Metabolic monitoring of the electrically stimulated single heart cell within a microfluidic platform
Wei
Cheng
a,
Norbert
Klauke
a,
Helen
Sedgwick
a,
Godfrey L.
Smith
b and
Jonathan M.
Cooper
*a
aBioelectronics Research Centre, Department of Electronics and Electrical Engineering, University of Glasgow, Glasgow, UK G12 8QQ. E-mail: jmcooper@elec.gla.ac.uk
bInstitute of Biomedical and Life Sciences, University of Glasgow, Glasgow, UK G12 8QQ
First published on 14th September 2006
Abstract
A device based on five individually addressable microelectrodes, fully integrated within a microfluidic system, has been fabricated to enable the real-time measurement of ionic and metabolic fluxes from electrically active, beating single heart cells. The electrode array comprised one pair of pacing microelectrodes, used for field-stimulation of the cell, and three other microelectrodes, configured as an electrochemical lactate microbiosensor, that were used to measure the amounts of lactate produced by the heart cell. The device also allowed simultaneous in-situ microscopy, enabling optical measurements of cell contractility and fluorescence measurements of extracellular pH and cellular Ca2+. Initial experiments aimed to create a metabolic profile of the beating heart cell, and results show well defined excitation-contraction (EC) coupling at different rates. Ca2+ transients and extracellular pH measurements were obtained from continually paced single myocytes, both as a function of the rate of cell contraction. Finally, the relative amounts of intra- and extra-cellular lactate produced during field stimulation were determined, using cell electroporation where necssary.
Introduction
The ability to analyse cell metabolic products in real time could enhance our understanding of cellular physiological processes, particularly those involved in inter- and intra-cellular communication between and within isolated cells. Typically, the release of small (sub-attomole) quantities of metabolites occurs rapidly, within less than a few hundred milliseconds of cell stimulation, making the dynamic monitoring of an individual cell a significant analytical challenge. Much of the recent activity in this area has focused on fluorescence measurements. As an alternative method, carbon-fibre electrodes have also been used to monitor electrochemically the cellular release of neurotransmitters,1 hormones2 and insulin,3 providing high temporal and spatial resolution metabolic analysis from a single cell.4–6 Despite the elegance of these approaches they have some general limitations, including the fact that only a small proportion of the total amount of metabolites released will reach the carbon fibre. In addition, such assays are often technically elaborate, as the carbon fibre has to be individually fabricated and manually positioned adjacent to the cells.Recent advances in lab-on-a-chip and other microtechnologies have attracted significant interest in the development of new microdevices for cell analysis, where precise control over the geometry of the sensor improves signal collection. The comparable size of single cells and microfluidic structures has provided an opportunity for more sensitive analysis, because the cell comprises a substantial fraction (often as much as 10%) of the volume being analysed.7–9
As a consequence, there has been a considerable interest in developing microsensors integrated within lab-on-a-chip structures for the analysis of single cells. For example, attempts to estimate the metabolic flux of cells using electrochemically-linked assays have been made10–13 and, most recently, a system to record membrane potential using microfluidics to trap single cells in a sub-nanolitre chamber has been described.14 There has, however, been substantially less work developing on-chip assays, where the cell’s metabolic and physiological function is manipulated in situ (including, for example, single-cell manipulation and intracellular monitoring,15–17 and single-cell perfusion or lysis followed by analysis of intracellular contents using chip-based electrophoresis18–20).
In this paper, studies of the physiology of single cardiomyocytes in a microfluidic system are presented. We describe an electrochemical biosensor, designed to measure extracellular lactate, which is combined with stimulating microelectrodes and integrated within a picolitre (pL)-scale microfluidic chamber. The device is used to stimulate the cell at pre-determined rates and explore the effect of making the cell “work” under different metabolic conditions. The microsystem also allows changes in cell length, pH and Ca2+ to be measured at the same time, providing details of the electrical and metabolic state of the heart cell. The measurements that we report of the metabolites released from the single heart cell will improve our understanding of cell metabolism during anoxia or ischemia.10–12
We chose to measure the single-cell production of lactate because it is an important cellular metabolite and of significant clinical interest in heart cell physiology. For example, lactate levels can be monitored to indicate the health of a cell, because damaged cells, or those in which oxygen supply is restricted, tend to produce larger amounts of lactate. Previously, the detection of lactate has been achieved using enzyme-linked electrochemical assays based on lactate oxidase. Although there have been attempts to immobilise the enzyme at both macroelectrodes21 and microelectrodes,22 less progress has been made on developing enzyme-modified integrated microbiosensors for monitoring lactate released from single cells within microfluidic devices.12
We have developed an immobilised microfabricated enzyme-modified sensor, used to determine the amounts of lactate produced from single cells in pL volumes with a detection limit of 4.8 fmol (equivalent to 7.4 µM). Within the microsystem, single cardiomycotes were forced to contract continuously using field stimulation (ca. 75 V cm−1) at rates between 0.5 and 2.0 Hz, as might be expected of the rabbit heart. Intracellular Ca2+ transients were monitored from continually paced single cardiomyocytes using Fluo-3 (a fluorescent Ca2+ indicator). Finally, the effect of continual pacing on the cell’s contractility and the extracellular pH in the restricted volume of the microchamber was investigated, and the results were interpreted in the context of lactate release.
Experimental
Reagents
All solutions were prepared using high-purity deionized water (Millipore Elix 10) and analytical reagent grade chemicals, without further purification. Unless otherwise stated, all chemicals were obtained from Sigma–Aldrich (Dorset, UK). Lactate solutions for sensor calibration were prepared as stock solutions in appropriate buffers immediately prior to use. The supporting electrolyte was 10 mM phosphate buffered saline, PBS (10 mM PBS contains 2.7 mM KCl and 137 mM NaCl, pH 7.4).Cell culture
Single adult rabbit ventricular myocytes were isolated from the left ventricle by perfusion of the heart with collagenase solution. Cells were maintained in Krebs Base Solution (KBS) containing 120 mM NaCl, 20 mM sodium N-hydroxyethylpiperazine-N′-2-ethane sulfonic acid, 5.4 mM KCl, 0.52 mM NaH2PO4, 3.5 mM MgCl2, 6H2O, 20 mM taurine, 10 mM creatine, 11.1 mM glucose, 0.1% BSA, and 1.8 mM CaCl2. The pH was adjusted to 7.4 with 100 mM NaOH. Extracellular medium was also used, which contained appropriate ionic concentrations to allow excitation–contraction of cells.Microfabrication
The microfabrication process, Fig. 1, was adapted from standard photolithographic methods including metal evaporation, electrochemical metal deposition and lift-off.10–12 Masks for microelectrodes and microfluidic structures were designed in L-Edit™ and produced as chrome-coated maskplates using a Leica EPBG5-HR electron beam writer (Leica, Germany). | ||
| Fig. 1 Schematic illustration of the major steps involved in the device fabrication: (1) the photoresist-coated substrate was exposed to UV radiation through a mask; (2) the photoresist was developed; (3) metals were deposited by evaporation; (4) the electrode pattern was generated by lift-off; (5) silver was deposited by evaporation; (6) spin-coating was used to generate an S1818 photoresist sacrificial layer; (7) the device was again exposed to UV radiation; (8) the SU-8 and S1818 layers were developed; (9) finally, AgCl was electrochemically deposited. | ||
The microstructure fabrication involved the deposition of the microelectrodes onto a glass slide, which had been sonicated and cleaned in Opticlear™, acetone and methanol. The clean slide was spin-coated with S1818 photoresist at 4000 rpm for 30 sec, baked for 30 minutes at 90 °C and then soaked for 15 minutes in chlorobenzene. The electrode pattern was transferred onto the photoresist layer by UV exposure through a mask, followed by resist development. Metals were then deposited by electron beam evaporation producing a multilayer electrode structure of Ti/Pd/Pt (10/10/100 nm). The 10 nm Ti metallic underlayer allowed for good adhesion of the Pt over-layer to the glass slide, whilst the 10 nm Pd layer acted as a diffusion barrier layer to ensure good electrochemistry by preventing Ti from diffusing into the Pt. Finally, the platinum over-layer was evaporated to produce a coherent 100 nm thick electrode layer. After metal deposition, the microelectrode pattern was realised by lift-off in acetone. Associated with the microelectrode array were alignment marks to enable subsequent microstructures including both microfluidic structures and the Ag|AgCl reference to be deposited.
One of the Pt electrodes (Fig. 2, B) was modified with an Ag|AgCl layer to provide a reference electrode. Silver was coated using the same basic microfabrication process described above, in which a second mask was used to define a 20 µm × 20 µm area, over the reference electrode, using an appropriate registration and alignment series. The AgCl layer was deposited galvanostically at +0.15 V in a solution of 0.1 M HCl (until the oxidation current decreased to background). During the deposition of AgCl, a coil of platinum wire was used as a common counter and the pseudo-reference electrode.
 | ||
| Fig. 2 Micrograph of the microelectrodes and 20 µm deep microchamber. The different functional microelectrodes are indicated as A, B, C, D and E (see text). A quiescent cardiomyocyte is also shown in the microchamber. | ||
Immediately prior to any further processing, a layer of S1818 photoresist was patterned over the microelectrode array as a sacrificial layer, protecting the electrodes from fouling by residues from subsequent processes. The microchamber and microfluidic structures were defined by photolithographically patterning an SU-8 negative resist. A 20 µm deep layer of SU-8 resist was patterned using the processing steps of spin-coating, pre-exposure baking, UV exposure through a mask, post-exposure baking and development. Five bonding pads, each associated with one of the five individual microelectrodes, were also exposed through the SU-8 layer.
A series of other microstructures were produced and used during this study, including the creation of micro-reservoirs for cell micro-culture. These comprised a poly(dimethylsiloxane) (PDMS) chamber of volume ca. 200 µl created by replica molding against an SU-8 master. Such structures were used as “holding tanks” for the selection and storage of cells immediately prior to measurements.
Enzyme immobilization
The enzyme lactate oxidase (LOX) catalyzes the oxidation of L-lactate in the presence of oxygen to produce pyruvate and hydrogen peroxide, eqn 1 and 2. In the device described in this paper, H2O2 was detected amperometrically, at a potential of +640 mv vs. Ag|AgCl.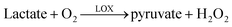 | (1) |
| H2O2 → 2H+ + O2 + 2e− +640 mV vs. Ag/AgCl | (2) |
Immediately prior to enzyme immobilization at the Pt working electrode (Fig. 2, A) the microfabricated electrochemical electrode was cleaned by scanning the applied potential between −1 V and +1 V in 100 mM H2SO4 at 1 V s−1, followed by washing in ultrapure water. The enzyme (20 µl solution containing 200 units per ml in PBS, pH 7.4) was pre-adsorbed onto the electrode surface by incubation overnight at 4 °C. A poly(o-phenylenediamine) film was deposited using cyclic voltammetry by scanning the applied potential at a rate of 50 mV s−1 for 6 min between 0.0 and +0.80 V vs. Ag|AgCl microreference in a supporting electrolyte solution of 50 mM PBS containing 50 mM KCl and 30 mM o-phenylenediamine. The growth of non-conducting polymer is self-limiting, and typically the resultant current fell to a steady state value within a minute, producing a film of defined thickness and composition. The polymer entrapped the enzyme locally at the working electrode, allowing the ready diffusion of electro-active species to the electrochemical interface. Inevitably some enzyme will be non-specifically adsorbed on other electrodes and within the microstructure, and much of this was removed by exhaustive washing prior to cell measurements. As a consequence, the majority of the measured current can be attributed to the locally immobilised enzyme. The device was stored under PBS buffer at 4 °C.
General experimental protcol
All experimental work was performed on a microscope stage, housed in a Faraday cage, onto which the microfabricated device was fixed. The five bonding pads on the microelectrodes (both electrochemical electrodes and stimulation electrodes) were wire-bonded to a PCB board and subsequently connected to the potentiostat and the stimulator. Throughout the course of an experiment, the temperature was maintained at 25 °C.pL-scale lactate dispensation
An in-house picolitre microinjection system12 was used for the reproducible dispensation of picolitre-scale volumes of calibrants into the chamber. Micropipettes of glass capillaries (Hilgenberg, Germany) with a 1.2 mm outer diameter and 0.96 mm inner diameter were pulled (PC-10, Narishige) to a tip, the diameter of which could be controlled to have a bore of between 1 and 100 µm. The volume injected by the microinjection system was calibrated by measuring the movement of the air/solution meniscus inside the filament using optical microscopy.12The pipette could be readily manoeuvred into the microchamber for low-volume dispensation using a three-axis micromanipulator (Leica, Germany). The whole operation was monitored using an inverted microscope (Zeiss, Axiovert). The microchamber was first filled with 0.2 µl of PBS, which was immediately covered with mineral oil to prevent the evaporation of bulk solution. An empty micropipette with an end bore of 40 µm was then inserted through the mineral oil layer into the droplet, so the buffer within the microchamber back filled the micropipette by capillary action. By carefully repeating the insertion and removal of the pipette, the total volume of the buffer could be readily controlled. To dispense pL-scale volumes, a second filament pipette with tip of ca. 1 µm diameter was filled with an appropriate stock solution (such as a calibration aliquot containing lactate). The injection pressure was 20 psi, and injection times of 20–1000 ms were used. There was a linear relationship between estimated injection volume and dispensation time over this range.
Single-cell selection
A 200 µl cell suspension was dropped into a PDMS “holding” microreservoir containing two platinum macroelectrodes, connected to a signal generator (isolated stimulator, Digitimer, Welwyn Garden City, UK). A 0.5 Hz square wave was used to field-stimulate the cells. The amplitude of the input signal was gradually increased. Single cells were selected if they contracted in response to a low amplitude stimulus (ca. 5 V cm−1) with regular and uniform shortening, as visually observed.Single-cell handling
The microsystem for single cell manipulation involved a micropipette (pulled capillary tip of diameter ca. 60 µm) coupled with a microfluidic connection to a 500 µl gas-tight microsyringe. The syringe was controlled by a micro-precision screw in a custom-built holder. The system was fixed on three-axis micromanipulators (for x-y-z movement) and filled with KBS buffer before cell handling.The micropipette was positioned above and close to the selected cell within the holding chamber and, by gentle expulsion and suction of the capillary, the cell was introduced into the capillary tip. The micropipette was then brought over the microchamber and a droplet containing the cell was deposited into the microchamber, such that the myocyte was aligned longitudinally to the axis of the chamber. The cell was observed to sediment to the bottom of chamber. As previously described, the volume of the medium was then reduced to the volume of the analytical microchamber by a second micropipette (tip diameter 40 µm).
Single-cell monitoring
Cells were loaded with Fluo-3 by incubation for ∼30 min in 20 µM Fluo-3 AM solution (Molecular Probes, Eugene, OR, USA) for the recording of intracellular Ca2+ transients. Sarcomere length and intracellular Ca2+ were both monitored using the fluorescence–contractility system of IonOptix (Milton, MA, USA). The Fluo-3 indicator was excited at 490 nm with a TILL monochromator (T.I.L.L. Photonics, Martinsried, Germany), which was mounted on a Zeiss Axiovert 200 (Zeiss, Germany) equipped with a 40× 0.80 N.A. Achroplan PH2 water immersion lens. The contractility of the cell was recorded by passing the light of the halogen lamp through a 680 nm bandpass filter and imaging the striation of the sarcomere, Fig. 2, onto a 240 Hz frame shift CCD (IonOptix, Milton, MA, USA). Length changes of the cell were recorded with the IonWizard Version 5.0 software and then converted to give a measure of relative cell length. Alternatively, the cell shortening was measured using off-line edge detection24 on recordings of the cell contraction. Sarcomere length of the cardiomyocyte is ca. 2.0 µm diastolic to 1.6 µm systolic, so 100% contraction with respect to the sarcomere length at rest is ca. 0.4 µm.Individual cells, Fig. 2, were stimulated within the device by applying a biphasic rectangular pulse of alternating polarity through the pair of stimulatory microelectrodes (Fig. 2, D and E) using a custom-built electric field stimulator coupled with a frequency generator. The amplitude, duration and frequency of applied potentials were controlled using instrumentation built in-house. The pH sensitive dye BCECF (10 µM, Molecular Probes, Eugene, OR, USA) was added to the microchamber to monitor the change in extracellular pH during field stimulation using the same filter set as for Fluo-3, described previously. The change in pH could arise either through the cell’s metabolic process or from electrolysis events owing to the stimulating microelectrodes (hydrolysis would be expected to occur at potentials >0.8 V vs. Ag|AgCl, and in all field stimulation experiments absolute applied potentials were maintained below this value).
A low-current potentiostat (CV-37, BAS) with data acquisition software was used to monitor the lactate produced from single cells. The lactate response was measured amperometrically at +640 mV vs. Ag|AgCl, recording the oxidation of enzymically produced H2O2, as previously detailed in eqn 1 and 2.
Electropermeabilisation
Single isolated cells were placed in the chamber and, following field stimulation, were electropermeabilised by the application of electric fields of 500 V cm−1 between the stimulation microelectrodes with 40 µs duration.Results and discussion
Design and fabrication
Fig. 2 shows the five-electrode system comprising: a three electrode micro-biosensor with working electrode (A), reference electrode (B), and counter electrode (C), together with the two stimulating electrodes (D, E). A single cardiomyocyte is shown within the microchamber. A number of issues arose during the design and fabrication of these electrodes that had to be addressed, particularly: metals with a sufficiently low overpotential for the oxidation of hydrogen peroxide, eqn 2, were used in order to minimise electrochemical interference with the cell (which would be expected to occur at large redox potentials); the physiochemical environment of the cell was maintained in the device; and finally, the spatially localised electrode geometry was designed to optimise the stimulation field and the signal-to-background of the sensor. In this latter respect, the counter (or return) electrode, Fig. 2, C, was designed to have a sufficiently large area to ensure that the redox processes at the working electrode were not rate-limiting. One further consideration was the potential toxicity of Ag+ associated with the Ag|AgCl reference electrode. In order to avoid compromising the cells, the reference electrode was designed to have a small size, so that there was no physical contact with the silver coating.Previous reports23,24 suggest that a more efficient stimulation is provided when the electrical axis of stimulation is parallel to the long axis of the cardiac cell, as measured by the reduced potential required for effective cell excitation. The two stimulatory electrodes were therefore fabricated within the microchamber so that the cell could be easily aligned parallel to the electrical field. A sufficiently low voltage stimulatory pulse was applied, avoiding both electrolysis and polarisation of the electrodes and minimising the ionic flux caused by stimulation that could interfere with measurements.
Characterization of microbiosensors
The first five cyclic voltammograms obtained during the oxidation of o-phenylenediamine in 50 mM PBS buffer (pH = 7.4) containing supporting electrolyte are shown in Fig. 3. The third scan shows that the oxidation current had almost decreased to background, indicating little further deposition of polymer film (i.e. the process was self-limiting as the polymer film is insulating). Determination of the amount of charge passed (as the sum of the integrals of scans 1 to 3) together with estimation of the molecular volume of the polymer suggests a film thickness of ca. 110 nm, consistent with the rapid responses when exposed to lactate, Fig. 4. | ||
| Fig. 3 Typical cyclic voltammograms showing the deposition of the polymer film (scan rate at 50 mVs−1) obtained using the microfabricated enzyme-modified sensor in PBS buffer (pH = 7.4) containing 30 mM o-phenylenediamine. The sensor surface area was 1800 µm2. | ||
After the adsorption of the enzyme and its subsequent entrapment in the polymer film, the sensors were characterised by the addition of standard aliquots of pL-amounts of L-lactate using the nanopipette system. The microchamber was first filled with 650 pL of PBS buffer (pH = 7.4) and, using pre-determined injection time intervals, 1.2 mM L-Lactate stock solution was injected to give final lactate concentrations of 7.4 µM, 18.5 µM , 36.9 µM, 55.4 µM, 101.5 µM, 221.5 µM and 443.1 µM. Each injection immediately resulted in a measurable electrochemical oxidation current with the signal returning to the background within 5–10 s, Fig. 4. The measured charge, Q, corresponded to total H2O2 oxidation, and showed a linear relationship to the quantity of injected lactate, x, up to a maximum of 101.5 µM (specifically, Q (nC) = 0.1835x (fmol), r = 0.993). The precision of measurement was investigated from four repetitive measurements with standard deviations <5.2%. To account for the non-Faradic current caused by the injection pulse perturbing the electrode double layer, two control experiments were performed involving the addition of identical volumes of buffer of the same ionic constitution, and the injection of lactate before the sensor was modified by enzyme. Although, in either case, there was a small background signal, the integral of total Q was always <2% of the equivalent titre of lactate, measured under identical experimental conditions.
 | ||
| Fig. 4 Representative current–time response to successive additions of pL lactate, as described in the text. The total charge generated is the integral of the current–time curve. The relationship between Q with respect to the amount of lactate added was linear between 4.8 and 66.0 fmol, with a correlation coefficient r of 0.993. The sensor surface area was 1800 µm2. | ||
A minimum detection limit of 4.8 fmol was obtained (equivalent to the detection of ca. 7.4 µM considering the chamber volume of 650 pL), which compared favourably with other immobilised macro- and microelectrode biosensor systems.21,22 It is also interesting to note the fast response of the sensors, especially when compared to that using free-enzyme sensors.12 This high temporal resolution and sensitivity is important for real-time monitoring of metabolites.
Characterisation of pH changes in the microfluidic system
To measure the local pH change during field stimulation, the microchamber was filled with the pH-sensitive dye BCECF (10 µM) buffered with 20 mM HEPES. Monophasic rectangular pulses of 4 ms with alternating polarity and amplitudes between 1.5 V (±0.75 V, field strength 75 V cm−1) and 2.5 V (field strength 125 V cm−1) were applied at 1.0 Hz to the stimulatory electrodes. It was noted that there was no change in the local pH under conditions of lowest field (75 V cm−1), although at increased field strengths there were small local changes. To confirm that no significant change occurred at the lowest field the voltage pulse was then applied continuously for 25 s and the change in BCECF emission was monitored, Fig. 5. It was noted, however, at high field strengths there was a rapid change in pH associated with local electrolysis.24 | ||
| Fig. 5 Time course of local pH change within the 650 pL microchamber buffered in 20 mM HEPES at field strength above the threshold for electrical stimulation of single myocytes. The change in emission of BCECF was related to the initial emission and plotted as the relative intensity against time. The pH was monitored from a region of 4 µm × 4 µm which was located 2 µm away from the initial anode within the microchamber. After 25 s of continuous pulsing, the polarity of the pulses was reversed. | ||
Single cell placement and contraction by field stimulation
After cell placement, there is ca. 1 min before cell adhesion begins. During this time, the cell was micro-manipulated into the desired orientation with respect to the axis of the stimulatory electrodes. Direct contact of the cardiomycote with the silver reference electrode resulted in cell hyper-contraction, so this was always avoided. Immediately after surface adhesion, the cell was forced to beat continuously by applying electrical fields across the stimulating electrodes.In common with previous reports, it was found that stimulating the myocyte along the length of the cell, as opposed to across its width, reduced the potential at which stimulation could be initiated.23,24
Intracellular Ca2+ during continuous pacing of the aerobic cell
Ca2+ transients from single, continually paced cardiomyocytes were recorded as a Fluo-3 emission. An isolated Fluo-3 loaded cardiomocyte was placed into the microchamber and contracted by stimulatory pulses at 0.5 Hz, followed by an increased pacing rate of 1 Hz, Fig. 6(a). Cell contraction was simultaneously recorded as the change of the sarcomere length (∼2.0 µm diastolic to ∼1.6 µm systolic), Fig. 6(b). The regular change of the sarcomere length at any given frequency demonstrated the cell’s ability to produce an action potential upon the electrical stimulation within the microfluidic structure. The cell also shortened in response to the rise in intracellular [Ca2+] and relaxed after the reuptake of the released Ca2+, indicating normal excitation-contraction coupling (EC coupling). The time course of the Ca2+ transients and the sarcomere length change were co-incident, and followed the stimulatory pulses (0.5 Hz and 1.0 Hz).![(a) Intracellular [Ca2+] transients from Fluo-3 indicator-loaded single cardiomyocytes stimulated at field strength of 75 V cm−1 and frequencies of 0.5 Hz and 1.0 Hz. The vertical axis gives the relative fluorescence intensity, and the vertical bars along the horizontal axis indicate the start of each pulse. (b) The relative mean sarcomere length recorded from the same cardiomyocyte within the microchamber, where 100% indicates the maximum relative degree of contraction with respect to the resting cell.](/image/article/2006/LC/b608202e/b608202e-f6.gif) | ||
| Fig. 6 (a) Intracellular [Ca2+] transients from Fluo-3 indicator-loaded single cardiomyocytes stimulated at field strength of 75 V cm−1 and frequencies of 0.5 Hz and 1.0 Hz. The vertical axis gives the relative fluorescence intensity, and the vertical bars along the horizontal axis indicate the start of each pulse. (b) The relative mean sarcomere length recorded from the same cardiomyocyte within the microchamber, where 100% indicates the maximum relative degree of contraction with respect to the resting cell. | ||
Monitoring of lactate from within the single aerobic cell
Permeabilisation of the cell plasma membrane was achieved by exposing the cell to large electric fields of short duration. This technique, known as electropermeabilisation, enabled the investigation of the intracellular chemistry of an isolated cell.25 The concentration of intracellular lactate usually present during aerobic respiration is ca. 2 mM within a cardiomyocyte of 10–20 pL volume.26 In order to investigate the intracellular lactate, a voltage pulse of 10 V with 0.04 ms duration was applied across the microelectrodes (separation of 200 µm) to irreversibly permeabilise the single cell. This resulted in cell hypercontraction, allowing intracellular lactate to diffuse into the extracellular surroundings. To understand the nature of the ionic currents resulting from the stimulation pulse, control experiments were carried out by applying the same electrical pulse but in the absence of the cell, Fig. 7(a). | ||
| Fig. 7 (a) The effect of applying short high voltage pulses (10 V, 0.04 ms) at 15 s, 30 s, and 45 s in the absence of the cell. (b) Typical responses to lactate released from an eletropermeabilised cell, (i) in the absence of pacing, no lactate is present in the extracellular space, (ii) electrochemical response as lactate is released from the single cardiomyocyte after eletropermeabilisation. The modified sensor was poised at 0.64 V vs. integrated Ag/AgCl microreference. | ||
Lactate was released from the cell immediately after the high pulse was applied. Fig. 7(b) shows a typical electrochemical response of lactate released from a single myocyte after electropermeablisation. Using the calibration curve, Fig. 4, we estimate that in total ca. 31 µM lactate was detected. Assuming that all the intracellular lactate was released and constrained within the microchamber, the average concentration of intracellular lactate was estimated to be 2.0 ± 0.1 mM (n = 3, assuming a myocyte volume of 10 pL), consistent with the normal values obtained from single healthy aerobic cells.26 In the future, this format will be used to monitor intracellular lactate released from the beating cell for different metabolic models, including those of hypoxia and ischemia, in order to provide a better understanding of the role of lactate in the control of both intracellular pH and cell metabolism.
Continual pacing and stimulated metabolism of the anaerobic cell
Single cardiomyocytes were continually stimulated in the microchamber by pulses of electrical field strength of 75 V cm−1 (a biphasic voltage of ±0.75 V giving 1.5 V per 200 µm) at 1.0 Hz. The effect of continual pacing on the contractility of single myocytes was investigated as the relative amplitude of cell shortening with respect to its length at rest, Fig. 8(a). Within the restricted extracellular volume of 650 pL, the myocyte was continually stimulated for 20 min, during which time the shortening amplitude decreased to ca. 25% of its initial (maximum) value. | ||
| Fig. 8 (a) Myocyte contractility measured as the change of the sarcomere length within the chamber when stimulated at electrical field strengths of 75 V cm−1 at 1.0 Hz. Single representative transients from a continuous record are shown. The values on the right-hand side represent the period from the development of steady-state shortening. (b) Recordings of extracellular pH during continuous contraction with field strength of 75 V cm−1 and a frequency of 1.0 Hz (as indicated through vertical bars which mark the point of electrical stimulation), using semi-quantitative single wavelength fluorescence of BCECF. (c) Simultaneous recording of cell contraction with a field strength of 75 V cm−1 and a frequency of 1.0 Hz, using edge detection measurement. | ||
The extracellular pH was monitored during 20 min of continuous cell contraction using semi-quantitative single wavelength fluorescence of BCECF, Fig. 8(b) and (c). Considering that there was no electrolysis acidification within the microchamber at the field strengths used, Fig. 5, we expect the acidification of extracellular space to be caused by efflux of lactate into the extracellular space. When glycolysis proceeds anaerobically, lactate acid production increases dramatically and accumulates within the intracellular space. Raised lactate and a fall in intracellular pH stimulate the lactate transporter that co-transports lactate and a proton. This electroneutral transport leads to the efflux of lactic acid from the cell which dissociates and acidifies the extracellular space.27 Data with cells equilibrated with the mitochondrial inhibitor CN− (as a model for ischemia) indicated that lactate acid production was considerably increased. Not only was the extracellular pH lower than control conditions, but the lactate signal after electroporation was ∼10× larger than the control (mean concentration of lactate = 18 mM). Detailed experimental studies showing the efflux of lactate into the extracellular space, either using field stimulated cells or by using a model for ischemia, where the cell’s glycolytic pathways are uncoupled using CN−, will form the basis of a further publication.
Finally, it was noted that the acidification of intracellular or extracellular space could also lead to a fall in the cell’s contractility. Renewal of the buffer balances the falling pH of the extracellular space, and thus the contractility partially recovers (to 60% that at rest), Fig. 8(a). The quantitative measurement of intracellular and extracellular pH during continuous stimulation will also be investigated in future studies to advance our understanding of the acidification mechanism.
The experiments described above, although technically exacting, typically allowed single cell measurements to be made every 30–60 minutes. In order to improve the rate of data acquisition in future we are currently developing an array sensor based upon our previous experience in multi-chamber microfluidic measurements.24
Summary
Using the contracting heart cell as a model and an enzyme-immobilised microfabricated sensor system, the experimental protocol described here provides a novel single-cell assay based format that can be used for monitoring intra- and extracellular dynamics, integrating electrochemical detection and fluorescence imaging under different metabolic conditions.Acknowledgements
The authors are grateful to the technicians in the Department of Electronics, particularly Mary Robertson and Helen McLelland for advice on the fabrication process. Aileen Rankin and Anne Ward (Biomedical Faculty, Glasgow University) are thanked for providing isolated rabbit cardiac cells. This work was supported by the IRC in Bionanotechnology (NK), the IRC in Proteomics Technology (HS), and the ORS scheme and the University of Glasgow (WC).References
- K. D. Kozminski, D. A. Gutman, V. Davila, D. Sulzer and A. G. Ewing, Anal. Chem., 1998, 70, 3123–3130 CrossRef CAS.
- C. D. Paras and R. T. Kennedy, Anal. Chem., 1995, 67, 3633–3637 CrossRef CAS.
- R. T. Kennedy, L. Huang and C. A. Aspinwall, J. Am. Chem. Soc., 1996, 118, 1795–1796 CrossRef CAS.
- L. Huang and R. T. Kennedy, Trends Anal. Chem., 1995, 14, 158–162 CrossRef CAS.
- W. H. Huang, D. W. Pang, H. Tong, Z. L. Wang and J. K. Cheng, Anal. Chem., 2001, 73, 1048–1052 CrossRef CAS.
- W. Z. Wu, W. H. Huang, W. Wang, Z. L. Wang, J. K. Cheng, T. Xu, R. Y. Zhang, Y. Chen and J. Liu, J. Am. Chem. Soc., 2005, 127, 8914–8915 CrossRef.
- H. Andersson and A. Van den Berg, Curr. Opin. Biotechnol., 2004, 15, 44–49 CrossRef CAS.
- H. Andersson and A. Van den Berg, Sens. Actuators, B, 2003, 92, 315–325 CrossRef.
- X. Lu, W. H. Huang, Z. L. Wang and J. K. Cheng, Anal. Chim. Acta, 2004, 510, 127–138 CrossRef CAS.
- C. D. T. Bratten, P. H. Cobbold and J. M. Cooper, Anal. Chem., 1997, 69, 253–258 CrossRef CAS.
- C. D. T. Bratten, P. H. Cobbold and J. M. Cooper, Anal. Chem., 1998, 70, 1164–1170 CrossRef CAS.
- X. X. Cai, N. Klauke, A. Glidle, P. H. Cobbold, G. L. Smith and J. M. Cooper, Anal. Chem., 2002, 74, 908–914 CrossRef CAS.
- P. Chen, B. Xu, N. Tokranova, X. J. Feng, J. Castracane and K. D. Gillis, Anal. Chem., 2002, 75, 518–524.
- A. A. Werdich, E. A. Lima, B. Ivanov, I. Ges, M. E. Anderson, J. P. Wikswo and F. J. Baudenbacher, Lab Chip, 2004, 4, 357–362 RSC.
- A. R. Wheeler, W. R. Throndset, R. J. Wheelan, A. M. Leach, R. N. Zare, Y. H. Liao, K. Farrell, I. D. Manger and A. Daridon, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS.
- W. H. Huang, W. Cheng, J. K. Cheng, D. F. Cui, Z. Zhang, D.-W Pang and Z.-L. Wang, Anal. Chem., 2004, 76, 483–488 CrossRef CAS.
- P. C. H. Li, L. D. Camprieu, J. Cai and M. Sangar, Lab Chip, 2004, 4, 174–180 RSC.
- M. A. McClain, C. T. Culbertson, S. C. Jacobson, N. L. Allbritton, C. E. Sims and R. M. Ramsey, Anal. Chem., 2003, 75, 5646–5655 CrossRef CAS.
- J. Gao, X. F. Yin and Z. L. Fang, Lab Chip, 2004, 4, 47–52 RSC.
- J. G. Shackman, G. M. Dahlgren, J. L. Petersa and R. T. Kennedy, Lab Chip, 2005, 5, 56–63 RSC.
- F. Palmisano, D. Centonze and P. G. Zambonin, Biosens. Bioelectron., 1994, 9, 471–479 CrossRef CAS.
- H. Frebela, G. C. Chemnitiusa, K. Cammanna, R. Kakerowb, M. Rospertb and W. Mokwab, Sens. Actuators, B, 1997, 43, 87–93 CrossRef.
- L. Tung, N. Sliz and M. R. Mulligan, Circ. Res., 1991, 69, 722–730 Search PubMed.
- N. Klauke, G. L. Smith and J. M. Cooper, Biophys. J., 2003, 85, 1766–1774 CrossRef CAS.
- J. Olofsson, K. Nolkrantz, F. Ryttsen, B. Lambies, S. Weber and O. Orwar, Curr. Opin. Biotechnol., 2003, 14, 29–34 CrossRef CAS.
- P. R. Moret, J. Weber, J. Haissly and H. Denolin, Lactate: Physiologic, Methodologic and Pathologic Approach, Springer-Verlag, Berlin, 1980, 16, 174–180 Search PubMed.
- J. I. Vandenberg, J. C. Metcalfe and A. A. Grace, Circ. Res., 1993, 72, 993–1003 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |