Fabrication and characterization of poly(methylmethacrylate) microfluidic devices bonded using surface modifications and solvents
Laurie
Brown
,
Terry
Koerner
,
J. Hugh
Horton
and
Richard D.
Oleschuk
*
Department of Chemistry, Queen's University, Kingston, Ontario, Canada K7L 3N6. E-mail: oleschuk@chem.queensu.ca
First published on 5th December 2005
Abstract
The fabrication of polymer microchips allows inexpensive, durable, high-throughput and disposable devices to be made. Poly(methylmethacrylate) (PMMA) microchips have been fabricated by hot embossing microstructures into the substrate followed by bonding a cover plate. Different surface modifications have been examined to enhance substrate and cover plate adhesion, including: air plasma treatment, and both acid catalyzed hydrolysis and aminolysis of the acrylate to yield carboxyl and amine-terminated PMMA surfaces. Unmodified PMMA surfaces were also studied. The substrate and cover plate adhesion strengths were found to increase with the hydrophilicity of the PMMA surface and reached a peak at 600 kN m−2 for plasma treated PMMA. A solvent assisted system has also been designed to soften less than 50 nm of the surface of PMMA during bonding, while still maintaining microchannel integrity. The extent to which both surface modifications and solvent treatment affected the adhesion of the substrate to the cover plate was examined using nanoindentation methods. The solvent bonding system greatly increased the adhesion strengths for both unmodified and modified PMMA, with a maximum adhesion force of 5500 kN m−2 achieved for unmodified PMMA substrates. The bond strength decreased with increasing surface hydrophilicity after solvent bonding, a trend that was opposite to what was observed for non-solvent thermal bonding.
Introduction
Since the development of the first microfluidic devices in the early 1990's, the field has rapidly expanded.1,2 Microfluidics has become popular as a result of the minute quantities of sample consumed during analysis and high throughput sampling capabilities of microfluidic devices.3 The first devices were fabricated as planar glass chips on which electrophoretic separations were performed.4,5 Since then, micro total analysis systems (μ-TAS) have been fabricated that carry out several sample preparation and analysis steps on a single chip.6Glass was the first substrate used to fabricate microfluidic chips because the photolithographic and chemical etching processes used to create these devices were well established in the microelectronics industry. The surface properties of glass were also well characterized allowing these devices to easily be tailored to several applications.2 More recently, polymers have been pursued as an alternative substrate for microfluidic devices because they offer a wider range of chemical and mechanical properties, and they are biocompatible.7 Microstructures can be inexpensively fabricated in polymers using several high throughput methods, including injection molding,8 soft lithography,9 laser ablation,10 X-ray photolithography11 and hot embossing.12 The low cost fabrication methods of microfluidic devices has had increasing relevance, as the need for single use, disposable microchips is required to prevent cross contamination during analysis of chemical and biological samples.7
Following fabrication of the microfluidic substrate, a cover plate must be bonded to the substrate to complete the fluidic conduits. Several methods of bonding rigid polymers have been explored, including thermal bonding,13,14 lamination,15 adhesives,16 solvents17–20 and surface modifications.21 To date, thermal bonding of polymeric microchips has been the most popular method. However, the bonds formed have a typical tensile strength of 100 kN m−2, and microchips must be handled with care to avoid cover plate delamination.22 Bonding of polymers with very small channels is difficult because the changes to the surface must be closely controlled. Ideally, the bond must be strong enough to withstand the requirements of the chemistry and attached instrumentation without delamination, the channel must not be clogged, and the channel integrity, shape and structure must not be altered during the bonding process.23
Here, we describe a robust, reproducible bonding method for poly(methylmethacrylate) (PMMA) microfluidic chips with small embossed features using a hot embosser. Surface modifications, including air plasma treatment, hydrolysis and aminolysis were performed on the PMMA substrate and were studied to determine if covalent and or hydrogen bonds between modified PMMA substrate and cover plates during bonding procedures increase substrate and cover plate adhesion. Solvent bonding of polymer microfluidic devices has also been explored as a viable method of increasing the substrate and cover plate adhesion. A solvent bonding method was developed for PMMA microchips that softened less than 50 nm of the polymer surface layers, allowing very strongly bonded microfluidic devices to be fabricated with no observable degradation of the channel features. The same surface modifications were again analyzed with this solvent bonding protocol to further characterize the transformations occurring at the surface of the polymer, and how this affects the adhesion strength. The surfaces were characterized using nanoindentation, spectroscopic and microscopic techniques. Nanoindentation was used to probe the effects of surface modification and solvent exposure on the mechanical properties of the surface. The bond strengths were measured, and compared to determine an optimized method of bonding PMMA substrates and cover plates.
Experimental
Materials and chemicals
Poly(methylmethacrylate) (PMMA) polymer sheets (150 mm × 150 mm × 1.5 mm and 150 mm × 150 mm × 2 mm) were purchased from Warehoused Plastic Sales Inc. (Toronto, ON, Canada) and cut to size before use. Sylgard 184 silicone elastomer and curing agent were purchased from Dow Corning Corporation (Midland, MI, USA) and used without modification. Conapoxy® FR-1080 high temperature epoxy was purchased as a kit including resin (glycidyl phenol/formaldehyde polymer), and hardener (nadic methyl anhydride, 2,4,6-tris(dimethylaminomethyl)phenol, bis[(dimethylamino)methyl]phenol), from Cytec Industries Inc. (Olean, NY, USA). Ethylenediamine, fluorescamine and methyl sulfoxide (DMSO) (HPLC grade) were purchased from Aldrich Chemicals (Milwaukee, WI, USA). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (WSC) was acquired from Fluka (Buchs, Switzerland). Methanol (ACS reagent grade), 2-propanol (ACS reagent grade), and acetone (ACS reagent grade) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Sulfuric acid (ACS reagent) was obtained from Fisher Scientific (Nepean, ON, Canada). Deionized water (18.2 MΩ) was purified by a MilliQ apparatus (Millipore, Bedford, MA, USA).Acid catalyzed hydrolysis of PMMA surfaces
PMMA was sonicated for 10 min in a 50% aqueous 2-propanol solution and dried under a stream of nitrogen to clean and hydrate the polymer surface. The PMMA was then submerged in 3 M sulfuric acid at 60 °C for 20 min (Fig. 1). After removal from the sulfuric acid solution, the PMMA was rinsed with copious amounts of water, followed by rinsing with 2-propanol and drying with nitrogen.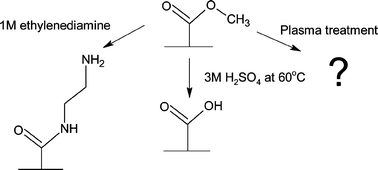 | ||
| Fig. 1 A schematic diagram of the surface modification performed on the surface of PMMA. | ||
Aminolysis of PMMA surfaces
PMMA was sonicated for 10 min in a 50% by volume solution of 2-propanol and water, following by drying under a nitrogen stream. The PMMA cover plates were then immersed in 1 M ethylenediamine in DMSO for 20 min at room temperature (Fig. 1). Following surface modification, the PMMA was rinsed with 2-propanol and dried with nitrogen.Air plasma modification of PMMA surfaces
PMMA was sonicated for 10 min in a 50% aqueous 2-propanol solution, and then dried under a stream of nitrogen. The clean and dry PMMA cover plates were placed in a PDC-001 air plasma chamber (Harrick, Ossining, NY, USA) and allowed to react for 10 s at 100 mTorr and 20 W (Fig. 1). After plasma modification was complete, the modified PMMA was then rinsed with 2-propanol and dried with nitrogen.Preparation of PMMA microfluidic chips
All microfluidic chips were prepared from PMMA (1.5 mm) by hot embossing using a HEX-01 hot embosser obtained from Jenoptik Microtechnik (Jena, Germany). An epoxy master clone was fabricated from a glass array of ‘double t’ microfluidic devices (Micralyne, Edmonton, AB, CA) using methods described previously.24 Briefly, a polydimethylsiloxane (PDMS) replica mold of the glass microfluidic array was fabricated into which a high temperature epoxy resin was poured, and cured according to manufacturer's instructions. The cured epoxy was removed from the PDMS mold and used as a stamp to fabricate microstructures in PMMA by hot embossing. The epoxy master and polymer substrate were aligned in the embossing chamber, the stamp and polymer were heated to 115 °C, and the chamber was evacuated. A force of 2500 N was applied for 12 min, after which time the chamber temperature was decreased to 50 °C. The chamber was opened and the epoxy master was de-molded from the substrate by hand. A PMMA plate was cut to cover the microfluidic chip and 2 mm access holes were drilled in this plate to allow access to the micro-channels. Surface modification was performed only on the cover plates of the microfluidic chips fabricated. Prior to bonding, both the substrate and cover plates were sonicated in a 50% aqueous 2-propanol solution for 10 min and dried under a nitrogen stream.Surface modification assisted bonding of PMMA microchips
The PMMA substrate and cover plates (modified or unmodified surfaces) were aligned on the plates of the hot embosser, the embossing chamber was evacuated, the temperature was brought to 85 °C, and a force of 5000 N was applied for 30 min. Following bonding the tool and substrate were cooled to 45 °C, the chamber was brought to atmospheric pressure and subsequently opened for removal of the bonded chip. It should be noted that this protocol does not bond unmodified PMMA, however, for comparison purposes, unmodified PMMA microchips were bonded in this manner as well. Optimized thermal bonding of unmodified PMMA microchips using hot embossing techniques occurred in an evacuated embossing chamber at a temperature of 105 °C and an applied force of 1000 N for 2 min.Solvent assisted bonding of PMMA microchips
Four drops of a solvent mixture (47.5% DMSO, 47.5% water, 5% methanol (ACS grade)) were evenly spread over the stamped polymer substrate, the cover plate (surface modified or unmodified PMMA) was placed in conformal contact and aligned. The two were then placed on the plates of the hot embosser and brought to 85 °C with a force of 3000 N applied for 30 min. Following bonding, the tool and substrate were cooled to 45 °C, the chamber was opened and the bonded chip was removed.Measurement of contact angles
PMMA was cut to 15 mm × 15 mm × 2 mm dimensions and the surface was modified as described above. A 2.0 µL volume of water was added to the surface of the PMMA, and a photograph of each droplet was taken immediately using a Nikon Coolpix 990 (Nikon, Toronto, Canada) digital camera. Advancing contact angles were measured from these photographs and reported values are the average of three separate droplets on each substrate.Quantitation of carboxy-terminated groups on PMMA surfaces
PMMA samples were cut to 15 mm × 15 mm × 2 mm dimensions. The PMMA was sonicated for 10 min in a 50% aqueous 2-propanol solution and dried under a nitrogen stream. These samples were modified using acid catalyzed hydrolysis or air plasma modification. Acid catalyzed hydrolysis was performed for up to 20 min at 60 °C, and air plasma modification was performed for up to 30 s at 100 mTorr and 20 W. After surface modification the samples were rinsed with copious amounts of water, rinsed with 2-propanol and dried under a stream of nitrogen. The modified PMMA samples were immersed in 3 mL of a N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (WSC) aqueous solution (10 mM) and allowed to react at room temperature for 2 h.25 The PMMA pieces were removed from the reaction solutions and the remaining concentration of WSC was measured using a Cary 3 UV/Vis spectrophotometer (Varian, Mississauga, ON, CA) at 215 nm to determine the extent of reaction of the modified PMMA with the carbodiimide solution. Each value reported is the average of at least three separate samples for each time tested. A calibration curve was prepared by measuring the absorbance of varying concentrations of WSC.Quantitation of amine-terminated groups on the surface of PMMA
PMMA samples (15 mm × 15 mm × 2 mm) were sonicated for 10 min in a 50% aqueous 2-propanol solution and dried with a nitrogen stream. These samples were modified in 1 M ethylenediamine in DMSO for up to 20 min at room temperature. After surface modification the samples were rinsed with water and 2-propanol followed by drying under a stream of nitrogen. The amine-modified PMMA samples were immersed in 3 mL of acetone for 30 min at room temperature to dissolve the surface layers of the polymer, followed by the addition of 1 mL of fluorescamine dissolved in acetone (0.1 mg mL−1) after the allotted period of time. Fluorescence measurements were performed using a 814 Photomultiplier Detection System (Photon Technology International, Lawrenceville, NJ, USA) with an excitation wavelength of 396 nm and fluorescence intensities were recorded at an emission wavelength of 475 nm.34 A calibration curve of varying concentrations of ethylenediamine solutions was prepared and the concentrations of amine functional groups on the surface of PMMA were calculated using this curve. Each value reported is the average of at least three separate samples for each time period tested.Measurement of cover plate adhesion strength
The tensile strength of the bond between the modified or unmodified cover plates and unmodified substrate plates was determined using an Instron 3369 tensile tester (Instron, Toronto, ON, CA). Two strips of PMMA (70 mm × 20 mm × 1.5 mm) were subjected to the same surface modification protocols as the fabricated microfluidic chips, as outlined above. The resulting strips were then overlapped (1.0–3.0 cm2) and then adhered to one another using different bonding conditions. The samples were then clamped into the tensile tester with grips set 114 mm apart, and pulled away from each other at a rate of 1.3 mm min−1.26 The force at which the bonded PMMA failed was measured and divided by the cross sectional area to obtain the tensile strength of adhesion. The reported values are the average of at least five samples per cross sectional area, and three cross sectional areas were tested for each method of bonding.Nanoindentation measurements on the surface of PMMA
PMMA pieces (10 mm × 10 mm × 2 mm) were modified using the surface treatments described above. Nanoindentation measurements were performed for each of these PMMA substrates, and solvent treated PMMA samples were prepared by immersing the modified PMMA pieces in 2 mL of the solvent mixture (47.5% DMSO, 47.5% water, 5% methanol) at 85 °C for 30 min to mimic the conditions of the hot embosser. A combined atomic force microscope (AFM)/nanoindentation system was used both for imaging of the samples and for determining the mechanical properties of the PMMA. All analyses were carried out using a Pico scanning probe microscope (SPM) (Molecular Imaging, Phoenix, AZ) scanner interfaced to a Nanoscope IIE Controller (Digital Instruments, Santa Barbara, CA) and Hysitron Triboscope low load test system (Hysitron Inc., Minneapolis, MN). The same tip was used for both imaging and indentation experiments. The tip that was used was a conical diamond tip with an effective radius of 1 µm. Due to the relatively large tip radius, the image resolution when using this indenter tip was about 100 nm—considerably larger than that achieved using a standard AFM tip (about 10 nm). The tip was suspended on a three plate capacitative transducer. Before indentation, the region of interest on the sample was imaged by monitoring the z-displacement of the central plate of the transducer by the feedback circuit of the AFM controller as in a standard contact mode AFM experiment. Force-displacement curves were generated by increasing the indenter load over a period of 5 s up to a maximum of 10–200 µN. The maximum load was held constant for 2 s, allowing the indenter velocity to drop low enough for any time-dependent plastic deformation to occur. After the hold, the load was removed from the sample at the same rate. Following the indentation cycle, the feedback loop was reset and the instrument returned to the imaging mode. Images of the indented region were acquired immediately. Each sample submitted was analyzed within 20 min of preparation.Reduced modulus values reported below were determined using the method previously described by Oliver and Pharr.27 In this case, a tangent is fitted to the unloading portion of the curve; the slope of the tangent fit is equal to the stiffness of the sample. The stiffness, S, is related to the reduced modulus, Er, of the system by the following:
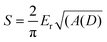 | (1) |
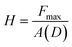 | (2) |
The tip area functions are unknown and were determined experimentally from eqn (1) by indenting into a standard of known modulus. A series of 20 indents at loads ranging from 10–1000 µN (and hence of varying contact depths) were made on a standard fused quartz substrate of Er = 69.6 Gpa. The resulting contact areas were then fit to a second order polynomial as the area function.
Results and discussion
Contact angle measurements of surface modified PMMA
The surface of PMMA was modified using three methods. Sulfuric acid modification is thought to catalyze the conversion of the terminal ester groups to carboxylic acid groups making the surface more hydrophilic. The chemistry occurring during plasma treatment is not as well understood. The plasma bombards the PMMA surface with high-energy gas molecules resulting in a variety of modifications occurring on the surface of the PMMA. From oxygen gas alone, it is thought that carboxyl groups, as well as oxides, acid anhydrides and aldehydes may form, which would also result in a more hydrophilic surface.28 Aminolysis was also performed on the PMMA surface, to generate primary amine functionality. The average measured advancing water contact angle for native unmodified PMMA is 76 ± 4°. This was in agreement with a previously published value of 75 ± 2° for unmodified PMMA surfaces.29 Water contact angles for each of the modified PMMA substrates (shown schematically in Fig. 1) were also measured (Table 1). The changes in water contact angles for each of the modified and unmodified PMMA samples clearly indicate that a chemical change had occurred during surface modification. The lower contact angles observed demonstrate that polarity, or hydrophilicity, of the PMMA surfaces increased30 with acid catalyzed hydrolysis and further yet with the addition of an amine functional group on the surface. This trend in hydrophilicity was as expected. After aminolysis with ethylenediamine, the PMMA has two additional electronegative nitrogen atoms on the surface, which would increase the polarity further from the hydrolyzed surface (Fig. 1). The modification by air plasma treatment results in the most hydrophilic surface. The hydrophilic nature of the air plasma treated PMMA may be due to the presence of several functional groups on the polymer surface, each contributing to the polarity of the surface.| Surface modification of PMMA | Advancing water contact angle/° | Tensile strength/kN m−2a | Tensile strength/kN m−2b |
|---|---|---|---|
| a The tensile strengths of PMMA bonded at 85 °C with unmodified and modified surfaces. b The tensile strengths of unmodified and modified PMMA bonded with the aid of a trisolvent system (47.5% DMSO, 47.5% water, 5% methanol). | |||
| Unmodified | 76 ± 4 | NA | (5.5 ± 0.3) × 103 |
| Acid-hydrolysis | 69 ± 1 | (0.3 ± 0.2) × 103 | (4.0 ± 0.3) × 103 |
| Plasma treatment | 35 ± 6 | (0.6 ± 0.3) × 103 | (2.6 ± 0.8) × 103 |
| Aminolysis | 57 ± 5 | (0.5 ± 0.2) × 103 | (3.6 ± 0.5) × 103 |
Quantitation of modified PMMA surfaces
In order to determine the extent of reaction and the amount of carboxylic acid groups on the surface of hydrolyzed PMMA, the polymer was modified with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (WSC), which binds selectively and quantitatively to carboxylic acid groups.31 After removal of the PMMA, the change in WSC concentration was measured. The number of carboxylic acid groups formed by acid catalyzed surface modification increases with time up to 20 min exposure to a concentration of 240 ± 70 nmol cm−2. No further increases in surface density were observed following longer exposures, which suggests the reaction is complete in this time. Plasma treatment showed a large increase in carboxylic acid functional groups to 210 ± 80 nmol cm−2 after only 10 s of exposure, however, at longer times showed a decrease in carboxylic acid functionality on the surface. With extended plasma treatment, these carboxylic acid groups may undergo further reaction by ionization and free radical formation resulting in a decrease in carboxylic acid functionality.32 As a result, 10 s was used for the remainder of the bonding experiments. The concentrations of carboxylic acid functional groups on the modified PMMA surfaces are approximations because a smooth surface morphology was assumed for all surface area calculations.Surface modification of PMMA to an amine terminus using a new, facile method was also studied. The method, commonly used to add amine functionality to the PMMA surface, is a reaction with a lithiated diamine. This chemistry is air sensitive, and the lithiated amines have limited lifetimes.33 In this study, a reaction was developed in which PMMA was immersed in a diamine solution in DMSO, an SN2 reaction promoting solvent, to obtain the same result. The quantitation experiments were performed using fluorescamine, a non-fluorescent molecule that reacts with primary amine groups to form a fluorescent compound. Any unreacted fluorescamine is quickly hydrolyzed to another non-fluorescent compound.34 A calibration curve of the fluorescamine reaction with known concentrations of ethylenediamine was prepared. It was assumed that fluorescamine reacted with both of the amine groups of ethylenediamine. The amine concentration on the modified PMMA surface increased up to 20 min of exposure time. At greater reaction times the surface of PMMA becomes opaque, and appears frosted and rough, rendering the PMMA useless for microchip fabrication. After 20 min, the amine concentration on the PMMA surface was 13 ± 5 nmol cm−2, which is slightly higher than the 6.85 ± 0.60 nmol cm−2 previously reported using lithiated amine chemistry.35 The difference in amine concentrations may be attributed to the increased surface roughness of the PMMA after 20 min of reaction time.
Thermal bonding of surface modified PMMA
Microfluidic chips were prepared from PMMA with cover plates modified with each of the methods described. The surface modifications were performed to determine if the presence of a more polar group on one surface of the PMMA microfluidic device may increase bond strength and stability of the device by the formation of hydrogen and/or covalent bonds between the PMMA surfaces. In this study, bonding was performed at 85 °C, however, no bonding of the unmodified PMMA was observed at this temperature. Thermal bonding usually requires heating the polymer to within a few degrees of the Tg to soften the surface and allow a bond to be formed.36 At 85 °C, no bond was formed when unmodified PMMA surfaces were used to fabricate a microfluidic device. This temperature was chosen because some softening of the surface may occur during surface modification, and at temperatures near the Tg, degradation of the channel features may occur. Therefore, any bonding occurring at this temperature must be caused by interactions of the modified/unmodified surfaces of the PMMA substrate and cover plates.Tensile strengths of the modified PMMA bonds increased with increasing hydrophilicity of the surface, with maximum bond strengths achieved for plasma modified microchips (Table 1). As the hydrophilicity increases, a greater number of possible hydrogen bonding sites will be available on the surface of the modified PMMA, which may explain the increase in bond strength. However, despite the increase in bond strength with surface modification, these polymeric microfluidic chips still must be handled with care to avoid bond delamination. A more durable bond was expected if covalent bonds had been formed between the polymer surfaces. The tensile strength of the optimized thermal bonding of PMMA is {1.0 ± 0.4} × 103 kN m−2, and although still not appreciable, is greater than the bonds that rely only on surface modification of the PMMA surfaces tested here.
Nanoindentation of modified PMMA
Nanoindentation experiments were performed for all of the surface modified PMMA substrates to determine the mechanical properties of the polymer surface. The load displacement curves obtained were used to determine the reduced modulus (essentially equal to the Young's modulus of the substrate) and hardness of the polymer as a function of indent depth.37The reduced modulus, and hardness of the unmodified PMMA are approximately constant at indentation depths between 0 and 200 nm. The reduced modulus of the PMMA was measured to be 5 ± 1 GPa (Fig. 2), comparable to the published value of 3.8 ± 0.5 GPa.37 A small increase in both reduced modulus and hardness at a depth of approximately 50 nm was observed. This result was reproducible for the unmodified PMMA but was not observed following any other surface treatment. The PMMA used for these experiments was fabricated for industrial purposes, and may contain modifiers or other impurities segregated in the near surface region that could cause the small increase in reduced modulus and hardness. The reduced modulus and, in the case of the plasma oxidized PMMA, the hardness changed quite dramatically when the surface of the PMMA was modified.
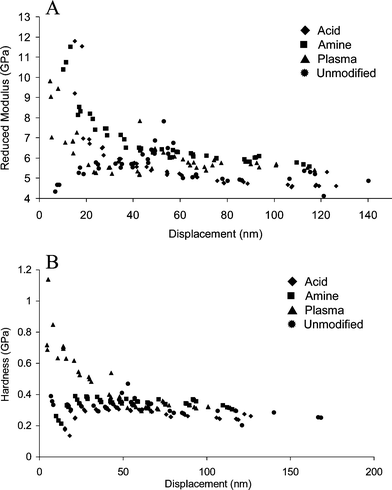 | ||
| Fig. 2 The reduced modulus (A) and hardness (B) of unmodified and modified PMMA substrates. | ||
The reduced modulus and hardness were used to characterize the extent of penetration of the surface modification procedures. The maximum depth of surface modification appears to be 50 nm in all cases. Any changes to the elasticity or hardness of the PMMA surface are observed at values less than this. The extent of this change to the properties of PMMA is ideal for microfluidic chip fabrication, where only the first few layers of molecules are modified, while the bulk properties of the polymer are maintained. The effects of these surface changes become negligible when microdevice channel features on the order of tens of microns are considered with respect to bonding.
The reduced modulus increases from 4 GPa for unmodified PMMA to approximately 12 GPa for each of the surface modifications performed (Fig. 2). The resulting decrease in elasticity is likely due to the presence of a hydrogen-bonded network formed by the presence of hydrophilic functional groups on the surface of the PMMA after surface modification.
In contrast, the only substrate that showed a significant increase in hardness was the plasma oxidized PMMA. Previous studies on plasma oxidized PDMS,38 showed an increase in modulus and hardness on these surfaces. This has been ascribed to the formation of a brittle layer of silicon oxides in the surface region. In the case of PMMA, the harder overlayer must arise from a formation of oxides, anhydrides and carboxylic acids in the surface region. The remaining modified surfaces were both exposed to solvent during the modification procedure. This may have led to a swelling and hence softening of these substrates during the modification procedure. Both show a very slight (<0.2 Gpa) decrease in hardness within the first 20 nm as compared to the unmodified PMMA, but this is close to the experimental error in the hardness determinations. The plasma treated PMMA resulted in the strongest bond formation for thermally bonded PMMA. Therefore, the presence of a highly polar surface to increase the number of chemical interactions occurring between the polymer substrates is likely more important than the mechanical properties of the surface when thermally bonding these materials.
Solvent assisted bonding of PMMA
In order to fabricate a PMMA microfluidic chip, the bond between the PMMA substrates must be durable. Several solvents were tested before an optimal solvent system and bonding protocol was found that allowed increased softening of the surface of the PMMA to aid in bond formation, and the maintenance of the channel features on the PMMA surface. A trisolvent bonding system has been developed consisting of a mixture of DMSO, methanol and water. The solubility parameters of PMMA, DMSO and methanol are 9.5, 12, and 14.5 (cal cm−3)1/2, respectively.39 While PMMA is not soluble in either DMSO or methanol at standard temperature and pressure,23 when subjected to increased temperatures and pressures, the polymer becomes slightly soluble in these solvents. Water, however, has a much higher solubility parameter of 23.4 (cal cm−3)1/2, and has no effect on the PMMA during hot embossing cycles.39 Water is added to the solvent mixture to increase the overall solubility parameter and maintain the channel structures during bonding. A cross sectional area of a solvent bonded unmodified PMMA microchip is shown in Fig. 3. The channel measures approximately 50 µm wide and 20 µm deep. The cross section confirms little degradation of the channel features after bonding with the solvent system developed. The tool used to cut the microfluidic chip caused the striations visible on the surface of the PMMA. | ||
| Fig. 3 The cross sectional area of a PMMA chip bonded with a trisolvent system consisting of 47.5% DMSO, 47.5% water and 5% methanol. The figure on the right is an SEM image of an unbonded channel which measures 50 µm wide and 20 µm deep. | ||
The tensile strengths of the solvent bonded PMMA microdevices were five to ten times greater than the thermally bonded PMMA microdevices (Table 1), forming a much more durable bond than thermally bonded PMMA devices. The tensile strengths were sufficiently high after solvent assisted bonding that the polymer microchips could not be delaminated without damage to the chip. The tensile strengths of solvent assisted bonding increased with increasing hydrophobicity. Unmodified PMMA microfluidic chips provided the strongest bond; whereas plasma treated PMMA had the lowest bond strength. The modification procedures may provide some protection to the PMMA surface when treated with the solvent system used for bonding by preventing the solvents from penetrating the surface.
Nanoindentation studies of solvent treated PMMA
Nanoindentation experiments were performed on these substrates to determine the changes occurring at the surface of the unmodified and modified PMMA as a result of the solvent treatment. These results are shown in Fig. 4. Regardless of previous treatment, immersion in solvent increases the reduced modulus at the surface to about 30 GPa. The previous modification history of the sample does not seem to be significant. In all cases, there is a sharp decrease back to bulk PMMA values within 50 nm of the surface. The effects of solvent treatment on reduced modulus are only significant at penetration depths of less than 50 nm, which again will not change the integrity of the micron scale channels.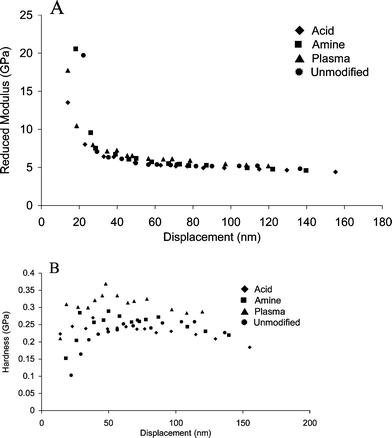 | ||
| Fig. 4 The reduced modulus (A) and hardness (B) of modified and unmodified PMMA substrates treated in a mixture of 47.5% DMSO, 47.5% water and 5% methanol at 85 °C for 30 min. | ||
The hardness of the PMMA samples is reduced when they are exposed to the solvent system, as seen in Fig. 4B. This is particularly significant for the plasma modified surface, the only sample that did not undergo previous exposure to solvent as a result of the first stage in the surface modification procedure. Thus, softening and decreased elasticity of the surface are the dominant trends observed following solvent exposure. The bond strength increases following solvent exposure. This may be due to the ability of the polymer surfaces to intermix within the softened layers, allowing a strong bond to form. The nanoindentation data give an indication of the effects of both the surface modification as well as the solvent on the ultimate bond strength of the PMMA microchips.
Conclusions
In this study, novel surface modification and solvent bonding protocols were developed as alternative methods for bonding PMMA microfluidic chips. Surface hydrophilicity was found to have a large effect on the strength of the bonds formed in both a thermally bonded and solvent bonded situation. The optimized solvent bonding system formed a very robust bond that did not interfere with the integrity of the channel features, and the microchips could be handled without any delamination. The adhesion strengths achieved are much greater than those of the best literature examples. The surface modifications performed would also allow the number of applications for polymer microchips to increase due to the large number of chemistries that can be performed with a surface carboxyl or amine group.Acknowledgements
We thank Genome Prairie (Enabling Technologies Project), the Natural Sciences and Engineering Research Council of Canada, Canadian Foundation for Innovation, Ontario Innovation Trust, Micralyne Inc. as well as Queen's University for financial support.References
- D. J. Harrison, A. Manz, Z. Fan, H. Ludi and H. M. Widmer, Anal. Chem., 1992, 64, 1926 CrossRef.
- D. J. Harrison, K. Fluri, K. Seiler, Z. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895 CrossRef CAS.
- J. Gao, J. Xu, L. E. Locascio and C. S. Lee, Anal. Chem., 2001, 73, 2648 CrossRef CAS.
- C. S. Effenhauser, A. Manz and H. M. Widmer, Anal. Chem., 1993, 65, 2637 CrossRef CAS.
- K. Seiler, D. J. Harrison and A. Manz, Anal. Chem., 1993, 65, 1481 CrossRef CAS.
- T. Vilkner, D. Janasek and A. Manz, Anal. Chem., 2004, 76, 3373 CrossRef CAS.
- T. R. Boone, A. J. Ricco, Z. H. Fan, H. Tan, H. H. Hooper and S. J. Williams, Anal. Chem., 2002, 74, 78A CAS.
- R. M. McCormick, R. J. Nelson, M. G. Alonso-Amigo, D. J. Benvegnu and H. H. Hooper, Anal. Chem., 1997, 69, 2626 CrossRef CAS.
- D. Qin, Y. Xia and G. M. Whitesides, Adv. Mater., 1996, 8, 917 CrossRef CAS.
- F. Dang, S. Tabata, M. Kurokawa, A. A. Ewis, L. Zhang, Y. Yamaoka, S. Shinohara, Y. Sinohara, M. Ishikawa and Y. Baba, Anal. Chem., 2005, 77, 2140 CrossRef CAS.
- Z. Meng, S. Qi, S. A. Soper and P. A. Limbach, Anal. Chem., 2001, 73, 1286 CrossRef CAS.
- Z. Chen, Y. Gao, J. Lin, R. Su and Y. Xie, J. Chromatogr., A, 2004, 1038, 239 Search PubMed.
- J. Chen, M. Wabuyele, H. Chen, D. Patterson, M. Hubert, H. Shadpour, D. Nikitopoulos and S. A. Soper, Anal. Chem., 2005, 77, 658 CrossRef CAS.
- A. Muck and A. Svatos, Rapid Commun. Mass Spectrom., 2004, 18, 1459 CrossRef CAS.
- S. Metz, R. Holtzer and P. Renaud, Lab Chip, 2001, 1, 29 RSC.
- F. Dang, S. Shinohara, O. Tabata, Y. Yamaoka, M. Kurokawa, Y. Shinohara, M. Ishikawa and Y. Baba, Lab Chip, 2005, 5, 472 RSC.
- R. Kelly, T. Pan and A. Woolley, Anal. Chem., 2005, 77, 3536 CrossRef CAS.
- A. Hiratsuka, H. Muguruma, K.-H. Lee and I. Karube, Biosens. Bioelectron., 2004, 19, 1667 CrossRef CAS.
- L. Kricka, P. Fortina, N. Panaro, P. Wilding, G. Alonso-Amigo and H. Becker, Lab Chip, 2002, 2, 1 RSC.
- H. Klank, J. Kutter and O. Geschke, Lab Chip, 2002, 2, 242 RSC.
- J. Behnisch, A. Hollaender, H. Blu, U. Heim and L. Mueller, World Pat., WO2001057119, 2001 Search PubMed.
- Y. Liu, D. Ganser, A. Schneider, R. Liu, P. Grodzinski and N. Kroutchinina, Anal. Chem., 2001, 73, 4196 CrossRef CAS.
- H. Becker and L. E. Locascio, Talanta, 2002, 56, 267 CrossRef CAS.
- T. Koerner, L. Brown, R. Xie and R. D. Oleschuk, Sens. Actuators, B, 2005, 105, 632 CrossRef.
- I.-K. Kang, D. K. Baek, Y. M. Lee and Y. K. Sung, Biomaterials, 1998, 36, 2331 CAS.
- Annual Book of ASTM Standards 2004, ASTM International, Philadelphia, PA, 2004, 08.01, D638–03 Search PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564 CrossRef CAS.
- J. Chai, F. Lu, B. Li and D. Y. Kwok, Langmuir, 2004, 20, 10919 CrossRef CAS.
- D. Raghavan, M. VanLandingham, X. Gu and T. Nguyen, Langmuir, 2000, 16, 9448 CrossRef CAS.
- C. Bain, J. Evall and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 7155 CrossRef CAS.
- Y. Yasaka and M. Tanaka, J. Chromatogr., B: Biomed. Appl., 1994, 659, 139 CrossRef CAS.
- J. Chai, F. Lu, B. Li and D. Y. Kwok, Langmuir, 2004, 20, 10919 CrossRef CAS.
- A. C. Henry, T. J. Tutt, M. Galloway, Y. Y. Davidson, C. S. McWhorter, S. A. Soper and R. L. McCarley, Anal. Chem., 2000, 72, 5331 CrossRef CAS.
- L. Locascio-Brown, A. L. Plant and R. A. Durst, Anal. Chim. Acta, 1990, 228, 107 CrossRef CAS.
- S. A. Soper, A. C. Henry, B. Vaidya, M. Galloway, M. Wabuyele and R. L. McCarley, Anal. Chim. Acta, 2002, 470, 87 CrossRef CAS.
- H. Becker and C. Gartner, Electrophoresis, 2000, 21, 12 CrossRef CAS.
- C. Klapperich, K. Komvopoulos and L. Pruitt, ASME J. Tribol., 2001, 123, 624 Search PubMed.
- G. Bar, L. Delineau, A. Hafele and M.-H. Whangbo, Polymer, 2001, 42, 3627 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley, NY, 1999, vol. VII, p. 675 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |