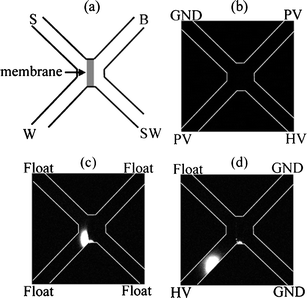 | ||
| Fig. 2 Schematic (a) and pictures (b–d) of a dialysis membrane in a microfluidic device. In (b), fluorescently labeled proteins are driven from S to SW (top left to bottom right). In (c), the concentrated plug of proteins at the membrane is apparent. In (d), the concentrated analyte is injected onto the analysis column (top right to bottom left). Reprinted with permission from Song et al.,40 copyright 2004 American Chemical Society. | ||
A third, and perhaps the most common, method used for sample preconcentration and cleanup is solid phase extraction (SPE).41–46 In SPE, hydrophobic analytes are adsorbed onto a solid hydrophobic medium, allowing hydrophilic contaminants to be rinsed away. The sample is then desorbed in a nonpolar elution buffer. SPE media are typically formed in microfluidic devices by means of (a) a packed bed of functionalized beads, (b) an in-situ polymerized porous monolith, or (c) a pre-formed porous membrane. Ramsey et al.44 implemented SPE in microfluidic channels by using a packed bed of beads and were able to detect samples with an initial concentration of 100 pM of analyte. Yu et al.45 reported forming two SPE beds by means of a monolithic polymer in microfluidic columns, for sample enrichment based on charge (ion exchange) and polarity (hydrophobic interactions). These devices were able to concentrate peptides and green fluorescent protein (GFP) by a factor of 1000, and were reproducible for hundreds of injections. Finally, Lion et al.46 integrated a hydrophobic PVDF membrane in a microfluidic device to concentrate analytes prior to analyzing with mass spectrometry. The method was demonstrated to be useful for extraction of proteins from high concentrations of urea, which would otherwise interfere with the analysis.
While SPE is used most often to enrich analytes based on general (e.g., hydrophobic) interactions, selective media can also be formed to enrich samples based on specific interactions. For example, Slentz et al.29 developed microfluidic devices packed with immobilized metal affinity chromatography (IMAC) beads to selectively concentrate histidine-rich peptides, and Mao et al.47 developed porous monolith-based methods to concentrate phosphorylated and glycosylated peptides.
Sample preconcentration and cleanup is a critical requirement for high-throughput proteomics analyses. It is unlikely that microfluidics-based preconcentration methods will be useful as stand-alone applications; however, the diversity of approaches that have been demonstrated bodes well for the development of integrated microfluidic-based proteomics methods (i.e., sample preconcentration combined with separations and mass spectrometry, etc.). In fact, an instrument with this style of integration has recently been commercialized by Agilent Technologies.48
Separations
High-resolution chemical separations is a baseline requirement for profiling proteomics, which makes it especially attractive for the lab-on-a-chip field. Separations was the first application to be implemented in microfluidics,49,50 and continues to be one of the most popular in academia and industry.48,51§ Although liquid-phase separation modes, such as capillary zone electrophoresis (CZE) and micellar electrokinetic chromatography (MEKC), dominate the microfluidics literature, recently, a trend has emerged for using solid-phase separation modes, such as capillary electrochromatography (CEC) or pressure driven chromatography. Although both liquid- and solid-phase media are useful for a wide range of applications, we contend that the latter may be better suited for two-dimensional separations (see discussion in the following section), and will be the most useful for proteomics applications using microfluidic devices.Microfluidic solid-phase separations media, like SPE media, are typically formed from either (a) a bed of packed, functionalized beads,52–54 or (b) an in-situ polymerized porous monolith.55–58 Both kinds of columns have been used for high-resolution separations of proteomic samples. For example, Xie et al.52 fabricated an on-chip pumping system integrated with a packed column of 3 µm C-18-functionalized beads. This device generated chromatograms with superior peak shape and resolution compared to those generated using a conventional capillary HPLC column. Although packed beads are more similar to conventional HPLC, polymeric monoliths are becoming the method of choice for microfluidics. Monolith fabrication is particularly well-suited for the planar, transparent device format; in addition, monoliths do not require the incorporation of frits, which are difficult to construct and can generate unwanted bubbles in microchannels.59 For example, Ro et al.55 reported the fabrication of a methacrylate-based reversed-phase monolithic column capable of separating complex mixtures of digest peptides. Fig. 3(a) shows an SEM image of a similar monolith,56 used to separate analytes for detection by ESI mass spectrometry. As an interesting aside, a third kind of solid-phase separation medium can be formed directly by means of microfabrication;60 such devices have been shown to be capable of separating analytes with comparable resolution and efficiencies to conventional techniques. A column formed in this manner is shown in Fig. 3(b). Though interesting, this method is not likely to become widely used, as it requires considerably more demanding microfabrication than the alternatives.
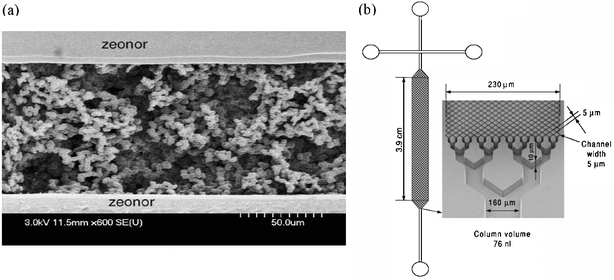 | ||
| Fig. 3 (a) An SEM of a methacrylate-based monolith in a plastic microchannel, adapted with permission from Yang et al.. Reproduced from ref. 56. (b) Solid phase separation medium formed from microfabricated posts rather than packed beads or monolith. Reprinted from Slentz et al.,60 copyright 2002, with permission from Elsevier. | ||
While the microfluidic separation methods described above are useful for many applications, they are likely not adequate for the rigorous demands of profiling proteomics. The complexity of proteomic samples requires the use of techniques capable of separating thousands of analytes. Methods with such large peak capacities can only be achieved when analytes are separated in multiple dimensions.¶ The most common macro-scale multidimensional separation technique is two-dimensional gel electrophoresis (2DGE). In 2DGE, analytes are separated by charge using isoelectric focusing (IEF) and molecular weight using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The limitations of 2DGE are discussed in the introduction; in an attempt to increase throughput, microfluidic implementations of 2DGE-like separations have recently been reported.61–65 Like 2DGE, these methods make use of separation dimensions that are positioned perpendicular to each other. For example, as shown in Fig. 4(a), Li et al.65 developed a polycarbonate device that enabled separation of proteins by IEF followed by PAGE. While the perpendicular geometry makes analyte transfer between dimensions easy (i.e., rotate 90°), it is not an ideal arrangement for high-throughput applications, as analytes become scattered across a plane, making analysis by a single detector very difficult.
 | ||
| Fig. 4 Two kinds of multidimensional separation geometries. (a) A parallel-type two-dimensional separation device. Reprinted with permission from Li et al.,65 copyright 2004 American Chemical Society. (b) An axial-type device. Reprinted with permission from Ramsey et al.,68 copyright 2003 American Chemical Society. | ||
An alternative macro-scale method for high peak capacity separations is two-dimensional column chromatography (2DCC), which is the basis for shotgun proteomics.13–15 Unlike 2DGE, in 2DCC, the two separation dimensions are positioned axial to one another, such that all analytes pass through the same physical space. This arrangement makes analysis by a single detector much more straightforward. A disadvantage of axial separations is that the transfer between dimensions is much more complex, requiring care to avoid re-combining analytes in the second dimension that were previously resolved in the first. This is typically accomplished by eluting bands of analytes stepwise from the first dimension and into the second (i.e., the first dimension is not used while the second dimension is active and vice versa).
Microfluidics has been used to implement axial-type two-dimensional separations.66–68 For example, as shown in Fig. 4(b), Ramsey et al.68 recently fabricated a microchip combining MEKC in the first dimension with CE in the second dimension; peak capacities for this method were shown to be comparable to 2DGE. A disadvantage for this (and for all microfluidic axial-type methods that have been reported66–68) is that it relies on liquid-phase separations, which are not convenient for stepwise elution between the first and second dimension. As a result, the reported techniques are limited to the use of very short second dimensions, and can sample only a fraction of total analyte (∼10%) onto the second dimension, reducing the sensitivity. Additionally, liquid-phase separation run buffers (especially for MEKC) are often not compatible with detection by mass spectrometry.
It is clear that, although separations in general is a strong application for microfluidics, the field is not yet capable of implementing methods with high peak capacities that are compatible with proteome profiling. This application is perhaps the most ripe for innovation in the field of microfluidics.
Interfaces with mass spectrometry
There is wide-spread interest in coupling microfluidic devices to ESI or MALDI mass spectrometry. Indeed, if a fully integrated method, comprising chemical processing, sample preconcentration and cleanup, and two-dimensional separations could be integrated with mass spectrometry detection, the result could revolutionize the field of proteomics. This revolution is on hold, however, as current microfluidic–mass spectrometry interfaces have not yet been widely adopted, as they continue to be plagued with technical challenges. Here, we survey the different kinds of interfaces reported in the literature, and discuss the most promising geometries and applications going forward.We contend that nanoelectrospray ionization is the most likely candidate for a robust interface between microfluidics and mass spectrometry. This assertion springs from the obvious similarities between the conventional technique of interfacing HPLC eluent to a spectrometer by means of pulled-glass nanospray tips, and the linear geometry of microfluidic channels. A variety of strategies for fabricating such devices have been reported, in which proteomic sample solutions are pumped through microchannels pneumatically or by electroosmotic flow (EOF) at ∼100–300 nL min−1. Samples are typically dissolved in low-pH buffers modified with organic solvents suitable for positive mode mass spectrometry, with detection limits in the fmol–amol range. These methods can be broadly classified by how the electrospray is generated, including: (1) direct spray from channels;46,69–72 (2) spray from mated, conventional tips;42,73–79 and (3) spray from microfabricated tips.43,52,54,80–86
Electrospray directly from a channel46,69–72 (i.e., the unmodified edge of a device) is the easiest approach for interfacing with mass spectrometry. The first microchannel–ESI interface was reported by Xue et al.,69 in which analyte was sprayed from the flat edge of a glass channel. While an important first step, the authors observed that performance was limited by eluent spreading at the interface, resulting from the non-tapered geometry and the hydrophilicity of the substrate. Lion et al.46 improved upon the former problem by tapering the edge of a polyimide substrate with scissors; Wang et al.72 countered the latter problem by integrating hydrophobic polytetrafluoroethylene (PTFE, or Teflon) surfaces at the device edge. Despite these advances, spraying directly from the edges of chips has been largely abandoned, as it seems that reduced sensitivity (less efficient sampling into the spectrometer), and decreased resolution when coupled to separations (eluent spreading at the edges) cannot be satisfactorily controlled.
The problems associated with direct spray from the edges of chips prompted the development of an alternative geometry for interfacing with mass spectrometry: mating microchannels to conventional pulled glass capillary tips.42,73–79 These devices are capable of generating mass spectra with sensitivities similar to those of conventional techniques. For example, Lazar et al.73 reported sub-attomole detection of peptides using a glass microfluidic device mated to a conventional electrospray tip; Chan et al.74 reported similar performance for a device formed from PDMS. Unfortunately, an unavoidable problem for this geometry is observed with coupling to separations: resolution is severely compromised as analytes pass through dead volumes in the interface between chip and capillary. As a result, this device geometry is not likely to be useful for the development of proteomics tools.
A third geometry for microfluidic–nanospray interfaces, microfabricated, tapered electrospray tips43,52,54,80–86 is the most promising geometry that has been reported. In fact, several devices with this configuration are now available commercially (for example, Advion Biosciences87 and Agilent Laboratories48). Schilling et al.84 micro-milled a nozzle in PMMA, and demonstrated exceedingly stable spray as a function of nozzle dimension. As shown in Fig. 5(a), this method results in a tip shape very similar to those found on conventional pulled-glass tips, but has no dead volume between channel and tip. Dahlin et al.85 used a mold to form integrated tips on PDMS devices; the tips were made conductive by the addition of graphite powder to the polymer, which enabled independent control of spray and separation potentials. Xie et al.52 used parylene to fabricate ESI tips on silicon microfluidic devices, enabling integrated liquid chromatography with mass spectrometry detection with comparable performance to conventional techniques. The one drawback for this geometry, however, is that these devices typically require complex, arduous cleanroom fabrication (i.e., many sequential photolithography steps), and are thus likely not viable for widespread use. Of the devices reported in the literature, the one developed by Yin et al.54 at Agilent Laboratories48(and characterized further by Fortier et al.53) is perhaps the most promising. This device, shown in Fig. 5(b), features an integrated nanospray tip formed by laser ablation of a polyimide substrate. When mated to an off-chip injection valve and pump, the device is capable of separations and tandem mass spectrometry detection (MS/MS) with characteristics (peak resolution, detection limits, background ion level, etc.) similar to those obtained by conventional macro-scale methods.
 | ||
| Fig. 5 Two kinds of microfabricated nanoelectrospray tips. (a) A tip mated to a microchannel formed by micromachining PMMA. Reproduced from ref. 84. (b) A polyimide microchannel device with integrated ESI tip formed by laser ablation. Reprinted with permission from Yin et al.,54 copyright 2005 American Chemical Society. | ||
An alternative mode to ESI for mass spectrometry analysis of proteomic samples is MALDI. Though the geometry of MALDI detection targets, which typically feature arrays of crystallized sample spots on an open surface, is not an obvious match for interfacing with microfluidics, several strategies for developing such interfaces have recently been developed. This work can be categorized in terms of device geometry, including: (1) conventional microfluidics in enclosed channels55,88–92 and (2) other microfabricated devices.93–99
Enclosed microchannels are by definition not accessible to laser desorption/ionization, which requires an open surface from which analytes can be sampled into the spectrometer. Brivio et al.90 circumvented this challenge by eluting bands of analytes from a device reservoir onto an open substrate, where they were dried and analyzed. Ro et al.55 used similar means to elute analytes from multiple columns onto a MALDI target, simultaneously. Brivio et al.89 recently improved on the original technique, by developing means to desorb analytes directly from enclosed channels through sub-micron pores in the device cover. Musyimi et al.91 employed a rotating ball to transfer analytes from polymer microchannels to a MALDI-MS system without compromising the vacuum required for mass spectrometry. In one of the most complete microfluidic systems developed for proteomics applications to date, Gustafsson et al.92 developed a MALDI interface for compact disk (CD)-based microfluidics devices (a technology in which reactions and separations are powered by centrifigual forces on a spinning device). In this method, analytes were delivered through channels to pre-formed open vias (holes in the substrate), where they were dried and interrogated by MALDI-MS.
The solutions that have been developed for interfacing MALDI-MS with enclosed microchannels are ingenious; however, it's not clear if such solutions are practical for widespread use. Several technologies that rely on microfabricated devices that are not “microfluidics” per se, may be a better match for MALDI-MS. For example, MALDI targets with lithographically patterned hydrophilic regions94,96 have become popular for forming spots with concentrated analytes (and increased signal-to-noise ratio (S/N)). Microfabricated piezoelectric dispensers are capable of depositing nanolitres of samples onto MALDI targets in parallel.93,95 Finally, a method called “digital microfluidics,” in which droplets are moved on an open surface by means of electrowetting and/or dielectrophoretic forces, has been used to process proteomic samples and form arrays of spots for analysis by MALDI-MS.97–99 As shown in Fig. 6, the technique has been used to perform in-situ sample cleanup on an open substrate, after which, samples are interrogated by MALDI-MS, with similar detection efficiencies, resolution, and S/N as conventional techniques.
 | ||
| Fig. 6 Video sequence (top-to-bottom) depicting digital microfluidics-based analysis of a sample containing insulin and urea. The large electrodes are used to move a water droplet to the dried spot, where it selectively dissolves the urea. Because the rinsing droplet primarily touches clean surfaces on the surrounding electrodes, it is easily moved away, leaving behind an (invisible) insulin film. Reproduced from ref. 99. | ||
Interfacing microfluidic devices with mass spectrometry is currently a popular research topic, which has generated a diverse and dynamic scientific literature. Although, in our opinion, the current geometries are not robust enough to compete with conventional, macro-scale techniques, it is clear that the technologies are becoming increasingly more practical and effective. We anticipate that in the near future, methods will emerge that will be applicable outside of the realm of the microfluidics community.
Conclusion
Significant challenges stand in the way of developing tools for high-throughput proteome profiling. Microfluidics, with its advantages in process integration and reduced reagent use and analysis time, is a logical technology to apply to this problem. We contend that the most important microfluidic functions for protein profiling are chemical processing, sample preconcentration and cleanup, chemical separations, and interfaces with mass spectrometry. One strategy will be to develop a fully integrated microfluidic method, comprising variations on each of these functions; such a method has not yet been developed. Another strategy will be to develop separate modules linked in an automated workstation. Some of the individual modules are ready; for example, microchannels with surface-adsorbed trypsin can be used to digest proteomic samples in seconds. Other modules have not yet been demonstrated; e.g., significant work must be accomplished in two-dimensional separations. In the final analysis, given the trajectory of interest and innovation in this marriage of proteomics applications and microfluidics technology, we speculate that microfluidics-based tools for high-throughput proteomics are indeed on the horizon, and will be realized within the next decade.Acknowledgements
Helpful comments from Jianhua Qin and Michael Watson are gratefully acknowledged. We thank the McLaughlin Centre for Molecular Medicine (MCMM) for financial support. ARW thanks the CRC for a Canada Research Chair.References
- J. C. Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans and R. A. Holt et al., The Sequence of the Human Genome, Science, 2001, 291, 1304–1351 CrossRef CAS.
- M. R. Wilkins, J.-C. Sanchez, A. A. Gooley, R. D. Appel, I. Humphrey-Smith, D. F. Hochstrasser and K. L. Williams, Progress with Proteome Projects: Why All Proteins Expressed by a Genome Should Be Identified and How to Do It, Biotechnol. Genet. Eng. Rev., 1995, 13, 19–50 Search PubMed.
- 6675 Entries Found in Scifinder Scholar™ Search of The “Chemical Abstracts Plus” Database on June 28, 2006 with Parameters: Keyword (as Written) “Proteomics”, Publication Year “2000–2005”, Document Type “Journal”.
- M. S. Lim and K. S. J. Elenitoba-Johnson, Proteomics in Pathology Research, Lab. Invest., 2004, 84, 1227–1244 Search PubMed.
- Proteomics' New Order, Nature, 2005, 437, 169–170 Search PubMed.
- D. Shalon, S. J. Smith and P. O. Brown, A DNA Microarray System for Analyzing Complex DNA Samples Using Two-Color Fluorescent Probe Hybridization, Genome Res., 1996, 6, 639–645 CrossRef CAS.
- A. Guttman, A. S. Cohen, D. N. Heiger and B. L. Karger, Analytical and Micropreparative Ultrahigh Resolution of Oligonucleotides by Polyacrylamide-Gel High-Performance Capillary Electrophoresis, Anal. Chem., 1990, 62, 137–141 CrossRef CAS.
- S. Fields, Proteomics: Proteomics in Genomeland, Science, 2001, 291, 1221–1224 CrossRef CAS.
- E. Apella, D. Arnott, K. Sakaguchi and P. J. Wirth, in Proteomics in Functional Genomics, ed. P. Jolles and H. Jornvall, Birkhauser Verlag, Basel, Germany, 2000, pp. 1–28 Search PubMed.
- P. Roepstorff, in Proteomics in Functional Genomics, ed. P. Jolles and H. Jornvall, Birkhauser Verlag, Basel, Germany, 2000, pp. 81–98 Search PubMed.
- K. Gevaert and J. Vandekerckhove, Protein Identification Methods in Proteomics, Electrophoresis, 2000, 21, 1145–1154 CrossRef CAS.
- D. N. Perkins, D. J. C. Pappin, D. M. Creasy and J. S. Cottrell, Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data, Electrophoresis, 1999, 20, 3551–3567 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, Large-Scale Analysis of the Yeast Proteome by Multidimensional Protein Identification Technology, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- J. K. Eng, A. L. Mccormack and J. R. Yates, An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef.
- T. Kislinger and A. Emili, Going Global: Protein Expression Profiling Using Shotgun Mass Spectrometry, Curr. Opin. Mol. Ther., 2003, 5, 285–293 Search PubMed.
- D. Figeys and D. Pinto, Proteomics a Chip: Promising Developments, Electrophoresis, 2001, 22, 208–216 CrossRef CAS.
- G. L. Gauthier and R. Grimm, Miniaturization: Chip-Based Liquid Chromatography and Proteomics, Drug Discovery Today Tech., 2006, 3, 59–66 Search PubMed.
- N. Lion, F. Reymond, H. H. Girault and J. S. Rossier, Why the Move to Microfluidics for Protein Analysis?, Curr. Opin. Biotechnol., 2004, 15, 31–37 CrossRef CAS.
- N. Lion, T. C. Rohner, L. Dayon, I. L. Arnaud, E. Damoc, N. Youhnovski, Z. Y. Wu, C. Roussel, J. Josserand and H. Jensen et al., Microfluidic Systems in Proteomics, Electrophoresis, 2003, 24, 3533–3562 CrossRef.
- L. Sage, Protein Biochips Go High Tech, Anal. Chem., 2004, 76, 137A–142A CAS.
- M. Uttamchandani, J. Wang and S. Q. Yao, Protein and Small Molecule Microarrays: Powerful Tools for High-Throughput Proteomics, Mol. Biosyst., 2006, 2, 58–68 RSC.
- K. Jahnisch, V. Hessel, H. Lowe and M. Baerns, Chemistry in Microstructured Reactors, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef.
- N. Gottschlich, C. T. Culbertson, T. E. McKnight, S. C. Jacobson and J. M. Ramsey, Integrated Microchip-Device for the Digestion, Separation and Postcolumn Labeling of Proteins and Peptides, J. Chromatogr., B, 2000, 745, 243–249 CrossRef CAS.
- Y. Huang, W. Shan, B. Liu, Y. Liu, Y. Zhang, Y. Zhao, H. Lu, Y. Tang and P. Yang, Zeolite Nanoparticle Modified Microchip Reactor for Efficient Protein Digestion, Lab Chip, 2006, 6, 534–539 RSC.
- Y. Liu, H. Lu, W. Zhong, P. Song, J. Kong, P. Yang, H. H. Girault and B. Liu, Multilayer-Assembled Microchip for Enzyme Immobilization as Reactor toward Low-Level Protein Identification, Anal. Chem., 2006, 78, 801–808 CrossRef CAS.
- L. J. Jin, J. Ferrance, J. C. Sanders and J. P. Landers, A Microchip-Based Proteolytic Digestion System Driven by Electroosmotic Pumping, Lab Chip, 2003, 3, 11–18 RSC.
- M. Slovakova, N. Minc, Z. Bilkova, C. Smadja, W. Faigle, C. Fütterer, M. Taverna and J.-L. Viovy, Use of Self Assembled Magnetic Beads for On-Chip Protein Digestion, Lab Chip, 2005, 5, 935–942 RSC.
- G. E. Yue, M. G. Roper, C. Balchunas, A. Pulsipher, J. J. Coon, J. Shabanowitz, D. F. Hunt, J. P. Landers and J. P. Ferrance, Protein Digestion and Phosphopeptide Enrichment on a Glass Microchip, Anal. Chim. Acta, 2006, 564, 116–122 CrossRef CAS.
- B. E. Slentz, N. A. Penner and F. E. Regnier, Protein Proteolysis and the Multi-Dimensional Electrochromatographic Separation of Histidine-Containing Peptide Fragments on a Chip, J. Chromatogr., A, 2003, 984, 97–107 CrossRef CAS.
- J. W. Cooper, J. Chen, Y. Li and C. S. Lee, Membrane-Based Nanoscale Proteolytic Reactor Enabling Protein Digestion, Peptide Separation, and Protein Identification Using Mass Spectrometry, Anal. Chem., 2003, 75, 1067–1074 CrossRef CAS.
- D. S. Peterson, T. Rohr, F. Svec and J. M. J. Fréchet, Enzymatic Microreactor-on-a-Chip: Protein Mapping Using Trypsin Immobilized on Porous Polymer Monoliths Molded in Channels of Microfluidic Devices, Anal. Chem., 2002, 74, 4081–4088 CrossRef CAS.
- K. Sakai-Kato, M. Kato and T. Toyo'oka, Creation of an On-Chip Enzyme Reactor by Encapsulating Trypsin in Sol-Gel on a Plastic Microchip, Anal. Chem., 2003, 75, 388–393 CrossRef CAS.
- J. Gao, J. Xu, L. E. Locascio and C. S. Lee, Integrated Microfluidic System Enabling Protein Digestion, Peptide Separation, and Protein Identification, Anal. Chem., 2001, 73, 2648–2655 CrossRef CAS.
- Y. Liu, R. S. Foote, S. C. Jacobson and J. M. Ramsey, Stacking Due to Ionic Transport Number Mismatch During Sample Sweeping on Microchips, Lab Chip, 2005, 5, 457–465 RSC.
- A. Wainright, S. J. Williams, G. Ciambrone, Q. Xue, J. Wei and D. Harris, Sample Pre-Concentration by Isotachophoresis in Microfluidic Devices, J. Chromatogr., A, 2002, 979, 69–80 CrossRef CAS.
- B. Jung, R. Bharadwaj and J. G. Santiago, Thousandfold Signal Increase Using Field-Amplified Sample Stacking for On-Chip Electrophoresis, Electrophoresis, 2003, 24, 3476–3483 CrossRef CAS.
- B. Jung, R. Bharadwaj and J. G. Santiago, On-Chip Millionfold Sample Stacking Using Transient Isotachophoresis, Anal. Chem., 2006, 78, 2319–2327 CrossRef CAS.
- R. S. Foote, J. Khandurina, S. C. Jacobson and J. M. Ramsey, Preconcentration of Proteins on Microfluidic Devices Using Porous Silica Membranes, Anal. Chem., 2005, 77, 57–63 CrossRef CAS.
- Y. Zhang and A. T. Timperman, Integration of Nanocapillary Arrays into Microfluidic Devices for Use as Analyte Concentrators, Analyst, 2003, 128, 537–542 RSC.
- S. Song, A. K. Singh and B. J. Kirby, Electrophoretic Concentration of Proteins at Laser-Patterned Nanoporous Membranes in Microchips, Anal. Chem., 2004, 76, 4589–4592 CrossRef CAS.
- R. D. Oleschuk, L. L. Shultz-Lockyear, Y. B. Ning and D. J. Harrison, Trapping of Bead-Based Reagents within Microfluidic Systems: On-Chip Solid-Phase Extraction and Electrochromatography, Anal. Chem., 2000, 72, 585–590 CrossRef CAS.
- J. Li, T. LeRiche, T.-L. Tremblay, C. Wang, E. Bonneil, D. J. Harrison and P. Thibault, Application of Microfluidic Devices to Proteomics Research: Identification of Trace-Level Protein Digests and Affinity Capture of Target Peptides, Mol. Cell. Proteomics, 2002, 1, 157–168 Search PubMed.
- A. P. Dahlin, S. K. Bergstrom, P. E. Andren, K. E. Markides and J. Bergquist, Poly(dimethylsiloxane)-Based Microchip for Two-Dimensional Solid-Phase Extraction-Capillary Electrophoresis with an Integrated Electrospray Emitter Tip, Anal. Chem., 2005, 77, 5356–5363 CrossRef CAS.
- J. D. Ramsey and G. E. Collins, Integrated Microfluidic Device for Solid-Phase Extraction Coupled to Micellar Electrokinetic Chromatography Separation, Anal. Chem., 2005, 77, 6664–6670 CrossRef CAS.
- C. Yu, M. H. Davey, F. Svec and J. M. J. Fréchet, Monolithic Porous Polymer for On-Chip Solid-Phase Extraction and Preconcentration Prepared by Photoinitiated In Situ Polymerization within a Microfluidic Device, Anal. Chem., 2001, 73, 5088–5096 CrossRef CAS.
- N. Lion, J.-O. Gellon, H. Jensen and H. H. Girault, On-Chip Protein Sample Desalting and Preparation for Direct Coupling with Electrospray Ionization Mass Spectrometry, J. Chromatogr., A, 2003, 1003, 11–19 CrossRef CAS.
- X. L. Mao, Y. Luo, Z. P. Dai, K. Y. Wang, Y. G. Du and B. C. Lin, Integrated Lectin Affinity Microfluidic Chip for Glycoform Separation, Anal. Chem., 2004, 76, 6941–6947 CrossRef CAS.
- HPLC-Chip, http://www.chem.agilent.com/Scripts/PDS.asp?lPage=38308, accessed on 08/04/2006.
- D. J. Harrison, A. Manz, Z. H. Fan, H. Ludi and H. M. Widmer, Capillary Electrophoresis and Sample Injection Systems Integrated on a Planar Glass Chip, Anal. Chem., 1992, 64, 1926–1932 CrossRef.
- A. Manz, D. J. Harrison, E. M. J. Verpoorte, J. C. Fettinger, A. Paulus, H. Ludi and H. M. Widmer, Planar Chips Technology for Miniaturization and Integration of Separation Techniques into Monitoring Systems: Capillary Electrophoresis on a Chip, J. Chromatogr., 1992, 593, 253–258 CrossRef CAS.
- Brio Cartridge, http://www.nanostream.com/products/brio/index.html, accessed on 08/04/2006.
- J. Xie, Y. N. Miao, J. Shih, Y. C. Tai and T. D. Lee, Microfluidic Platform for Liquid Chromatography-Tandem Mass Spectrometry Analyses of Complex Peptide Mixtures, Anal. Chem., 2005, 77, 6947–6953 CrossRef CAS.
- M. H. Fortier, E. Bonneil, P. Goodley and P. Thibault, Integrated Microfluidic Device for Mass Spectrometry-Based Proteomics and Its Application to Biomarker Discovery Programs, Anal. Chem., 2005, 77, 1631–1640 CrossRef CAS.
- N. F. Yin, K. Killeen, R. Brennen, D. Sobek, M. Werlich and T. V. van de Goor, Microfluidic Chip for Peptide Analysis with an Integrated HPLC Column, Sample Enrichment Column, and Nanoelectrospray Tip, Anal. Chem., 2005, 77, 527–533 CrossRef CAS.
- K. W. Ro, J. Liu and D. R. Knapp, Plastic Microchip Liquid Chromatography-Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Using Monolithic Columns, J. Chromatogr., A, 2006, 1111, 40–47 CrossRef CAS.
- Y. Yang, C. Li, J. Kameoka, K. H. Lee and H. G. Craighead, A Polymeric Microchip with Integrated Tips and In Situ Polymerized Monolith for Electrospray Mass Spectrometry, Lab Chip, 2005, 5, 869–876 RSC.
- D. J. Throckmorton, T. J. Shepodd and A. K. Singh, Electrochromatography in Microchips: Reversed-Phase Separation of Peptides and Amino Acids Using Photopatterned Rigid Polymer Monoliths, Anal. Chem., 2002, 74, 784–789 CrossRef CAS.
- D. S. Reichmuth, T. J. Shepodd and B. J. Kirby, Microchip HPLC of Peptides and Proteins, Anal. Chem., 2005, 77, 2997–3000 CrossRef CAS.
- S. Le Gac, C. Cren-Olivé, C. Rolando, J. Carlier and J. C. Camart, Monoliths for Microfluidic Devices in Proteomics, J. Chromatogr., B, 2004, 808, 3–14 CrossRef CAS.
- B. E. Slentz, N. A. Penner and F. E. Regnier, Capillary Electrochromatography of Peptides on Microfabricated Poly(Dimethylsiloxane) Chips Modified by Cerium(IV)-Catalyzed Polymerization, J. Chromatogr., A, 2002, 948, 225–233 CrossRef CAS.
- X. X. Chen, H. K. Wu, C. D. Mao and G. M. Whitesides, A Prototype Two-Dimensional Capillary Electrophoresis System Fabricated in Poly(Dimethylsiloxane), Anal. Chem., 2002, 74, 1772–1778 CrossRef CAS.
- A. E. Herr, J. I. Molho, K. A. Drouvalakis, J. C. Mikkelsen, P. J. Utz, J. G. Santiago and T. W. Kenny, On-Chip Coupling of Isoelectric Focusing and Free Solution Electrophoresis for Multidimensional Separations, Anal. Chem., 2003, 75, 1180–1187 CrossRef CAS.
- Y. C. Wang, M. N. Choi and J. Y. Han, Two-Dimensional Protein Separation with Advanced Sample and Buffer Isolation Using Microfluidic Valves, Anal. Chem., 2004, 76, 4426–4431 CrossRef CAS.
- A. Griebel, S. Rund, F. Schönfeld, W. Dömer, R. Konrad and S. Hardt, Integrated Polymer Chip for Two-Dimensional Capillary Gel Electrophoresis, Lab Chip, 2004, 4, 18–23 RSC.
- Y. Li, J. S. Buch, F. Rosenberger, D. L. DeVoe and C. S. Lee, Integration of Isoelectric Focusing with Parallel Sodium Dodecyl Sulfate Gel Electrophoresis for Multidimensional Protein Separations in a Plastic Microfludic Network, Anal. Chem., 2004, 76, 742–748 CrossRef CAS.
- R. D. Rocklin, R. S. Ramsey and J. M. Ramsey, A Microfabricated Fluidic Device for Performing Two-Dimensional Liquid-Phase Separations, Anal. Chem., 2000, 72, 5244–5249 CrossRef CAS.
- N. Gottschlich, S. C. Jacobson, C. T. Culbertson and J. M. Ramsey, Two-Dimensional Electrochromatography/Capillary Electrophoresis on a Microchip, Anal. Chem., 2001, 73, 2669–2674 CrossRef CAS.
- J. D. Ramsey, S. C. Jacobson, C. T. Culbertson and J. M. Ramsey, High-Efficiency, Two-Dimensional Separations of Protein Digests on Microfluidic Devices, Anal. Chem., 2003, 75, 3758–3764 CrossRef CAS.
- Q. F. Xue, F. Foret, Y. M. Dunayevskiy, P. M. Zavracky, N. E. McGruer and B. L. Karger, Multichannel Microchip Electrospray Mass Spectrometry, Anal. Chem., 1997, 69, 426–430 CrossRef CAS.
- R. S. Ramsey and J. M. Ramsey, Generating Electrospray from Microchip Devices Using Electroosmotic Pumping, Anal. Chem., 1997, 69, 1174–1178 CrossRef CAS.
- N. Lion, V. Gobry, H. Jensen, J. S. Rossier and H. H. Girault, Integration of a Membrane-Based Desalting Step in a Microfabricated Disposable Polymer Injector for Mass Spectrometric Protein Analysis, Electrophoresis, 2002, 23, 3583–3588 CrossRef CAS.
- Y. X. Wang, J. W. Cooper, C. S. Lee and D. L. DeVoe, Efficient Electrospray Ionization from Polymer Microchannels Using Integrated Hydrophobic Membranes, Lab Chip, 2004, 4, 363–367 RSC.
- I. M. Lazar, R. S. Ramsey, S. Sundberg and J. M. Ramsey, Subattomole-Sensitivity Microchip Nanoelectrospray Source with Time-of-Flight Mass Spectrometry Detection, Anal. Chem., 1999, 71, 3627–3631 CrossRef CAS.
- J. H. Chan, A. T. Timperman, D. Qin and R. Aebersold, Microfabricated Polymer Devices for Automated Sample Delivery of Peptides for Analysis by Electrospray Ionization Tandem Mass Spectrometry, Anal. Chem., 1999, 71, 4437–4444 CrossRef CAS.
- D. M. Pinto, Y. B. Ning and D. Figeys, An Enhanced Microfluidic Chip Coupled to an Electrospray QSTAR Mass Spectrometer for Protein Identification, Electrophoresis, 2000, 21, 181–190 CrossRef CAS.
- B. L. Zhang, F. Foret and B. L. Karger, High-Throughput Microfabricated CE/ESI-MS: Automated Sampling from a Microwell Plate, Anal. Chem., 2001, 73, 2675–2681 CrossRef CAS.
- S. H. Chen, W. C. Sung, G. B. Lee, Z. Y. Lin, P. W. Chen and P. C. Liao, A Disposable Poly(Methylmethacrylate)-Based Microfluidic Module for Protein Identification by Nanoelectrospray Ionization-Tandem Mass Spectrometry, Electrophoresis, 2001, 22, 3972–3977 CrossRef CAS.
- Y. Z. Deng, J. Henion, J. J. Li, P. Thibault, C. Wang and D. J. Harrison, Chip-Based Capillary Electrophoresis/Mass Spectrometry Determination of Carnitines in Human Urine, Anal. Chem., 2001, 73, 639–646 CrossRef CAS.
- S. Ssenyange, J. Taylor, D. J. Harrison and M. T. McDermott, A Glassy Carbon Microfluidic Device for Electrospray Mass Spectrometry, Anal. Chem., 2004, 76, 2393–2397 CrossRef CAS.
- G. A. Schultz, T. N. Corso, S. J. Prosser and S. Zhang, A Fully Integrated Monolithic Microchip Electrospray Device for Mass Spectrometry, Anal. Chem., 2000, 72, 4058–4063 CrossRef CAS.
- J. Kameoka, R. Orth, B. Ilic, D. Czaplewski, T. Wachs and H. C. Craighead, An Electrospray Ionization Source for Integration with Microfluidics, Anal. Chem., 2002, 74, 5897–5901 CrossRef CAS.
- J. Xie, Y. N. Miao, J. Shih, Q. He, J. Liu, Y. C. Tai and T. D. Lee, An Electrochemical Pumping System for On-Chip Gradient Generation, Anal. Chem., 2004, 76, 3756–3763 CrossRef CAS.
- M. Svedberg, M. Veszelei, J. Axelsson, M. Vangbo and F. Nikolajeff, Poly(dimethylsiloxane) Microchip: Microchannel with Integrated Open Electrospray Tip, Lab Chip, 2004, 4, 322–327 RSC.
- M. Schilling, W. Nigge, A. Rudzinski, A. Neyer and R. Hergenröder, A New On-Chip ESI Nozzle for Coupling of MS with Microfluidic Devices, Lab Chip, 2004, 4, 220–224 RSC.
- A. P. Dahlin, M. Wetterhall, G. Liljegren, S. K. Bergstrom, P. Andren, L. Nyholm, K. E. Markides and J. Bergquist, Capillary Electrophoresis Coupled to Mass Spectrometry from a Polymer Modified Poly(Dimethylsiloxane) Microchip with an Integrated Graphite Electrospray Tip, Analyst, 2005, 130, 193–199 RSC.
- L. Licklider, X. Wang, A. Desai, Y. Tai and T. Lee, A Micromachined Chip-Based Electrospray Source for Mass Spectrometry, Anal. Chem., 2000, 72, 367–375 CrossRef CAS.
- ESI Chip®, http://www.advion.com/products_nanomate.php, accessed on 08/04/2006.
- Y. X. Wang, Y. Zhou, B. M. Balgley, J. W. Cooper, C. S. Lee and D. L. DeVoe, Electrospray Interfacing of Polymer Microfluids to MALDI-MS, Electrophoresis, 2005, 26, 3631–3640 CrossRef CAS.
- M. Brivio, N. R. Tas, M. H. Goedbloed, H. J. E. Gardeniers, W. Verboom, A. van den Berg and D. N. Reinhoudt, A MALDI-Chip Integrated System with a Monitoring Window, Lab Chip, 2005, 5, 378–381 RSC.
- M. Brivio, R. H. Fokkens, W. Verboom, D. N. Reinhoudt, N. R. Tas, M. Goedbloed and A. van den Berg, Integrated Microfluidic System Enabling (Bio)Chemical Reactions with on-Line MALDI-TOF Mass Spectrometry, Anal. Chem., 2002, 74, 3972–3976 CrossRef CAS.
- H. K. Musyimi, J. Guy, D. A. Narcisse, S. A. Soper and K. K. Murray, Direct Coupling of Polymer-Based Microchip Electrophoresis to Online MALDI-MS Using a Rotating Ball Inlet, Electrophoresis, 2005, 26, 4703–4710 CrossRef CAS.
- M. Gustafsson, D. Hirschberg, C. Palmberg, H. Jornvall and T. Bergman, Integrated Sample Preparation and MALDI Mass Spectrometry on a Microfluidic Compact Disk, Anal. Chem., 2004, 76, 345–350 CrossRef CAS.
- D. P. Little, T. J. Cornish, M. J. O'Donnell, A. Braun, R. J. Cotter and H. Koster, MALDI on a Chip: Analysis of Arrays of Low-Femtomole to Subfemtomole Quantities of Synthetic Oligonucleotides and DNA Diagnostic Products Dispensed by a Piezoelectric Pipette, Anal. Chem., 1997, 69, 4540–4546 CrossRef CAS.
- R. L. Gundry, R. Edward, T. P. Kole, C. Sutton and R. J. Cotter, Disposable Hydrophobic Surface on MALDI Targets for Enhancing MS and MS/MS Data of Peptides, Anal. Chem., 2005, 77, 6609–6617 CrossRef CAS.
- S. Ekstrom, D. Ericsson, P. Onnerfjord, M. Bengtsson, J. Nilsson, G. Marko-Varga and T. Laurell, Signal Amplification Using “Spot-on-a-Chip” Technology for the Identification of Proteins via MALDI-TOF MS, Anal. Chem., 2001, 73, 214–219 CrossRef CAS.
- T. Redeby, J. Roeraade and A. Emmer, Simple Fabrication of a Structured Matrix-Assisted Laser Desorption/Ionization Target Coating for Increased Sensitivity in Mass Spectrometric Analysis of Membrane Proteins, Rapid Commun. Mass Spectrom., 2004, 18, 1161–1166 CrossRef CAS.
- A. R. Wheeler, H. Moon, C.-J. Kim, J. A. Loo and R. L. Garrell, Electrowetting-Based Microfluidics for Analysis of Peptides and Proteins by Matrix Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI-MS), Anal. Chem., 2004, 76, 4833–4838 CrossRef CAS.
- A. R. Wheeler, H. Moon, C. A. Bird, R. R. O. Loo, C.-J. Kim, J. A. Loo and R. L. Garrell, Digital Microfluidics with In-Line Sample Purification for Proteomics Analyses with MALDI-MS, Anal. Chem., 2005, 77, 534–540 CrossRef CAS.
- H. Moon, A. R. Wheeler, R. L. Garrell, J. A. Loo and C.-J. Kim, Integrated Digital Microfluidic Chip for Multiplexed Proteomic Sample Preparation and Analysis by MALDI-MS, Lab Chip, 2006, 6, 1213–1219 RSC.
Footnotes |
| † (1) Lyse cells and wash, (2) acidify and digest by cyanogen bromide (overnight), (3) denature in urea, (4) reduce in dithiothreitol (DTT), (5) alkylate in iodoacetamide, (6) dilute and digest by Lys-C (overnight), (7) dilute and digest by trypsin (overnight), and (8) purify and concentrate by solid phase extraction. |
| ‡ Chemical reactions in microfluidic devices are associated with a high surface area-to-volume ratio and reduced diffusion length of the reactants. Although these issues are the subject of discussion, several experimental results described in the literature have shown that reactions can be made to proceed faster in microfluidic devices. |
| § For example, Agilent sells chips for the separation of analytes using capillary electrophoresis, and Nanostream sells microfluidic devices for liquid chromatography. |
| ¶ Peak capacity is defined by L/w, where L is the separation channel length and w is the average analyte bandwidth. In multidimensional separations, the overall peak capacity is the product of the capacities of each dimension. |
| This journal is © The Royal Society of Chemistry 2006 |