Handheld recirculation system and customized media for microfluidic cell culture†
Nobuyuki
Futai
a,
Wei
Gu
ab,
Jonathan W.
Song
a and
Shuichi
Takayama
*ac
aDepartment of Biomedical Engineering, University of Michigan, 2115 Gerstacker Bldg., 2200 Bonisteel Blvd., Ann Arbor, Michigan 48109-2099, USA. E-mail: takayama@umich.edu
bDepartment of Chemical Engineering, University of Michigan, 3074 H. H. Dow Bldg., 2300 Hayward St., Ann Arbor, Michigan 48109-2136, USA
cDepartment of Macromolecular Science & Engineering, University of Michigan, 2541 Chemistry Bldg., 930 N. University Ave., Ann Arbor, Michigan 48109-1055, USA
First published on 5th December 2005
Abstract
A palm-sized microfluidic recirculation system and customized media enable simplified long-term culture and imaging of cells. The combination of bare Braille display modules, a leveled monolithic surface for complete chip mounting, and a transparent heater improved portability, mechanical stability and optical accessibility. Modification of basal culture media with Leibovitz's L-15 medium enabled an incubator-free culture of carbonate-dependent cells by eliminating the need for exogenous carbon dioxide. This capability is demonstrated through time-lapse recording of proliferation of C2C12 myoblasts and MC3T3-E1 osteoblasts for over 2 weeks in ambient atmosphere without medium exchange. The method opens up new possibilities for portable cell culture and for long-term continuous visual monitoring of cells.
Many point-of-need cell culture applications such as modular cell cultures embedded in complex optical systems and handheld cellular sensors would benefit from portable, self-contained, maintenance-free, and long-term cell culture systems. Most optically accessible long-term cell culture systems described to date1,2 including some microfluidic systems,3–6 however, require an external media supply usually demanding a tank and a pump, CO2 supply, and accompanying interconnects and tubing.
Previously, we have presented some cell culture systems using commercially-available refreshable Braille displays and the microfluidic recirculation architecture eliminating the need for large external media pumps and tanks.7,8 In these systems, microfluidic channels have a deformable PDMS membrane (thickness: 100 ∼ 200 µm) at the bottom and channels with a smooth curved wall. The channel acts as a valve when the membrane is pressed by a pin of a Braille display and deformed. Three (or more) pins on a channel placed in series act as a pump by a peristaltic actuation sequence of the Braille pins such as OOC → OCC → CCO → COO, where O(Open) denotes a retracted pin, and C(Closed) a raised pin pushing the membrane up.
Although the devices are useful as self-contained cell culture systems that integrate all the needed fluid on a silicone rubber chip, the setups for cell culture still required an externally controlled atmosphere to maintain optimal temperature and CO2 concentration. Heating of the whole setup including the medium reservoir is not only unnecessary but degrades the medium ingredients and causes visually obvious evaporation (approximately a third of the reservoir evaporates in 3 days). Thus the previous setups required medium exchange every 1∼3 days. In addition, the convenience of these setups are limited since it is both necessary for the microfluidic chips to be separated from the opaque Braille display to observe them using transmitted light microscopy and for the microchannels to be realigned with the Braille pins after each such observation. The detachment of the chip from the Braille pin array disturbed the valving and pumping conditions resulting in unwanted mixing of fluids between channels originally insulated by Braille pins.
Here we present an improvement of Braille-pin-based cell culture system by use of a monolithic and functional “fingerplate” with accompanying media formulations for handheld cell cultures. It eliminates the need for an incubator or an external CO2 source to maintain cell culture and precludes the need for chip removal/realignment to perform optical observations. This system allows culture of highly CO2-dependent cells continuously in a non-37 °C, non-CO2-rich, non-humidified and non-sterile atmosphere with real-time cell imaging. The ability of dynamic and multiplied microfluidic control (such as flow segmentation, multiple laminar flow generation, and mixing) is one of the common features of Braille-based microfluidic systems, and there is no change in the present microfluidic device that reduces the advantages.
Fig. 1A shows the structure of a custom cell culture system. The system consists of (1) four 2 × 4-pin Braille display modules, (2) a machined aluminum monolithic fingerplate with two hold-down clamps, (3) a transparent heater unit, and (4) a battery-powered control circuit with Universal Serial Bus interface (see Fig. 1 of the ESI†). The monolithic fingerplate ensures stable chip mounting with sealing of the bottom membrane surface of the Braille-compatible PDMS microfluidic chip.8 Note that the chip mounting surface of a commercially available Braille display may not be level because each character cell (usually consisting of 8 pins) has its own fingerplate that is separated from adjacent fingerplates. The resulting non-flat surfaces can sometimes cause unwanted valving in some parts of the chip. Two hold-down clamps are used to fix the microfluidic chip instead of rubber bands or weights used in the previous studies. The positions of these hold-down clamps can be adjusted to match the thickness of the microfluidic chip by sliding the clamps along the stainless steel pole shafts installed perpendicularly on the monolithic fingerplate. The clamps constrain the displacement of the microfluidic chip without preloading to the chip that limits the deformation of the microchannels by Braille pins. Fig. 1B shows schematic views of the microfluidic chip that has four individual microfluidic loops for long-term cell cultures. Each loop has one U-shaped segment gathered at one cell cultivation area distant from Braille pins. In this setup, the cell cultivation area is accessible to transmitted light microscopy because the cultivation area is placed beyond the edge of the fingerplate (Fig. 1A). To implement a heated observation area, a slide glass with a flexible transparent heater was attached flush to the fingerplate. The cultivation area is in full contact with the surface of the slide glass and is heated through the slide glass. Heating the PDMS chip through the glass slide makes the temperature more uniform at the PDMS surface compared to applying the transparent heater directly to the chip. Temperature is controlled by a digital temperature controller and a thermocouple glued to the surface of the slide glass. It heats only the cultivation/observation area and reduces evaporation or degradation of the medium contained in the microfluidic chip. The overall custom system can be made for less than the cost of ready-made refreshable Braille displays designed for the blind because (1) most of the parts in the device are commercially available at low cost (typically $1,200 per system including an eight 8-pin actuator array for Braille display) and (2) it is unlikely that a long line of Braille characters (typically 16–46 8-pin actuator array) is needed for operating one microfluidic chip.
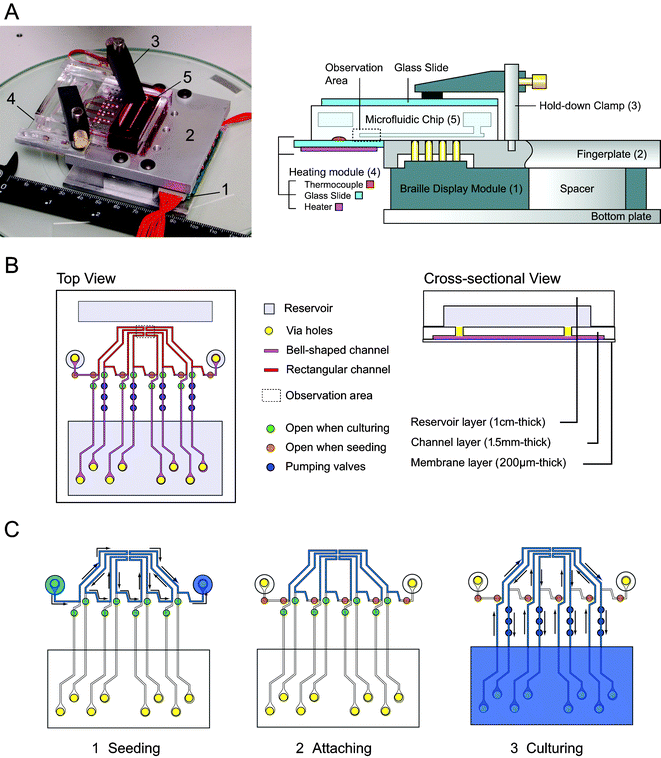 | ||
| Fig. 1 A handheld Braille display-based microfluidic cell culture system. (A) (left) A photograph of the whole system placed on a diascopic stand. 1. Six 8-pin Braille cells; 2. Aluminum fingerplate; 3. Two hold-down clamps to fix the PDMS chip; 4. Transparent heating unit; 5. PDMS microfluidic chip. (right) The configuration of a PDMS microfluidic chip installed on the Braille setup. The chip is fixed on a flat surface consisting of the fingerplate and the heater unit. The hold-down clamps are used to fix the chip by holding it down to the fingerplate. The Braille modules are fixed on the bottom plate so that the pins are aligned with the holes of the fingerplate. (B) Schematic representation of the microfluidic chip. The chip has four individual channel-reservoir loops for cultivation and one cell seeding line connecting all the loops. Cells flow and are cultured in the channels that have conventional rectangular sidewalls, whereas the channels to be deformed by the Braille pins have bell-shaped sidewalls. The region that includes the observation area is placed on the transparent heater unit. The microfluidic chip consists of three layers: reservoir layer, channel layer, and membrane layer.7,8 The channel layer has grooves at the bottom that compose the ceiling of the microfluidic channel, and punched via holes that connect the channels and the reservoirs. (C) Working scheme of the microfluidic chip for cell culture. (1) Cell seeding by injection of cell suspensions to one of the seeding ports. The cell suspension flows in cultivation areas in the channels and reaches another seeding port. (2) The Braille pins stop flow in the cultivation area to promote cell adhesion for 20 minutes–1 hour. (3) Cell culture. The valve configuration is then changed to enable perfusion of the four channels in the observation area. The channels extend beyond the opaque fingerplate to enable optical observation of cell cultivation areas by transmitted light microscopy. | ||
We could image the cultivation area and monitor cell growth simply by putting the entire setup on the stereoscope stage with a long working distance with 6×–10× magnification and oblique illumination. The objective did not conflict with the setup because of the large working distance of the stereo microscope. On the other hand, the setup can also be observed with an inverted microscope if (1) the center of the objective lens is placed immediately below the cell culture area, and (2) the working distance of the objective lens is larger than the total thickness of the PDMS membrane layer and the transparent heating unit. To satisfy (1), the channel running from the Braille pins to the cultivation area needs to be longer than the radius of the objective to give optical access. As for (2), objectives with a working distance over 2 mm (typically max. 20×) can be used with the transparent heater unit. An objective with shorter working distance requires a thinner transparent heater than the present one. Fig. 4 in the ESI† shows an example of a phase-contrast image of cells taken on a custom Braille display fitted to an inverted microscope.
We have also developed a modified basal cell culture media for long-term cell culture outside a CO2 incubator with continuous light exposure associated with long-term imaging. These modified media partially contain Leibovitz's L-15 medium and have the following advantageous characteristics: (1) high stability to light exposure, (2) limited but sufficient pH stability particularly in microfluidic cell cultures, and (3) sufficiently high NaHCO3 to satisfy immediate cellular needs for carbonates. We note that NaHCO3 is a necessary nutrient for many cell types9 but is a cause of basic shift of pH, which is harmful to cells, and that HEPES increases both pH stability and phototoxity. The addition of L-15 medium enables reduced use of NaHCO3 and HEPES because: (1) the high concentration of sodium pyruvate increases endogenous production of CO2;10 (2) free base amino acids enhances buffering capacity;10 (3) the partial replacement of glucose with galactose helps suppress pH drops due to rapid glycolysis.10 The decreased HEPES, high pyruvate, low riboflavin, and low phenol red concentrations used in the medium also improves stability against light exposure.11 These modifications of media when combined with the characteristics of small-volume microfluidic cell culture, which increases endogenous CO2 and other cell-generated products,12 enable incubator-free culture and long-term continuous optical imaging.
We tested long-term culture of C2C12 myoblasts and MC3T3-E1 osteoblasts with the microfluidic recirculation system filled with the modified media. Fig. 2 shows the first 6 days of a long-term culture experiment of the two cell types at high and low initial cell seeding densities. The results show that the combination of microfluidic channels, Braille-cell based recirculation system, and the modified media can support long-term cell culture at both low and high cell densities. Fig. 2C shows how different height channels affect myotube formation of C2C12 cells. From Day 6 to Day 8, relatively short myotubes (10–100 µm long) were formed in parallel in the 15 µm- and 30 µm- height channel, whereas subsequent fusion into larger myotubes (>100 µm) were more pronounced in the 30 µm-height channel than in the 15 µm-height channel. The effect of channel height on the mobility of myoblasts/myotubes was observed in the time-lapse recording of C2C12 (see ESI Fig. 3†). In the time-lapse recording of the 30 µm-height channel, some myoblasts and myotubes could migrate over another myotube and then fuse onto one of the myotubes. On the other hand, in the recording of the 15 µm-height channel, some grown myotubes contacted to the ceiling of the channel, so that the migration of some myoblasts or myotubes were blocked by the ceiling. In addition, the smaller channel heights, which are comparable to the size of the grown myotubes in Fig. 2C, restricted myotube mobility to increase myotube alignment but decreased myoblast fusion, resulting in smaller myotubes in the 15 µm-height channel. Thus, a unique effect of low-height microfluidic channels on myotube formation is that decreased spatial allowance decreases the migration of myotubes and the chance to form larger myotubes. These observations may explain why myoblasts do not differentiate or fuse well on some scaffolds and substrates.4,13
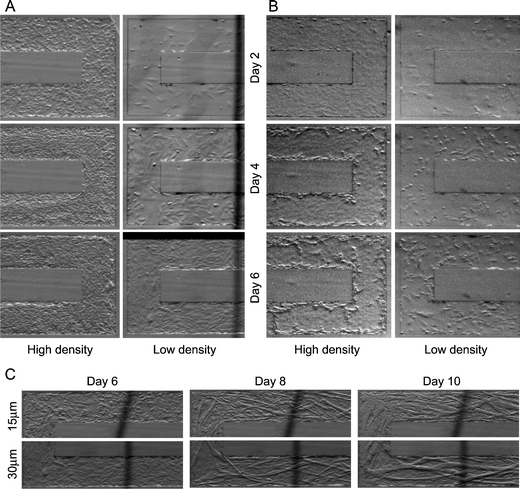 | ||
| Fig. 2 Time-lapse recording of cell proliferation and myotube formation in microfluidic channels (300 µm width, rectangular sidewalls). In all cultures, cells were seeded at Day 1. Culture media (2 ml per one channel) were not changed during culture. Actual recording rate was 10 min frame−1. (A) Proliferation of C2C12 myoblasts. The growth medium used was the modified formulation of DMEM and L15 (1∶1). At high density, cells were confluent at Day 2; then multilayering of cells occurred, resulting in a high packed culture. Cell migration upstream7 was seen after Day 6. Using the same medium, the cell growth from low initial cell density was also supported but no steady migration upstream was observed. (B) Proliferation of MC3T3-E1 osteoblasts. The growth medium used was the modified formulation of MEM α and L15 (1∶1). At high density, cells reached confluence on Day 2, multilayering started at the sidewalls on Day 4 and 6. In low density, cell growth rate was relatively slow but still steady, and reached near confluence at Day 6. In both initial densities, cell migration upstream was seen from Day 1 to Day 3. (C) Differentiation of C2C12 myoblasts induced by media replacement from growth medium to differentiation medium at Day 6 of the culture. The height of microfluidic channels does not greatly affect the speed of differentiation (number of multi-nucleated myotubes), but affects the size and alignment of the myotubes. Dark lines seen in these pictures are a defocused image of the copper wire in the transparent heater. | ||
This cell culture system has improved portability, reduced atmospheric requirements, and can maintain intact cell culture with minimal maintenance such as medium replacement. In addition to portability due to the vibration-, tilting-, and contamination-resistant nature of the Braille-driven monolithic PDMS chip, some of the notable improvements in this design compared to the commercially-available Braille displays used in our previous studies7,8 are (1) improved mechanical fixation of the microfluidic chip to enable stable valving and pumping, (2) local heating of the cell cultivation area to minimize evaporation and degradation of culture media in the reservoir, and (3) optical accessibility of cell cultivation area by transmitted light microscopy.
In this study, the setup with cells already inside was transported from a sterile cell culture hood in one part of a building to microscopes and other non-sterile places in another part of the building with no special carrying case. The setup is battery-powered and is stable when it is carried in a car or to other buildings as long as the setup is kept dark, clean, windless, and protected from sharp shocks (typically, simply placing the setup in a corrugated board case). For extensive field use, a more rugged and insulated container (compared to a corrugated board case), larger battery, and more clamps that apply to the PDMS chip will be needed, although the design of the Braille device and PDMS chips inside do not have to be changed.
These methods should find many applications in cell and developmental biology where long-term time-lapse recording or culture of cells is required. In addition, the present setup is especially advantageous in cell cultures which require long-term cell culture in a medium that has lower storage stability in elevated temperature and extensive medium changes have been required in conventional protocols. The small, self-contained, and portable nature of the system also opens new opportunities for point-of-need cell culture applications such as development of handheld cellular biosensors.14
Methods
Fluid control and heating system
A 48-pin Braille display module (SC9; KGS, Saitama, Japan) was used for fluid actuation. The Braille display module is controlled with a computer via Universal Serial Bus (USB) through a finger-sized standalone custom controller circuit board (Olimex; Plovdiv, Bulgaria). The Braille modules and the controller circuit were powered by a one-cell Lithium-ion battery for short durations (∼2 hours) or for longer durations by USB bus powering. Aluminum parts including a fingerplate, spacers, and bottom plate were fabricated by conventional machining.To fabricate a heater unit, a 250 µm-thick transparent heater made with a meandering line of a 30 µm-diameter copper wire insulated by sandwiching with polyester sheets (H15227; Minco, Minneapolis, MN) was bonded on a 75 mm × 25 mm × 1 mm glass slide (2947; Fisher) using cyanoacrylate adhesive, and a wire thermocouple (5TC-TT-J; Newport, Santa Ana, CA) was bonded to another surface of the glass slide using 2-component epoxy glue. A PID controller (E5GN; Omron; Schaumburg, IL) was used for temperature control.
Microfluidic chip for cell culture
The poly(dimethylsiloxane) (PDMS) microfluidic chip was fabricated by soft lithographic methods previously described.8 The main differences in the chip design from previous studies7,8 were (1) mixed channel design of multiple thickness and sidewall shapes, (2) extended cell cultivation channels for optical access, and (3) two seeding ports (0.2 ml capacity) in the reservoir layer to improve cell seeding (Fig. 1B). The fabrication processes of molds containing 30 µm-height bell-shaped cross-section, 30 µm- and 15 µm-height conventional rectangular cross-sections of the channels are outlined as follows: SU-8 50 (Microchem, Newton, MA) was spun on an acetone-cleaned 65 mm × 48 mm coverglass (Fisher) and patterned using 10 mW cm−2 collimated i-line from the photoresist side and diffused i-line from the substrate side through two pre-aligned 10000 dpi photoplotted films (CAD/Art Services, Poway, CA). The widths of the channels were all 300 µm regardless of the channel cross-sectional profile, using the compensation of photomask apertures shown in a previous study.15 After the fabrication processes, the completed microfluidic chip was sterilized by a germicidal lamp for 30 min, and 0.1 ml of human fibronectin (F2006; Sigma) solution (100 µg ml−1 in water) was incubated in the channels at 37 °C for 2 days to improve the biocompatibility of PDMS surfaces. The solution was injected to the seeding ports using a 30-gauge hypodermic needle, which were used for subsequent access to the microfluidic chip including injection, suction, and venting. Another detail that improves biocompatibility is to mix the PDMS well and to cure it for long periods of time (over 24 hours) at higher (75–120 °C) temperatures.General cell culture
C2C12 murine myoblasts were cultured in Dulbecco's Modified Eagle's Medium (DMEM; 11960; Gibco) with 15%v/v fetal bovine serum (FBS; 10082; Gibco), 1%v/v antibiotic–antimicotic (15240; Gibco) and 1%v/v GlutaMAX™-I Supplement (35050; Gibco). MC3T3-E1 murine osteoblasts were cultured in MEM α Medium powder (without ascorbic acid; Gibco) dissolved in distilled water with 10%v/v FBS and 1%v/v antibiotic–antimicotic. Both stock cultures were maintained under 5% CO2 on 100 mm cell culture dishes (Falcon), and passaged by dissociation with 0.25% trypsin-EDTA (25200; Gibco). Cell suspensions for seeding were made by centrifugation of dissociated cells at 100g for 1 minute.Reduced-CO2-dependence customization of medium
For C2C12 cells, DMEM powder (12100; Gibco) dissolved in distilled water, buffered with NaHCO3 and 2-[4-(2-hydroxyethyl)-1-piperadinyl] ethansulfonic acid (HEPES). The concentrations of NaHCO3 and HEPES in DMEM were 20 mM. The buffered DMEM was mixed with Leibovitz's L-15 medium (21083; Gibco) in a 1∶1 ratio. The modified basal media were supplemented as described above. For MC3T3-E1 cells, MEM α powder was used instead of DMEM, and the concentrations of NaHCO3 and HEPES in MEM α were 10 mM.Microfluidic cell culture
The media reservoir of the microfluidic chip was filled with 4 ml of the modified medium with venting immediately before starting cell culture inside the chip. Before filling, fibronectin solution in the reservoir layer level was suctioned, while the solution in the channels and the via holes connecting the channels and the reservoirs was not removed to prevent aeration. The microfluidic chip was then fixed on the cell culture device with pin alignment. Cell suspension sampled in a 1 ml syringe was injected into one of the seeding ports, while the other port is vented by another needle. During seeding, Braille pins actuated to a valve such that the cell suspension flows in all the cultivation areas and does not enter pumping channels (Fig. 1C). The cell suspension was allowed to flow by pressure through the syringe for 30 seconds. Then all valves were closed to stop the flow and the needles in the ports were removed.The culture device with the seeded microfluidic chip was placed on a diascopic light stand (C-DSS; Nikon) in room atmosphere (non-humidified, 25 °C, and 0.04% CO2). A stereoscope installed on the light stand was used for observation of the cells. Stereoscopes used for time-lapse recording of C2C12 and MC3T3-E1 were SMZ1500 and SMZ800 (Nikon), respectively. After placing the device on the light stand, the heating unit was powered and left to stand for 30–60 minutes to allow the cells to attach. Typically C2C12 cells attached to the coated surface within 20 minutes after seeding, while MC3T3-E1 attached within 40 minutes. The medium was then circulated by pulsatile pumping using three pins repeating a pattern of COO → COC → OCC → CCO (O: open, C: closed) at a 1 Hz refresh rate. The average volumetric flow rates generated by the sequence were obtained from the measurement of velocity of 2 µm-diameter fluorescent beads (FP-2045-2; Spherotech, Libertyville, IL). The measured average volumetric flow rate was approximately 50 nl min−1 regardless of channel height. Cell behaviors were recorded by time-lapse video microscopy for 8–21 days depending on the cell types and the initial seeding density. Fusion of C2C12 myoblasts into myotubes was induced by changing the medium in the media reservoir to the differentiation medium. The differentiation medium contains 2%v/v horse serum (26050; Gibco) instead of FBS. All pumps were stopped and all valves were closed during the medium change. Medium was not changed during cell culture except at this myotube fusion induction.
Acknowledgements
We thank Dr Brian Johnson and Prof. Mark Burns for use of clean room facilities, Dr James Foulke at the Center for Ergonomics, Univ. of Michigan for use of the machine shop facility, and the US Army Research Laboratory and the US Army Research Office under contract/grant number DAAD19-03-1-0168, the National Science Foundation (BES-0238625), and the NASA BioScience and Engineering Institute (NNC04AA21A) for funding.References
- G. Diaz, R. Isola, A. M. Falchi and A. Diana, BioTechniques, 1999, 27, 292 CAS.
- C. L. Ho, T. Y. Mou, P. S. Chiang, C. L. Weng and N. H. Chow, BioTechniques, 2005, 38, 267 CrossRef CAS.
- A. Prokop, Z. Prokop, D. Schaffer, E. Kozlov, J. Wikswo, D. Cliffel and F. Baudenbacher, Biomed. Microdevices, 2004, 6, 325–339 CrossRef CAS.
- A. Tourovskaia, X. Figueroa-Masot and A. Folch, Lab Chip, 2005, 5, 14–19 RSC.
- E. Leclerc, Y. Sakai and T. Fujii, Biochem. Eng. J., 2004, 20, 143–148 CrossRef CAS.
- P. J. Hung, P. J. Lee, P. Sabounchi, R. Lin and L. P. Lee, Biotechnol. Bioeng., 2005, 89, 1–8 CrossRef CAS.
- W. Gu, X. Y. Zhu, N. Futai, B. S. Cho and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15861–15866 CrossRef CAS.
- J. W. Song, W. Gu, N. Futai, K. A. Warner, J. E. Nor and S. Takayama, Anal. Chem., 2005, 77, 3993–3999 CrossRef CAS.
- A. Itagaki and G. Kimura, Exp. Cell Res., 1974, 83, 351–360 CrossRef CAS.
- A. Leibovitz, Am. J. Hyg., 1963, 78, 173–183 Search PubMed.
- W. H. Siegel and T. Pritchett, Biopharm.-Appl. T. Bio., 2000, 13, 65–68 Search PubMed.
- G. M. Walker, H. C. Zeringue and D. J. Beebe, Lab Chip, 2004, 4, 91–97 RSC.
- P. Clark, G. A. Dunn, A. Knibbs and M. Peckham, Int. J. Biochem. Cell Biol., 2002, 34, 816–825 Search PubMed.
- K. H. Gilchrist, V. N. Barker, L. E. Fletcher, B. D. DeBusschere, P. Ghanouni, L. Giovangrandi and G. T. A. Kovacs, Biosens. Bioelectron., 2001, 16, 557 CrossRef CAS.
- N. Futai, W. Gu and S. Takayama, Adv. Mater., 2004, 16, 1320–1323 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig.1, Block diagram of the circuit for Braille device control and power management; Fig.2, Long-term maintenance-free cell culture in microfluidic channels; Fig.3, Time-lapse recording of myotube formation shown in Fig. 2C; Fig.4, Phase contrast image of murine bone marrow cultured on a modified Braille setup. See DOI: 10.1039/b510901a |
| This journal is © The Royal Society of Chemistry 2006 |