Optimisation of the synthesis and modification of CdTe quantum dots for enhanced live cell imaging†
Stephen J.
Byrne
a,
Serena A.
Corr
a,
Tatsiana Y.
Rakovich
a,
Yurii K.
Gun'ko
*a,
Yury P.
Rakovich
b,
John F.
Donegan
b,
Siobhan
Mitchell
c and
Yuri
Volkov
c
aThe School of Chemistry, Trinity College, University of Dublin, Dublin 2, Ireland. E-mail: igounko@tcd.ie
bThe School of Physics, Trinity College, University of Dublin, Ireland
cThe Department of Clinical Medicine, Trinity College, University of Dublin, Ireland
First published on 12th June 2006
Abstract
We report the preparation and luminescence enhancement of thioglycolic acid (TGA) stabilised CdTe quantum dots (QDs) for use as live cell imaging tools in THP-1 macrophage cells. Short irradiating times utilising a high powered Hg lamp resulted in increases in luminescence efficiencies of up to ∼40% and permit significantly enhanced live imaging of the THP-1 cellular components. It was found that the TGA-stabilised QDs traverse the cell membrane, illuminating the cytoplasm and decorating the nuclear membrane. These studies highlight the potential use of photoetched CdTe QDs as probes for specific in vitro labelling.
Introduction
Advances in molecular medicine require the optimum detection of individual biomolecules, cell components and other biological entities. Traditional methods for detecting biological compounds in vivo and in vitro rely mostly on the use of radioactive markers or fluorescent dyes as tags (e.g. fluorescein, ethidium, methyl coumarin, rhodamine, etc.).1 However, these have a number of chemical and physical limitations which include the cost and complexity of methods utilizing multiple fluorescent dyes and the deterioration of their fluorescence intensity upon prolonged exposure to excitation light (photobleaching). In addition, the differences in the chemical properties of standard organic fluorescent dyes make multiple, parallel assays quite impractical since different chemical reactions may be involved for each dye used in the variety of applications of fluorescent labels.Fluorescent semiconductor (II–VI) nanocrystals, often referred to as quantum dots (QDs), represent a material of condensed matter possessing a characteristic spectral emission, which is tunable to a desired energy by selection of the particle size, size distribution and composition of the nanocrystal.2,3 QDs present a great potential for biolabelling applications due to their stable and narrow size-dependent emission which can be observed and measured spectroscopically by using fluorescent confocal microscopy. All of this makes QDs very promising for fluorescent biological imaging4–6 and biofunctionalisation.7,8
Nanocrystal stability is of paramount importance for their use in such instances and consequently, surface structure and reconstruction will play a crucial role. To enhance the quantum yield (QY) of the QDs, epitaxial-type shells of a semiconducting material with a larger band gap, such as ZnS or CdS9,10 have been grown on the surface to effectively cap the defects present. The transfer of QDs possessing large QYs which are stable in organic media to an aqueous phase via ligand exchange11 and photooxidation of the surface via irradiation12–14 have also been used to produce stable luminescent water soluble QDs. QDs possess considerable advantages over conventional organic dyes for live cell imaging and diagnostics15 due to their higher degree of photostability, multiplexing ability, tuneable emission spectra and broadband excitation over a wide wavelength range. Much work in this area has centered on the conjugation of highly luminescent QDs to proteins, peptides and other biological molecules and their subsequent addition to live cell cultures to elucidate the mechanistic pathways of their movement.16–18
However, limited studies into the use of unmodified thiol stabilised CdTe QDs for use as biological imaging reagents have been explored.18 Here we report the preparation and synthetic optimisation of thioglycolic acid (TGA) stabilised CdTe QDs, their luminescence enhancement by photoetching, PL lifetime data, Raman spectra and investigations into their use for live human macrophage cell imaging.
Experimental
General procedures
Aluminium telluride (Al2Te3) was obtained from Cerac Inc. All other chemicals and reagents were obtained from Sigma-Aldrich. All manipulations in the preparation and addition of precursors were carried out under vacuum or argon by standard Schlenk techniques. Absorption spectra were recorded using a Cary 50 Conc. UV–vis spectrophotometer. Photoluminescence (PL) measurements were performed on a Cary Eclipse fluorescence spectrometer. A Heittich Universal 32 machine at 3500 rpm was used for centrifugation.The Raman spectra were excited by a 488 nm line of an Ar+ laser with power in the range of 1 mW. A micro-Raman spectrometer (Renishaw-1000) equipped with 20× objectives and a cooled CCD camera was used in the experiments. The spectral resolution of the spectrometer was about 1 cm−1. Each spectrum was averaged over 20 measurements with an accumulation time of 20 s.
Luminescence decays were measured using time-correlated single photon counting (Time-Harp, PicoQuant). The samples were excited by 480 nm picosecond pulses generated by a PicoQuant, LDH-480 laser head controlled by a PDL-800B driver. The setup was operated at an overall time resolution of ∼150 ps. Decays were measured to 3000–5000 counts in the peak and reconvoluted using non-linear least squares analysis (FluoFit, PicoQuant), using an equation of the form:  , where τi are the PL decay times. The pre-exponential factors αi, were taken into account by normalisation of the initial point in the decay to unity. The quality of fit was judged in terms of a χ2 value (with a criterion of less than 1.1 for an acceptable fit) and weighted residuals.
, where τi are the PL decay times. The pre-exponential factors αi, were taken into account by normalisation of the initial point in the decay to unity. The quality of fit was judged in terms of a χ2 value (with a criterion of less than 1.1 for an acceptable fit) and weighted residuals.
TGA-Stabilised CdTe QD preparation and a modified 22 factorial statistical study
CdTe QDs were prepared according to the published procedure.12 100 ml of millipore water (18 MΩ) was degassed by bubbling argon for approximately 1 hour. Initially Cd(ClO4)2·6H2O (0.737 g, 1.76 mmol) and thioglycolic acid (TGA) (0.324 g, 3.51 mmol) [Cd–thiol ratio 1 ∶ 2)] were added and the pH was adjusted to 11.2–11.3 by the addition of a 2 M NaOH solution. Argon was further bubbled through the solution for approximately 30 minutes. H2Te gas, generated from Al2Te3 (0.128 g, 0.29 mmol) [Cd–Te ratio 2 ∶ 1] via dropwise addition of a 0.5 M H2SO4 solution, was bubbled under a slow argon flow for approximately 10 minutes. The solution was subsequently refluxed for 100 minutes. The samples were stored in darkness for a period of 24 hours before the absorption and luminescence spectra were taken. Subsequently, the ratios were adjusted as described in Table 1 and the experimental procedure was repeated. Room temperature luminescence efficiencies were determined by comparing the nanocrystal integrated emission with that of Rhodamine 6G in absolute ethanol with a quantum efficiency of 95%.| Cd ∶ Te ratio | Cd ∶ TGA ratio | QY after 100 min reflux | FWHM/nm | |
|---|---|---|---|---|
| A | 1.5 | 1.5 | 10% | 52 |
| B | 1.5 | 2.5 | 7% | 50 |
| C | 2.5 | 1.5 | 15% | 44 |
| D | 2 | 2 | 16% | 44 |
| E | 2 | 3 | 8% | 51 |
| F | 3 | 2 | 13% | 46 |
| G | 2.5 | 2.5 | 9% | 49 |
| H | 3 | 3 | 8% | 50 |
Photoetching
TGA-Stabilised CdTe QDs were diluted with 3 cm3 millipore water (particle conc. ∼10−5 M).19 These solutions (under constant stirring) were irradiated with a water-filtered, high pressure mercury (Hg) lamp (in air) [the field of irradiation was 350–1000 nm]. Luminescence and absorption spectra were taken to monitor any optical effects.Intracellular visualisation using confocal fluorescence microscopy
The human THP-1 monocyte cell line was obtained from the European Collection of Animal Cell Cultures (ECACC, Salisbury, UK). Cells were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine L−1, 100 µg penicillin ml−1 and 100 mg streptomycin ml−1, and incubated at 37 °C in 5% CO2. To induce the monocyte to macrophage differentiation, THP-1 cells were cultured in the presence of 100 ng ml−1 phorbol 12-myristate 13-acetate (PMA) for 72 hours. Cells were then washed three times with HBSS before use. THP-1 cells were incubated with a 1 in 100 dilution with the QDs. Live cell imaging was performed in Lab-Tek chambered coverglass slides (Nunc). Images were acquired by fluorescence microscopy (Nikon Eclipse TE 300) and on the UtraView Live Cell Imager confocal microscopy workstation (Perkin-Elmer Life Sciences, Warrington, UK) (Nikon Eclipse TE 2000-U). Processing was performed using UltraView LCI.Results and discussion
Optimisation of CdTe QD synthesis
The standard water based synthesis developed by Gaponik et al.12 was utilised as our synthetic route. In order to fine tune the resultant nanocrystal quantum yield (QY) we have carried out a statistical analysis of the different reaction conditions and parameters to further allow us to optimise the production of CdTe QDs. This was done by implementing a modified 22 factorial design, where the main factors involved are the metal to metal and the stabiliser to metal ratios present in the reaction.This type of experimental design allows us to vary the reaction conditions systematically, enabling us to determine both the effect of these main factors and any interaction between them. The experimental data are summarised in Table 1.
Fig. 1 shows the room temperature absorption spectra taken for the various QD solutions. As shown, certain reaction conditions are congruent with clearly resolved bands in the absorption spectrum (C, D, F), thus indicating a sufficiently narrow QD size distribution within the sample. Others show a less structured spectrum indicative of a certain degree of inhomogeneity within the sample (A, B, E, H, G). This can be further emphasized by the FWHM (full width at half maximum) values taken from the luminescence spectra (see Fig. 2), which show that the well resolved absorption peaks also possess the narrowest luminescence spectra. It is evident that the conditions which produce the QDs with greater quantum efficiencies also have narrower size distributions and also less surface defects as seen via the symmetrical nature of the excitonic emission (see Fig. 2) A small red-shift at the emission maxima can also be identified in samples E and A, thus indicating that over the same reflux period the varying conditions promote different nanocrystal growth in the same region.
 | ||
| Fig. 1 Room temperature absorption spectra of TGA-stabilised CdTe QDs taken 24 hours following QD synthesis. | ||
 | ||
| Fig. 2 Normalised room temperature emission spectra (λex 450 nm) of TGA-stabilised CdTe QDs taken 24 hours following QD synthesis (see Table 1 for references). | ||
The initial conditions (metal and stabiliser ratios) were chosen and then varied as a function of one another throughout eight experiments (Fig. 3). The response measured was the resultant QY and consequently the FWHM can also be determined from the emission spectra (Fig. 2) The design was carried out according to Table 1 and these values were used to construct a 3-D surface response plot from the resultant data (Fig. 4) From this plot the optimal ratio levels can be easily identified from the colour map surface. According to these data the highest QYs can be achieved at a Cd ∶ Te ratio of 2.1 ∶ 1 and a Cd ∶ TGA ratio of 1.75 ∶ 1. Thus, efficiencies and FWHM of TGA-capped CdTe QDs are directly proportional to both the stabilizer and metal ratios and the highest quality of nanocrystals can be controlled by the incorporation of the required stoichiometric amounts of the precursors. Repeat experiments were carried out at this optimum ratio producing an overall QY for the solutions of 20%. Previous reports have indicated that a QY of up to 65% (red spectral region) and 30% (green spectral region) were achievable.20,21 However, this increased QY requires low precursor concentration resulting in lower reaction yields. Decreased pH values have also been investigated producing similar results.21,22 While the values for the green region of the spectrum compare favourably with our results we wished to investigate the impact of photoetching as a luminescence enhancer and also investigate the visual impact of increasing the QY in live cells.
 | ||
| Fig. 3 Graphical representation of the modified 22 statistical factorial design. Each point represents a metal to metal and metal to stabiliser ratio. Numbers in bold represent the Cd ∶ Te ratios while those in italics represent the thiol ∶ Cd ratio. | ||
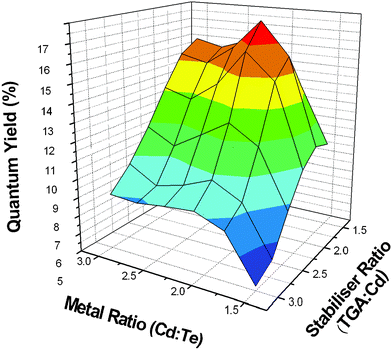 | ||
| Fig. 4 Statistical surface response curve for TGA capped-CdTe QDs with variations in the metal and TGA stabilizer ratios. | ||
Thus, a QY of 20% was our starting point from which we sought to improve the efficiencies further.
Improvement of quantum yield by photoetching
In terms of quantum efficiencies, non aqueous synthesis of QDs produce nanocrystals with superior luminescent properties than those of their water based counterparts.23,24 However, transfer of these dots to an aqueous phase results in a reduction in the overall luminescence efficiency of the QDs.11,25 In an effort to improve the QY of TGA-stabilised CdTe QDs we have employed the statistical analysis shown previously and analysed the effects of illumination on the resultant QY. Photoetching of aqueous CdTe QDs can aid in improving the luminescence properties of the solutions while maintaining the integrity of the dots themselves. This improvement of the aqueous luminescence of the QDs can somewhat enable us to achieve a long existing goal of highly luminescent and highly stable water soluble QDs.Photoetching has been explored by various groups yielding positive results and substantial increases in luminescence. Bao et al.13 using a low pressure Hg lamp witnessed a substantial increase in PL QY up to ∼85% over a 20 day illumination period at room temperature. Post-preparative size-selective precipitation and selective photochemical etching have also been proposed12 as methods providing an increase in luminescence quantum efficiency of the QDs of up to 40%. This QY increase has been explained by removal of the dangling bond associated with unsaturated Te atoms present on the surface, which act as hole traps. These and other defect states are broader, less pronounced and appear red shifted compared to that of the band-gap emission.26 At low temperature these states can be clearly resolved and identified27 however at room temperature they appear to cumulatively add to the intrinsic emission resulting in spectral broadening, increased FWHM values and decreased symmetry of the PL spectra. Upon illumination, these defects can be removed or reduced and superior QYs can be achieved. Dissolution of these highly defective QDs of a poorer quality from solution results in a QY increase accompanied by a blue shift in the PL emission wavelength.
As our aim was to investigate CdTe QDs in living cells, we would require the smallest possible sizes, so this growth mechanism of QDs would be ineffective. Also we wanted to achieve an increase in the PL intensity through sacrifice of the smaller and poorer quality dots and possibly even reduce the sizes of the dots throughout the sample.
However, unlike photoetching experiments carried out by and Bao et al.,13 where illumination times stretched from hundreds of hours to days of illumination, our experiments were undertaken in accordance with previously reported procedures12 aimed at obtaining samples for Raman and intracellular studies.
Fig. 5 shows the absorption and normalised room temperature PL spectra of TGA-capped CdTe QDs irradiated for 30 minutes by an Hg lamp. We can see that the enhancement in luminescence (up to ∼40% QY) is accompanied by only a slight increase in the absorption spectrum (Fig. 5a) in the region of the first electronic transition. In Fig. 5 we can also see that the luminescence spectra (Fig. 5b) recorded during photoetching are blue-shifted (by up to ∼8 nm), while the absorption maximum position remains constant. This substantial increase in QY and blue-shift can be attributed to the previously discussed dissolution of dots throughout the sample medium. While these poorer and defect-rich dots are removed the overall crystallite size distribution of the sample is reduced, but their removal also results in fewer dots present adding to the increased QY up to its maximum attainable value.
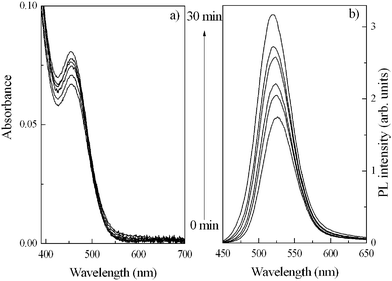 | ||
| Fig. 5 Absorption and normalised room temperature PL spectra (λex = 425 nm) for photoetched TGA-capped CdTe QDs. | ||
One direct way to understand the role of surface defect states in the photoinduced PL enhancement of semiconductor QDs is to monitor the QY of photoetched samples along with the PL decay dynamics. Unlike CdSe QDs whose time-resolved dynamic properties affected by photoetching have been thoroughly examined28 CdTe QDs are much less studied.
Fig. 6 shows the PL decay curves measured at the respective emission peak wavelengths for CdTe QDs whose spectra are presented in Fig. 5. The PL decay curves for each sample can be successfully simulated using a biexponential function with a shorter lifetime equal to 4–5 ns, and longer one equal to 23–27 ns. This multiexponential behaviour is almost universal in the PL dynamics of colloidal II–VI QDs.27–33 The shorter lifetime can be attributed to the intrinsic recombination of populated core states,34,35 while the longer lifetime can be associated with the involvement of surface states in the carrier recombination process.36 The shorter lifetime observed in our experiments correlates with theoretically obtained values of ∼3 ns taking into account the screening of electro-magnetic fields inside the QDs.37 With regard to the longer-lifetime component, the poor overlap of the carrier wavefunctions is trapped by the surface states, with hole wavefunctions from the internal core states accounting for lifetimes longer then 10 ns.36 In our case, values of longer lifetimes were found to systematically increase with irradiation time (i.e. in conjunction with PL efficiency) clearly indicating a delocalization of electrons following the photoexcitation and involvement of surface states in their recombination. However, observed biexponential PL decays give no way of deducing the quantitative information on the character of the recombination parameters of luminescence in QDs. Therefore, to gain a good understanding of PL modification dynamics during photoetching, an estimate of the average lifetime was obtained from the time in which emission intensity drops to 1/e of the initial value, τ.29,30 This allowed us to obtain a singular decay parameter, which can be considered as an effective average lifetime. Deducing τ from the PL decay curves presented in Fig. 6 and taking into account QY(Φ) values of the photoetched samples, we can calculate nonradiative (nnr) and radiative (kr) constants using the photophysical equations:
 | (1) |
 | (2) |
 | (3) |
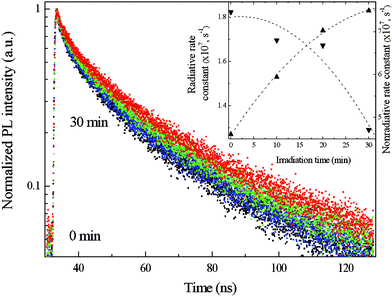 | ||
| Fig. 6 Four PL decay curves for TGA-capped CdTe QDs (whose spectra are presented in Fig. 5) recorded during the photoetching process. The insert shows radiative and nonradiative rate constants calculated by eqn (1) and (3) respectively. The dashed lines are simply guides to the eye. | ||
The insert in Fig. 6 shows estimated effective nonradiative coefficients in comparison with radiative rate constants. There is a clear correlation between the efficiency of luminescence (Fig. 5), increased radiative rate constants and substantial suppressing of nonradiative transitions.
As it has been suggested,12 all these phenomena observed in our experiments can be accredited to the photodegradation of excess and surface reacted TGA molecules and their subsequent incorporation into a CdS shell around the QDs. This epitaxial shell caps the defect surface states, thus reducing the number of nonradiative pathways and increasing the PL intensity, but also results in a decrease in the size of the dot core as indicated by a blue-shift in the PL spectra. Although very plausible, this suggestion was never put to the experimental test.
In order to gain a deeper insight into the mechanism of surface modification during photoetching we have utilized Raman spectroscopy. The high sensitivity of Raman spectra to surface reconstruction was demonstrated recently for CdSe QDs nanocrystals capped with an organic ligand38 and inorganic capping shell39 as well as QDs embedded in glass or polymer matrices.40,41 It was also shown that a strong dependence of the frequency of longitudinal optical (LO) phonons on the thickness of the QD shell, can be used as a tool to control the shell thickness with accuracy to 0.5 of a monolayer.39
In our Raman experiments CdTe QDs were deposited from their aqueous solution on a Si wafer. Because of the high quantum efficiency of the QDs, resonant Raman spectra were superimposed on a broad luminescence background. This background has been subtracted in all spectra presented below in order to show more clearly the Raman signal itself.
The group of lines shown in Fig. 7 can be well fitted by three Lorentzians with variable amplitudes, peak positions, and FWHM. The more pronounced peak in both spectra can be assigned to the fundamental LO phonon mode of CdTe. The Stokes shift of this peak, ΩLO = 164 cm−1, is smaller than the corresponding bulk value of 168 cm−142 by 4 cm−1. The shift originates from two sources: a red-shift due to confinement of the optical phonons,43 and a blue-shift caused by lattice contraction.38 The 15 cm−1 width of the LO phonon line for CdTe QDs in our experiment reflects the size distribution of the QDs (about 12% as estimated from the spectral width of the absorption peak). The presented Raman spectra also show a signal in the region of the first overtone (band at 320 cm−1) belonging to the CdTe, which partly overlaps the peak centered at 275 cm−1. This last peak shows an increasing intensity after photoetching of the aqueous solution of CdTe QDs indicating the contribution of the crystallographic phase which cannot be assigned, either to the CdTe core or the TGA capping layer. Considering the presence of excess surface TGA molecules in our samples and an increase in the intensity of the 275 cm−1 peak after photoetching, we can assume that this peak is caused mainly by the formation of a ternary Cd–S–Te interlayer between the CdTe core and the thiol stabilizing TGA group.
 | ||
| Fig. 7 Raman spectra of aqueous TGA-capped CdTe QDs before photoetching (a) and after photoetching (b). The solid curves are the result of the fitting procedure by the Lorentzian peaks. | ||
The observed changes in the Raman spectra after photoetching can originate from the elimination (etching) of defect states—most probably unsaturated surface tellurium atoms44—and the further formation of a Cd–S–Te crystallographic structure which is associated with an increase in intensity of the corresponding LO phonon peak. It is well known that depending on the contribution of the CdTe or CdS crystallographic phases, phonon modes of a ternary compound can span the spectral region between 250 and 305 cm−1.45 A significant broadening of this peak (FWHM = 26.9 cm−1) may suggest a strong strain effect at the interface, due to the lattice mismatch between CdTe and CdS. In fact, the existence of a ternary phase on the surface of the CdTe core should cause the appearance of two additional peaks in Raman spectra: one belonging to the ternary compound, and another signal in the region of the fundamental CdS LO phonon frequency (305 cm−1). Therefore it may be suggested that the band observed in both Raman spectra at 320 cm−1 consists of two strongly overlapped peaks. The first one may indeed be assigned to the first CdTe overtone, as it was suggested above. The maximum of this peak is expected to be detected at 328 cm−1. The second peak may belong to the CdS LO phonon although this peak can also be shifted from its expected position due to quantum confinement and strain. Strong broadening of the band observed at 320 cm−1 (FWHM = 46.7 cm−1) may justify this possibility.
Results of our Raman studies along with observed strong increases in QY and radiative rate (Figs 5 and 6 respectively) reinforce the theory that TGA-capped dots contain a mixed phase of CdTe and sulfur on the surface and that increases in PL intensity are a result of light-induced incorporation of surface thiol molecules into this phase. This increase must result from a further amalgamation of Cd(S) on the surface and an efficient reduction in the nonradiative pathways (insert in Fig. 6). It is worth noting that the obtained evidence of light-induced growth of a CdS shell and/or Cd–S–Te phase on the surface of CdTe QDs is of great importance for the application of these QDs as biological labels for intracellular imaging in vitro. It has long been known that one of the mechanisms employed by living organisms (such as bacteria and yeasts) to counter the toxic effects of free cadmium ions involves complexation of the metal ion by peptides or proteins incorporating sulfide ions46–48 or even by peptide-assisted growth of CdS QDs.49
CdTe QD imaging in live THP-1 cells
We have investigated the uptake and cellular localisation of TGA-capped CdTe QDs in live THP-1 cells. THP-1 cells were differentiated to macrophages as described in the Experimental and the QDs (particle conc. 10−5 M)19 were added at a 1 in 100 dilution followed by incubation for the indicated times at 37 °C. Vigorous washing with PBS followed the incubation period to detach the loosely bound QDs. We assessed the ability of TGA-stabilised QDs to act as suitable labels for biological imaging and their potential for penetration of the cell membrane. Fig. 8 shows intense binding of the QDs to the cell surface following a coincubation time of only five minutes. The highlighted cell borders visible in Fig. 8(b) are decorated by a patchy QD distribution. | ||
| Fig. 8 Phase contrast (A) and fluorescent images (B) of TGA-stabilised QDs adhering to the macrophage cell surface following a short (5 minute) coincubation treatment. | ||
Further experimentation, Fig. 9, shows that the TGA-stabilised QDs are internalized by the macrophages and sequestered to the cytoplasm within 10 minutes. A diffuse pattern of distribution of the QDs is visible within the cytoplasm. Interestingly, following the extended 10 minute coincubation time, the QDs previously adhering to the cell membrane are internalized and now exhibit a diffuse cytoplasmic and an enhanced nuclear membrane localisation.
 | ||
| Fig. 9 TGA-stabilised QDs showing intracellular localisation following a 10 minute coincubation time in THP-1 cells. | ||
We then evaluated the effects of photoetching on the behavior and properties of the TGA-capped QDs within the cell cultures. Fig. 10 shows the intensely fluorescent photoetched QDs displaying a cytoplasmic location within the THP-1 cells. These QDs possess far greater luminescent properties than their non etched counterparts (Figs 8 and 9) and while they display the same cellular compartmentalization, they exhibit a far greater optical enhancement of the cell region making them ideal tools for biomedical imaging. Extended incubation times (12 hours, 24 hours) revealed no deleterious effects on cell viablility (data not shown).
 | ||
| Fig. 10 Photoetched TGA-stabilised QDs showing intracellular localisation following a 10 minute coincubation time in THP-1 cells. | ||
These photoetched QDs permit a greater detailed examination of the nuclear membrane and further studies are required to investigate and employ this phenomenon.
Conclusions
We have shown that for TGA-stabilised QDs, the luminescence QY can be increased significantly, up to ∼40%, enabling their use as enhanced biolabelling agents. This has been achieved by the utilisation of the correct stoichiometric precursor amounts and irradiation of the QD solutions on a smaller than previously reported time scale (tens of minutes).12,13 Employing steady-state and time-resolved PL spectroscopy in combination with Raman spectroscopy we have addressed the issue of how the surface states influence the recombination efficiency of charge carriers in photoetched colloidal CdTe QDs. Our results contribute to the understanding of the role of surface ligands, which is critical to the design of stable, high-quantum-yield QDs.We have also investigated the potential use of TGA-stabilised QDs and their highly luminescent photoetched counterparts as future bioimaging tools. We demonstrate the role of TGA-stabilised CdTe QDs as excellent live cell imaging agents as they exhibit strong luminescence and excellent photostability while maintaining cell viability over a number of extended incubation periods. In addition, the ability of TGA-stabilised CdTe QDs to traverse the cell membrane of macrophages is a formidable quality that may potentially be harnessed for imaging and therapeutics. Modulating the delivery of QDs to subcellular locations in living cells opens a myriad of potential applications ranging from drug delivery to the examination of intracellular processes.
Acknowledgements
The authors wish to thank Alexander Eychmüller for aiding our expertise in this field. Financial support of this work by Enterprise Ireland is gratefully acknowledged.References
- J. C. Politz, Trends Cell Biol., 1999, 9, 284 CrossRef CAS.
- W. J. Parak, D. Gerion, T. Pellegrino, D. Zanchet, C. Micheel, C. S. Williams, R. Boudreau, M. A. LeGros, C. A. Larabell and A. P. Alivisatos, Nanotechnology, 2003, 14, R15 CAS.
- A. P. Alivisatos, Nat. Biotechnol., 2004, 22, 47 CrossRef.
- M. Green, Angew. Chem., Int. Ed., 2004, 43, 4129 CrossRef CAS.
- X. Gao, Y. Cui, R. M. Levenson, L. W. K. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS.
- M. Brunchez, Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS.
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861 CrossRef CAS.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS.
- J. J. Li, Y. A. Wang, W. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. Peng, J. Am. Chem. Soc., 2003, 125, 12567 CrossRef CAS.
- B. O. Dabbousi, J. Roderiguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463 CrossRef CAS.
- A. R. Clapp, I. L. Medintz, B. R. Fisher, G. P. Anderson and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 1242 CrossRef CAS.
- N. Gaponik, D. V. Talapin, A. L. Rogach, K. Hoppe, E. V. Shevchenko, A. Kornowski, A. Eychmuller and H. Weller, J. Phys. Chem. B, 2002, 106, 7177 CrossRef CAS.
- H. Bao, Y. Gong, Z. Li and M. Gao, Chem. Mater., 2004, 16, 3853 CrossRef CAS.
- A. A. Bol and A. Meijerink, J. Phys. Chem. B, 2001, 105, 10203 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- Z. Lin, S. Cui, H. Zhang, Q. Chen, B. Yang, X. Su, J. Zhang and Q. Jin, Anal. Biochem., 2003, 319, 239 CrossRef CAS.
- B. Ballou, B. C. Lagerholm, L. A. Ernst, M. P. Bruchez and A. S. Waggoner, Bioconjugate Chem., 2004, 15, 79 CrossRef CAS.
- N. N. Mamedova, N. A. Kotov, A. L. Rogach and J. Studer, Nano Lett., 2001, 1, 281 CrossRef CAS.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854 CrossRef CAS.
- C. Li and N. Murase, Chem. Lett., 2005, 34, 92 CrossRef CAS.
- L. Li, H. Qian, N. Feng and J. Ren, J. Lumin., 2006, 116, 59 CrossRef CAS.
- H. Zhang, Z. Zhou, B. Yang and M. Gao, J. Phys. Chem. B, 2003, 107, 8 CrossRef CAS.
- S. F. Wuister, A. van Houselt, C. de Mello Donegá, D. Vanmaekelbergh and A. Meijerink, Angew. Chem., Int. Ed., 2004, 43, 3029 CrossRef CAS.
- E. Hao, H. Sun, Z. Zhou, J. Liu, B. Yang and J. Shen, Chem. Mater., 1999, 11, 3096 CrossRef CAS.
- S. Jeong, M. Achermann, J. Nanda, S. Ivanov, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2005, 127, 10126 CrossRef CAS.
- S. F. Wuister, F. van Driel and A. Meijerink, J. Lumin., 2003, 102–103, 327 CrossRef CAS.
- A. M. Kapitonov, A. P. Stupak, S. V. Gaponenko, E. P. Petrov, A. L. Rogach and A. Eychmueller, J. Phys. Chem. B, 1999, 103, 10109 CrossRef CAS.
- X. Wang, L. Qu, J. Zhang, X. Peng and M. Xiao, Nano Lett., 2003, 3, 1103 CrossRef CAS.
- T. Franzl, D. S. Koktysh, T. A. Klar, A. L. Rogach and J. Feldmann, Appl. Phys. Lett., 2004, 84, 2904 CrossRef CAS.
- S. F. Wuister, F. van Driel and A. Meijerink, Phys. Chem. Chem. Phys., 2003, 203, 1253 RSC.
- S. F. Wuister, I. Swart, F. van Driel, S. G. Hickey and C. de Mello Donegá, Nano Lett., 2003, 3, 503 CrossRef CAS.
- E. P. Petrov, F. Cichos, E. Zenkevich, D. Starukhin and C. von Borczyskowski, Chem. Phys. Lett., 2005, 402, 233 CrossRef CAS.
- M. Nirmal, C. B. Murray and M. G. Bawendi, Phys. Rev. B, 1994, 50, 2293 CrossRef CAS.
- V. I. Klimov, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Phys. Rev. B: Condens. Matter, 1999, 60, 13740 CrossRef CAS.
- J. Y. Zhang, X.-Y. Wang and M. Xiao, Opt. Lett., 2002, 27, 1253 CAS.
- M. G. Bawendi, P. J. Carroll, W. Wilson and L. Brus, J. Chem. Phys., 1992, 96, 946 CrossRef CAS.
- B. L. Wehrenberg, C. Wang and P. Guyot-Sionnest, J. Phys. Chem. B, 2002, 106, 10634 CrossRef CAS.
- J.-Y. Zhang, X.-Y. Wang, M. Xiao, L. Qu and X. Peng, Appl. Phys. Lett., 2002, 81, 2076 CrossRef CAS.
- A. V. Baranov, Y. P. Rakovich, J. F. Donegan, T. S. Perova, R. A. Moore, D. V. Talapin, A. L. Rogach, Y. Masumoto and I. Nabiev, Phys. Rev. B: Condens. Matter, 2003, 68, 165306 CrossRef.
- J. J. Shiang, A. V. Kadavanich, R. K. Grubbs and A. P. Alivisatos, J. Phys. Chem., 1995, 99, 17417 CrossRef CAS.
- Y.-N. Hwang, S. Shin, H. L. Park, S.-H. Park, U. Kim, H. S. Jeong, E.-J. Shin and D. Kim, Phys. Rev. B, 1996, 54, 15120 CrossRef.
- M. L. Scheepers, R. J. Meier, L. Markwort, J. M. Gelan, D. J. Vanderzande and B. J. Kip, Vib. Spectrosc., 1995, 9, 139 CrossRef CAS.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 110, 468 CrossRef.
- H. Borchert, D. V. Talapin, N. Gaponik, C. McGinley, S. Adam, A. Lobo, T. Moller and H. Weller, J. Phys. Chem. B, 2003, 107, 9662 CrossRef CAS.
- A. Fischer, L. Anthony and A. D. Compaan, Appl. Phys. Lett., 1998, 72, 2559 CrossRef CAS.
- D. W. Ow, Dev. Biol., 1993, 29, 213.
- R. N. Reese and D. R. Winge, J. Biol. Chem., 1988, 263, 12832 CAS.
- R. K. Mehra and D. R. Winge, J. Cell Biochem., 1991, 45, 30 CAS.
- C. T. Dameron, R. N. Reese, R. K. Mehra, A. R. Kortan, P. J. Carroll, M. L. Steigerwald, L. E. Brus and D. R. Winge, Nature, 1989, 338, 596 CrossRef CAS.
Footnote |
| † The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2006 |