[60]Fullerene–perchlorotriphenylmethide anion triads. Synthesis and study of photoinduced intramolecular electron-transfer processes†
Stéphanie
Chopin
a,
Jack
Cousseau
*a,
Eric
Levillain
a,
Concepció
Rovira
*b,
Jaume
Veciana
*b,
Atula S. D.
Sandanayaka
c,
Yasuyuki
Araki
c and
Osamu
Ito
*c
aLaboratoire CIMMA, UMR CNRS 6200, University of Angers, 2 Bd Lavoisier, 49045 Angers Cedex, France. E-mail: jack.cousseau@univ-angers.fr
bInstitut de Ciencia de Materials de Barcelona, C.S.I.C., Campus de la UAB, 08193 Bellaterra, Spain. E-mail: vecianaj@icmab.es; cun@icmab.es
cInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan. E-mail: ito@tagen.tohoku.ac.jp
First published on 17th November 2005
Abstract
For the first time an anionic donor, the perchlorotriphenylmethide anion (PTM−), has been covalently bonded to C60, generating the C60–(PTM−)2 triad that is reversibly oxidized to the corresponding C60–perchlorotriphenylmethyl radical triad C60–(PTM˙)2. For the triad C60–(PTM−)2, photoinduced charge-separation can be confirmed to occur via the excited singlet states of the C60 moiety and the PTM anion in polar and nonpolar solvents from quenching of their fluorescence intensities in the region of 700–750 nm and 560–630 nm, respectively. The charge-separation state was confirmed by the nanosecond transient absorption spectra in the visible and near-IR spectral regions. After charge-separation, back electron transfer takes place with a lifetime of about 80 ns. Steady-state concentration of the highly persistent PTM radical was observed after repeated laser light irradiation.
Introduction
Since C60 has been revealed as a very attractive electron acceptor with unique photo-physical and electrochemical properties,1–3 considerable efforts have been devoted in recent years to develop systems in which C60 is covalently linked to electron donors,4–15 in addition to the blend systems consisting of C60 and donors.16–23 Donor–acceptor systems including C60 are of particular interest, because they exhibit characteristic electronic properties in the excited states with interesting photo-physical properties attributed to the small reorganization energy of C60 in electron transfer processes due to its spherical rigid molecular shape.24 Thus, a lot of research has been conducted for a better understanding of photoinduced electron-transfer processes in dyads and triads including C60.4–15 These phenomena open the potential applications in the realization of new artificial photosynthetic systems, molecular electronic devices, and photovoltaic cells.4,6,7Among the wide variety of donor molecules that have been covalently linked to C60, most donors are neutral molecules such as aromatic amines, carotene, tetrathiafulvalenes, ferrocenes, porphyrins, phthalocyanines, oligothiophenes, π conjugated phenyl oligomers, etc. These dyads generated the radical anion–radical cation pairs, some of them showing quite persistent charge-separated state in polar solvents, since the charge-recombination processes enter the inverted region of the Marcus parabola.
In the present study, for the first time an anionic donor, the perchlorotriphenylmethide anion (PTM−), has been covalently bonded to a C60 derivative, generating the C60–perchlorotriphenylmethide anion triad 1 shown in Fig. 1.
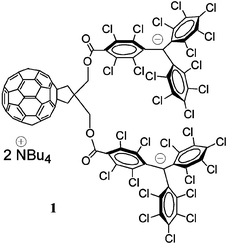 | ||
| Fig. 1 Molecular structure of C60–(PTM−)2. | ||
Triphenylmethide anions have been reported to be quite stable when the hydrogen atoms of the triphenyl groups are substituted by chlorine atoms.25 The stability is not only due to the electron-withdrawing character of the substituents, but also to an effective steric shielding of the central carbon wrapped by the six bulky chlorine atoms in the ortho positions of the phenyl rings. In addition, PTM anions are reversibly oxidized to highly persistent perchlorotriphenylmethyl radicals (PTM˙) at low potential (around −0.55 V, versus Fc/Fc+) being therefore very good donors.26
For the target triad C60–(PTM−)2 (1) we expected the occurrence of an electron transfer from the anionic part to the photoexcited C60 moiety, generating the neutral perchlorotriphenylmethyl radical (PTM˙) and the radical anion of the C60 moiety. This represents a completely new situation in this kind of donor–acceptor systems since the charge-recombination takes place between the C60 radical anion and a neutral radical, which may show quite different behavior from the usual charge-separated states generating oppositely charged species.
Results and discussion
Synthesis of C60–(PTM−)2
The synthesis of the target triad 1 consisting of two donor units, the perchlorinated triphenylmethide anions, and the C60 moiety as the acceptor unit, was achieved by a multi-step synthetic procedure in which the bis-acylation of the C60-diol 427 using the PTM acid chloride 3 is the key step. The new acid chloride derivative 3 was obtained in good yield (80%) following the same procedure used in the synthesis of its radical counterpart28 through refluxing 4-[bis(2,3,4,5,6-pentachlorophenyl)methyl]-2,3,5,6-tetrachlorobenzoic acid29 in an excess of thionyl chloride. We carried out the esterification reaction in refluxing CH2Cl2 under an argon atmosphere in the presence of dimethylaminopyridine (DMAP). Note that this reaction affords compound 5, the precursor of the target compound 1, in quite good yield (∼60%), although two bulky groups are attached to the fullerene moiety through the neighboring alcohol groups. In fact the yield is very similar to that obtained in the esterification of C60-diol 4 with a TTF-acid chloride with long alkyl chains that afford high flexibility to the linkage.30 As shown in Scheme 1 the deprotonation of the bis α-H precursor 5 yields quantitatively the target triad 1, by converting the non electroactive α-H triphenylmethane addends to electroactive carbanions that are very good donors. Compound 1 was isolated as the pure tetrabutylammoniun salt by washing the dark purple precipitate with hexane several times.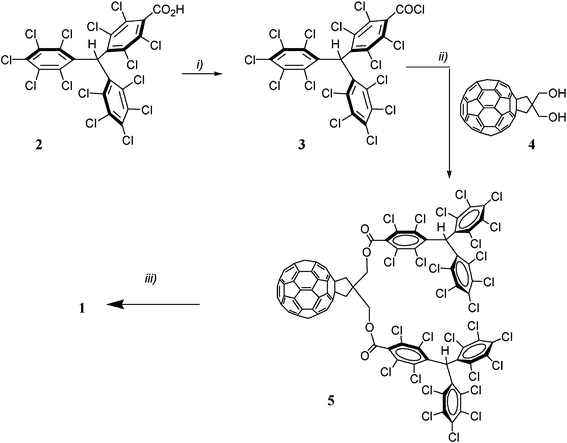 | ||
| Scheme 1 Synthesis of C60–(PTM−)2(NBu4+)2. Conditions: i) SOCl2, reflux; ii) DMAP, CH2Cl2, 35 °C; iii) NBu4OH, THF, room temperature. | ||
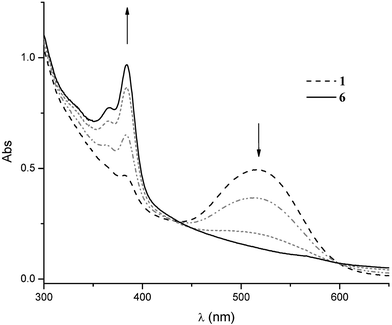
According to the donor characteristics of bisanion 1, when a CH2Cl2 solution of this salt containing an excess of AgNO3 as oxidant is shaken for a few minutes, the bistriphenylmethyl radical 6 was obtained as a deep red solid in quantitative yield after filtration (Scheme 2). The reaction was followed by UV-Vis spectroscopy; Fig. 2 shows the evolution of the spectra over time. The disappearance of the intense band centered at 518 nm corresponding to the PTM− anion26e was accompanied by the increasing of the characteristic bands of the PTM radical at 364 and 384 nm.26 The presence of two isosbestic points at 435 and 600 nm is clear evidence of the clean transformation of C60–(PTM−)2 (1) into C60–(PTM˙)2 (6).
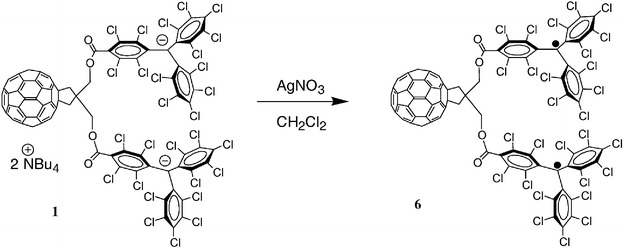 | ||
| Scheme 2 Synthesis of C60–(PTM˙)2 (6). | ||
Bisradical 6 was characterized by EPR spectroscopy. The X-band isotropic spectrum of 6 in toluene–CH2Cl2 at room temperature consists of a signal with a central intense line and small satellite overlapped lines corresponding to the coupling of the unpaired electron with the naturally abundant 13C isotope at the α and aromatic positions of the PTM˙ addends. The giso value, 2.0023, as well as the isotropic coupling constant values, a1 (13Cα) = 30 G and a2 (13Carom) = 12 G, are usual values for the monoradicals of the PTM family.26 The spectrum of bisradical 6 in frozen toluene–CH2Cl2 solution does not show the characteristic fine structure nor the forbidden Δms = 2 transition characteristic of the triplet species. All these features indicate that the two radical units in the C60–(PTM˙)2 triad are not interacting.
Electrochemical measurements
The redox behavior of all new compounds was studied by cyclic voltammetry and the PTM−[18-crown-6]+ salt and pristine C60 were also studied under the same conditions as the reference compounds. As expected, triad 5, in which only the C60 moiety is electroactive, presents the characteristic reversible reduction waves of the substituted C60 compounds. By contrast, unexpected results were obtained in the cyclic voltammogram of the triad C60–(PTM−)2, 1, which contains two different electroactive moieties. In all conditions assayed, only one reversible oxidation wave appears clearly (see ESI† Fig. SI1 and SI2) whereas the waves expected for the C60 moiety are quite weak. The wave corresponding to the redox characteristics of the anionic PTM moieties appears at the same potential as the reference PTM−[18-crown-6]+, indicating that there is no influence of one anionic moiety on the other and that there is no interaction with the C60 moiety.The reversibility observed in the cyclic voltammogram of compound 1 is in accordance with the stability of the free bisradical C60(PTM˙)2, 6. In Table 1, the E1/2 values for C60, C60–(PTM−)21, PTM−[18-crown-6]+ and C60–(PTMH)25 are summarized. Weak reduction peaks of the C60 unit in charged triad 1 were always observed under different conditions, the phenomenon being independent of the concentration, excluding an intermolecular phenomenon, the nature of the electrode (platinum, gold and carbon vitreous), excluding a phenomenon of adsorption to the electrode, and the solvent (CH2Cl2 and o-dichlorobenzene (o-DCB)). The unexpected behavior of compound 1 is not understood and new studies on other negative charged compounds are underway in order to know if the negative charges present in the addends of C60 can be responsible for this.
In order to calculate, from the Weller equations,31 the free-energy changes for charge-separation (ΔGCS) and charge-recombination (ΔGCR) for C60–(PTM−)2 the first reduction potential (Ered) value of the C60 moiety and the first oxidation potential (Eox) of the PTM− moiety are needed. From these Eox and Ered values, the ΔGCS and ΔGCR values were calculated using eqn (1)–(3):
| −ΔGCS = ΔE0-0 − (−ΔGCR) | (1) |
| −ΔGCR = Eox − Ered + ΔGS | (2) |
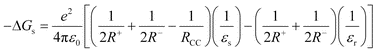 | (3) |
| Solvent | −ΔGSCS/eVa | −ΔGTCS/eVa | −ΔGCR/eVa |
|---|---|---|---|
| a Calculated from eqns (1)–(3) employing ΔE0-0 = 1.72 eV for 1C60*, ΔE0-0 = 1.52 eV for 3C60*, Eox = −0.52 V for PTM−, and Ered = −1.12 V for C60vs. Fc/Fc+ in o-DCB–CH3CN 95 : 5. R+ = 7.4 Å for PTM−, and R− = 4.7 Å for C60, and RCC = 10 Å.32 Permittivities of toluene, o-DCB, and PhCN are 2.38, 9.93, 25.2, and 37.5 respectively. | |||
| Toluene | 0.92 | 0.70 | 0.82 |
| o-DCB | 1.26 | 1.06 | 0.46 |
| PhCN | 1.33 | 1.13 | 0.39 |
| CH3CN | 1.34 | 1.14 | 0.38 |
Steady-state absorption measurements
Steady-state absorption spectra of C60–(PTMH)25, C60–(PTM−)21, and PTM− in PhCN are shown in Fig. 3. For C60–(PTMH)2, the peaks at 700 and 430 nm bands are attributed to the characteristic bands of the functionalized C60. In the case of C60–(PTM−)2, the broad absorption band at 518 nm is characteristic of the PTM− moiety, as shown in Fig. 2. The absorption bands of the C60 moiety appear at 700 nm and in the shorter wavelength than 400 nm. Since this spectrum is almost the same as the one resulting from the summed spectrum of the components PTM− and C60, an appreciable interaction between moieties may not be present in the ground state. For the transient absorption measurements, a 355 nm laser light was used, which excites the C60 moiety about 90% and 10% of to the PTM− moiety. For time-resolved fluorescence measurement, a 400 nm laser light was used, which excites both the C60 moiety (75%) and the PTM− moiety (25%).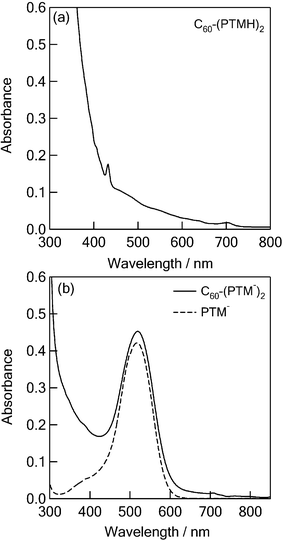 | ||
| Fig. 3 Steady-state absorption spectra of (a) C60–(PTMH)2 (0.1 mM) in PhCN and (b) C60–(PTM−)2 and PTM− (0.025 mM) in PhCN. | ||
Fluorescence measurements
Steady-state fluorescence spectra of PTM−, C60–(PTMH)2, C60–(PTM−)2 in toluene and o-DCB are shown in Fig. 4. The fluorescence peak (λf) for C60–(PTM−)2 appeared at 600 nm (Fig. 4b), which is almost the same as that of PTM− (Fig. 4a). Therefore, the origin of the observed fluorescence at 600 nm of C60–(PTM−)2 is attributed to the PTM− moiety. From the cross point of the fluorescence band and absorption band after normalizing both intensities, the lowest excited singlet energy (E0-0) of the PTM− moiety was estimated to be 2.1 eV.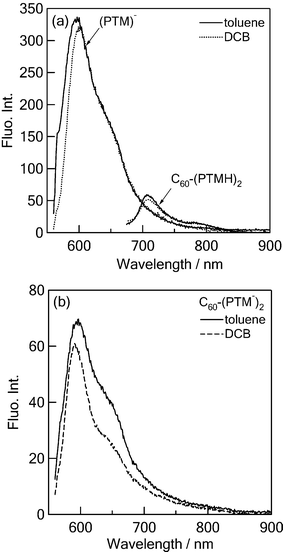 | ||
| Fig. 4 Steady-state fluorescence spectra of (a) PTM− (0.05 mM) and C60–(PTMH)2 (0.05 mM), and (b) C60–(PTM−)2 (0.05 mM) in toluene and o-DCB; λex = 550 nm. | ||
The fluorescence of the C60 moiety would be expected to appear at 720 nm as shown for C60–(PTMH)2 (Fig. 4a). In the case of C60–(PTM−)2, however, the fluorescence of the C60 moiety may be hidden by the huge fluorescence tail of 1(PTM−)*. Further quantitative analyses on the fluorescence properties were carried out based on fluorescence lifetime measurements.
Time-resolved fluorescence spectra and fluorescence lifetimes
The peak position and shape of time-resolved fluorescence spectra of 1(PTM−)* (see ESI†) are quite similar to those of the steady-state spectra. Within the fluorescence lifetime, the spectral shape is almost the same. The time profiles of the fluorescence peak position are shown in Fig. 5. The fluorescence time profile of 1(PTM−)* showed slow fluorescence decay obeying a single exponential function, yielding a fluorescence lifetime of 4100 ps in toluene and o-DCB.20a In PhCN, a shorter lifetime (1000 ps) was evaluated, suggesting differences in the excited state of 1(PTM−)* in this polar solvent from that in nonpolar and less polar solvents. | ||
| Fig. 5 Fluorescence time profiles at 560–630 nm in o-DCB and PhCN; λex = 400 nm. | ||
Also for C60–(PTMH)2, the time-resolved fluorescence spectra (see ESI†) are in good agreement with the steady-state fluorescence spectrum. Within the fluorescence lifetime of C60–(PTMH)2 the spectral shape is unchanged. Time profiles of the fluorescence intensity at the peak position in toluene and o-DCB are shown in Fig. 6. In slightly polar o-DCB, the fluorescence lifetime of the 1C60* moiety is almost the same to that in non-polar toluene within experimental errors, in which both decays are the same as that of pristine 1C60*, as expected for a compound with no donor moieties that can not present charge-separation.
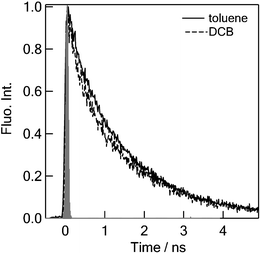 | ||
| Fig. 6 Fluorescence decays of C60–(PTMH)2 at 700–750 nm in toluene and o-DCB; λex = 410 nm. | ||
Time profiles of the fluorescence peak position of C60–(PTM−)2 in toluene and o-DCB are shown in Fig. 7. Slight blue shift of the florescence peak was observed in the time-resolved fluorescence spectra of C60–(PTM−)2 (see ESI†) on going from toluene to o-DCB, which is the same trend as observed in steady-state measurements. The fluorescence lifetimes of the 1(PTM−)* moiety in C60–(PTM−)2 at 560–630 nm (Fig. 7) are quite shorter than that of 1(PTM−)* in all studied solvents (Fig. 5). This finding indicates that a quenching process from the 1(PTM−)* moiety to the C60 moiety takes place for C60–1(PTM−)*(PTM−). Even in nonpolar solvents like toluene, the fluorescence decay rate of the 1(PTM−)* moiety in C60–1(PTM−)*(PTM−) was faster than that of the isolated 1(PTM−)*, it is possible to consider that both energy transfer and charge separation occur via1(PTM−)*.
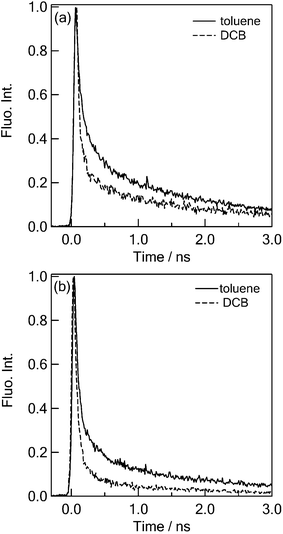 | ||
| Fig. 7 Fluorescence time profiles at (a) 560–630 nm and (b) 710–750 nm, respectively, in toluene and o-DCB; λex = 400 nm. | ||
From the (τf)sample value of the 1(PTM−)* moiety in C60–(PTM−)2, the intramolecular quenching rate-constant (kSq) was evaluated using eqn (4):
| kSq = (1/τf)sample − (1/τf)ref | (4) |
| Solvent | τ f b/ps via1(PTM−)* | k S q a/s−1via1(PTM−)* | Φ S q a via 1(PTM−)* | τ f b/ps via1C60* | k S CS a/s−1via1C60* | Φ S CS a via 1C60* |
|---|---|---|---|---|---|---|
| a Calculated by using eqn (4) and (5). The values of (τf)ref were employed to be 4130 ps for PTM− and 1390 for C60. b Fluorescence decays of 1(PTM−)* and 1(C60)* were fitted with biexponential functions and the kqS, ΦqS, kSCS and ΦSCS values were calculated from the shorter lifetimes with the major fraction. c Effect of impurity in PhCN may be included. | ||||||
| Toluene | 150 (70%) | 6.4 × 109 | 0.96 | 100 (85%) | 9.3 × 109 | 0.93 |
| o-DCB | 79 (85%) | 1.3 × 1010 | 0.98 | 83 (94%) | 1.1 × 1010 | 0.94 |
| PhCN | 257 (70%)c | (3.7 × 109)c | (0.94)c | 95 (88%) | 9.8 × 109 | 0.93 |
The fluorescence quenching quantum yields of (ΦSq) via the 1(PTM−)* moiety in C60–(PTM−)2 in polar and nonpolar solvents were evaluated from eqn (5):
| ΦSq = [(1/τf)sample − (1/τf)ref]/(1/τ)sample | (5) |
In PhCN, the fluorescence decay rate was slower than those in toluene and o-DCB, probably because of the presence of some impurity in PhCN, which was quite difficult to eliminate.
By contrast with the fluorescence decay of the C60–(PTMH)2 moiety yielding the fluorescence lifetime (τf) of 1390 ps in toluene (Fig. 6),20 the time profiles of the fluorescence at 720 nm of the C60 moiety in C60–(PTM−)2 (Fig. 7b) show biexponential decay in o-DCB and toluene. The major component decayed with the τf values in the 83 ps (94%)–100 ps (85%) region, whereas the minor slow-decay components gave τf values of 1300 ps. The τf values of the major components are summarized in Table 3.
The difference between the τf values evaluated from the main fluorescence decays in 710–750 nm in polar and nonpolar solvents can be attributed predominantly to charge separation via the 1C60* moiety in C60–(PTM−)2 generating the radical ion-pair states (C60˙−–(PTM˙)(PTM−)). From the τf value of the 1C60* moiety in C60–(PTM−)2, the intramolecular charge-separation rate constants (kSCS) in polar and nonpolar solvents were evaluated from eqn (4) as summarized in Table 3. Thus, the kSCS values for the C60–(PTM−)2 were evaluated to be (9.3–11.0) × 109 s−1 which indicate that charge separation via the 1C60* moiety is an effective process in polar and nonpolar solvents. The quantum yields of charge separation (ΦSCS) via the 1C60* moiety in C60–(PTM−)2 were evaluated from eqn (5). Thus, ΦSCS = 0.93–0.94 were calculated for the C60–(PTM−)2 (Table 3) in polar and nonpolar solvents, respectively, which indicates that the charge separation via the 1C60* moiety is the main process, overwhelming the intersystem crossing (ISC) process to the 3C60* moiety even in toluene.
Nanosecond transient absorption measurements
Transient absorption spectra observed upon nanosecond laser excitation (355 nm) of C60–(PTMH)2 in toluene are shown in Fig. 8. The 355 nm laser light excites the C60 and PTMH moieties. The broad absorption bands were observed in the region of 600–850 nm, which is the absorption region of 3C60* (peak = 700 nm).20e The decay time profile is quite slow on the time scale of 1.5 µs. Even the PTMH moiety was excited, the energy transfer gives the 1C60* moiety, which is finally converted to 3C60* by ISC.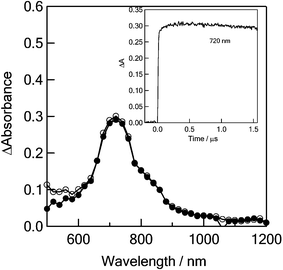 | ||
| Fig. 8 Nanosecond transient absorption spectra of 0.1 mM C60-(PTMH)2 observed by 532 nm laser irradiation at 100 ns (●) and 1000 ns (○) in PhCN. Inset: Absorption–time profiles at 720 nm. | ||
Transient absorption spectra observed by the nanosecond laser excitation (355 nm) of C60–(PTM−)2 in toluene are shown in Fig. 9. Broad absorption bands were observed in the region of 600–1200 nm, which can be attributed to the absorption of the C60˙− moiety,20e although the absorption bands are broader compared with the pristine C60˙− and other C60˙− derivatives. This broadening may be caused by the interaction between the C60˙− moiety and the (PTM˙)(PTM−) moiety, in which anion, radical and halogen atoms may be the candidate of the interactions. From the decay time-profile at 1020 nm, the charge-recombination rate constant (kCR) between C60˙− and PTM˙ of C60˙−–(PTM˙)(PTM−) was evaluated to be 1.23 × 107 s−1, from which the lifetime (τRIP) of the charge-separated state was calculated to be 81 ns at room temperature. Since the 355 nm laser light predominantly excites the C60 moiety, the process photoinduced by the excitation of PTM− can be neglected.
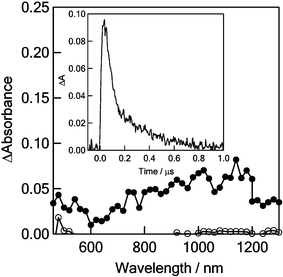 | ||
| Fig. 9 Nanosecond transient absorption spectra of 0.05 mM C60–(PTM−)2 observed by 355 nm laser irradiation at 100 ns (●) and 1000 ns (○) in toluene. Inset: Absorption–time profiles at 1020 nm. | ||
Similarly, transient absorption–time profiles at 1020 nm were observed in o-DCB and PhCN (see ESI†), although the transient absorption spectra were distorted in o-DCB and PhCN by laser irradiation (see ESI† Fig. SI-8). From the decay time-profiles at 1020 nm, the charge-recombination rate constants (kCR) were evaluated to be 1.30 × 107 and 1.72 × 107 s−1 for o-DCB and PhCN, from which the τRIP values of the charge-separated states were calculated to be 77 and 58 ns, respectively, at room temperature as listed in Table 4.
| Solvent | k CR/s−1 | τ RIP/ns |
|---|---|---|
| Toluene | 1.23 × 107 | 81 |
| o-DCB | 1.30 × 107 | 77 |
| PhCN | 1.72 × 107 | 58 |
The solvent polarity effect on the τRIP values was small; for polar and nonpolar solvents, almost similar lifetimes (τRIP) are found. However, C60˙−–(PTM˙)(PTM−) in toluene gives a long lifetime.
Repeated laser irradiation
By excitation of C60–(PTM−)2 with the 355 nm light, the absorption band of PTM− decreased; instead, a sharp peak appeared at 390 nm (Fig. 10), which is attributed to the PTM˙ moiety (see Fig. 2). This observation indicates that after the photoinduced charge-separation the stable C60–(PTM˙)(PTM−) species was generated, probably because the C60˙− moiety may be quenched by some reactions such as donation of an electron to the impurity in the solution.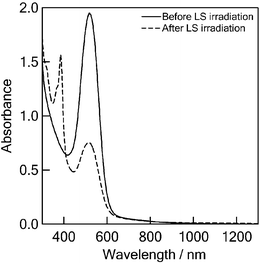 | ||
| Fig. 10 Steady-state absorption spectra of 0.10 mM C60–(PTM−)2 in PhCN before and after 355 nm laser light (LS) irradiation. | ||
Energy diagram
Fig. 11 shows an energy diagram of C60–(PTM−)2 when PTM− and C60 are excited. Energy levels of the radical ion-pairs C60˙−–(PTM˙)(PTM−) are deduced from Table 2. In toluene, o-DCB and PhCN, the charge-separation takes place via C60–1(PTM−)*(PTM−) and 1C60*–(PTM−)2 as indicated by the weak fluorescence intensity and the short fluorescence lifetime, since the energy levels of C60˙−–(PTM˙)(PTM−) are lower than these excited singlet states even in toluene. Thus, the generation of 3C60*–(PTM−)2 through the ISC process from 1C60*–(PTM−)2 is not possible, because the ΦSCS values are almost unity in all solvents. Furthermore, with excitation of PTM−, an energy-transfer process is also possible from the 1(PTM−)* moiety to generate the 1C60* moiety.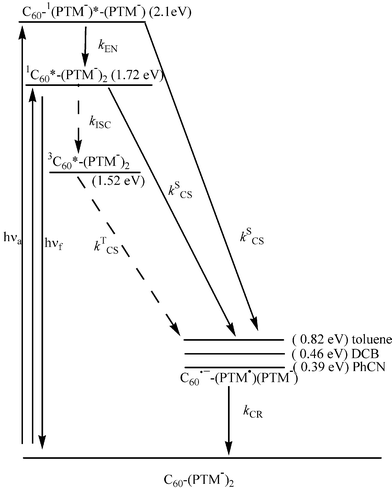 | ||
| Fig. 11 Schematic energy diagram for electron-transfer processes of C60–(PTM−)2. | ||
Summary
We have succeeded, for the first time, in synthesizing a C60 based triad which bears an anionic donor as addend, the perchlorotriphenylmethide anion (PTM−), thus generating the C60–(PTM−)2 triad 1. For this triad, photoinduced charge separation via both C60–1(PTM−)*(PTM−) and 1C60*–(PTM−)2 were observed in polar and nonpolar solvents through photoexcitation at room temperature. The efficiency and rate of the charge-separation process are quite high. The longest lifetime for C60˙−–(PTM˙)(PTM−) was 81 ns in toluene at room temperature, which is quite long and comparable with other C60 based dyads and triads such as C60–fluorene–diphenylamine,33 C60–bridge–dimethylaniline,5 C60–extended TTF34 or C60–long flexible bridge-TTF.30 Even though the properties of C60 perchlorotriphenylmethide anion based triads are not so good like the best ones found in some C60–porphyrins and C60–chlorines based dyads,36 the present study provides a new possibility for functional fullerenes based on the perchlorotriphenylmethide anion, which can be added to other known functional materials based on the parent perchlorotriphenylmethyl radical that works as an acceptor and has permitted the development of multifunctional switchable molecular systems.26e,35 We believe that the present study gives valuable information not only about the photo-induced electron-transfer chemistry of fullerene-triad systems, but also about the materials science of perchlorotriphenylmethide anion functional compounds.Experimental
General information
Reagents were purchased from commercial suppliers and used without further purification. Compounds 2, PTM−[18-crown-6]+, and 4 were synthesized as previously described.25,27,28 All solvents were spectrophotometric grade and were distilled before use. Column chromatography was performed using Merck silica gel 60. The NMR spectra were recorded on a Bruker Avance DRX 500 spectrometer (500 MHz for 1H, 125.5 MHz for 13C). IR spectra were run on a FT-IR spectrometer BIO-RAD FTS 155, the solid compounds being studied in KBr pellets. UV-Vis spectra were recorded on a Cary 5 E Varian Spectrometer. MALDI-TOF mass spectra were obtained on a Bruker Biflex III spectrometer, equipped with a N2 laser (337 nm) using dithranol as a matrix. The ESI-MS spectra were obtained with a JEOL, JMS 700 B/E mass spectrometer.Electrochemical measurements
Oxidation potentials (Eox) and reduction potentials (Ered) were measured by a voltammetric analyzer (EGG PAR 273A) in a conventional three-electrode cell equipped with Pt working electrodes and counter electrodes. A silver wire served as a quasi-reference electrode; its potential was checked against the ferrocene/ferrocenium couple (Fc/Fc+) before and after each experiment, at a scan rate of 100 mV s−1. In each case, the solution contained ∼0.5 mM sample and 0.1 M tetra n-butylammonium hexafluorophosphate (Bu4NPF6) dissolved in a mixture of o-dichlorobenzene–acetonitrile 95 : 5 or other solvents. The experiments were performed in a glove box.Synthesis of 4-[bis(2,3,4,5,6-pentachlorophenyl)methyl]-2,3,5,6-tetrachlorobenzoyl chloride (3)
A solution of 4-[bis(2,3,4,5,6-pentachlorophenyl)methyl]-2,3,5,6-tetrachlorobenzoic acid (2)29 (149.7 mg; 0.194 mol) in thionyl chloride (3 mL) was refluxed for 24 h. Excess SOCl2 was removed under reduced pressure. The resulting solid was purified by chromatography (silica gel, chloroform). Compound 3 was obtained in 80% yield as a beige solid. IR (KBr): 675, 764, 810, 1780 (COCl) cm−1. Anal. Calc. for C20HCl15O2: C, 29.84; H, 0.12; Cl, 66.06. Found: C, 29.67; H, 0.14; Cl, 66.36%.Synthesis of 2,2-bis{4-[bis(2,3,4,5,6-pentachlorophenyl)methyl]-2,3,5,6-tetrachlorobenzoyloxymethyl}-1,2-propano-1,2-dihydro[60]fullerene (5)
A mixture of compound 427 (68.3 mg; 0.083 mmol), compound 3 (150 mg; 0.190 mmol) and dimethylaminopyridine (35.5 mg; 0.290 mmol) in freshly distilled dichloromethane (10 mL) was stirred under argon atmosphere at 38 °C for 5 days. The solvent was evaporated and the residue was purified by column chromatography (silica gel, hexane then hexane–CH2Cl2 2 ∶ 1). Compound 5 was obtained in 59% yield as a brown solid. 1H NMR (250 MHz, CDCl3): 3.97 (s, 4H, CH2–C60), 5.41 (s, 4H, CH2–O–CO–), 7.06 (s, 2H, CH). IR (KBr): 527(C60), 810 (C–Cl), 1749 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1. UV-Vis (CH2Cl2): λmax (ε) = 430 (4625), 326 (32800), 305 (38900), 255 nm (167000 L mol−1 cm−1). MS (MALDI-TOF): m/z calcd for the maximum peak of C105H10O4Cl28 = 2327.17, observed maximum [M]+ = 2327.25.
O) cm−1. UV-Vis (CH2Cl2): λmax (ε) = 430 (4625), 326 (32800), 305 (38900), 255 nm (167000 L mol−1 cm−1). MS (MALDI-TOF): m/z calcd for the maximum peak of C105H10O4Cl28 = 2327.17, observed maximum [M]+ = 2327.25.
Synthesis of C60–(PTM−)2(NBu4)2 (1)
Tetrabutylammonium hydroxide, 40 wt% solution in water, ∼1.5 M (31.5 µl; 47.25 µmol) was added to a solution of compound 5 (20 mg; 8.59 µmol) dissolved in freshly distilled THF (2.5 mL). This mixture immediately became garnet-red and was stirred for 2 hours in a light protected place. The compound precipitates after addition of deoxygenated distilled hexane. The resulting solid was purified by centrifugation, and washed several times with deoxygenated distilled hexane. The garnet-red compound 1 was obtained quantitatively. IR (KBr): 518 (C60), 810 (C–Cl), 1738 (CO) cm−1. UV (THF), λmax (ε) = 518 nm (75000 L mol−1 cm−1). MS (ESI−): m/z calcd for the maximum peak of C105H8O4Cl28 = 1163.58, observed maximum 1162.45.
Steady-state measurements
Steady-state absorption spectra in the visible and near-IR regions were measured on a Jasco V570 DS spectrophotometer. Steady-state fluorescence spectra were measured on a Shimadzu RF-5300 PC spectrofluorophotometer equipped with a photomultiplier tube having high sensitivity in the 700–800 nm region.Time-resolved fluorescence measurements
The time-resolved fluorescence spectra were measured by single photon counting method using a streakscope (Hamamatsu Photonics, C4334-01) as a detector and the laser light (second harmonic generation (SHG), 400 nm) of a Ti ∶ sapphire laser (Spectra-Physics, Tsunami 3950-L2S, 150 fs fwhm) as an excitation source.36 Lifetimes were evaluated with software attached to the equipment.Nanosecond transient absorption measurements
Nanosecond transient absorption measurements were carried out using THG (355 nm) of a Nd ∶ YAG laser (Spectra-Physics, Quanta-Ray GCR-130, 5 ns fwhm) as an excitation source. For transient absorption spectra in the near-IR region (600–1200 nm) and the time-profiles, monitoring light from a pulsed Xe lamp was detected with a Ge-APD (Hamamatsu Photonics, B2834). For the measurements in the visible region (400–1000 nm), a Si-PIN photodiode (Hamamatsu Photonics, S1722-02) was used as a detector.20,36Acknowledgements
The present work was supported by a Grant-in-Aid on Scientific Research on Priority Areas (417) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Dirección General de Investigación, Spain (Project BQU2003-00760), DGR, Generalitat de Catalunya (Centre de Referencia CeRMAE and Project 2001SG00362), EU COST D14 and CNRS (France).References
- C. S. Foote, Top. Curr. Chem., 1994, 169, 347 CAS.
- Fullerene, Chemistry, Physics and Technology, ed. K. M. Kadish and R. S. Ruoff, Wiley-Interscience, New York, 2000 Search PubMed.
- Fullerene and Related Structures, ed. A. Hirsch, Springer, Berlin, 1999 Search PubMed.
- (a) P. A. Liddell, J. P. Sumida, A. N. Macpherson, L. Noss, G. R. Seely, K. N. Clark, A. L. Moore, T. A. Moore and D. Gust, Photochem. Photobiol., 1994, 60, 537 Search PubMed; (b) H. Imahori, S. Cardoso, D. Tatman, S. Lin, L. Noss, G. R. Seely, L. Sereno, C. Silber, T. A. Moore, A. L. Moore and D. Gust, Photochem. Photobiol., 1995, 62, 1009; (c) D. D. Kuciauskas, S. Lin, G. R. Seely, A. L. Moore, T. A. Moore, D. Gust, T. Drovetskaya, C. A. Reed and P. D. W. Boyd, J. Phys. Chem., 1996, 100, 15926 CrossRef CAS.
- (a) R. M. Williams, M. N. Zwier and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 4093 CrossRef CAS; (b) R. M. Williams, M. Koeberg, J. M. Lawson, Y.-Z. An, Y. Rubin, M. N. Paddon-Row and J. W. Verhoeven, J. Org. Chem., 1996, 61, 5055 CrossRef CAS; (c) K. G. Thomas, V. Biju, M. V. George, D. M. Guldi and P. V. Kamat, J. Phys. Chem. A, 1998, 102, 5341 CrossRef CAS; (d) K. G. Thomas, V. Biju, D. M. Guldi, P. V. Kamat and M. V. George, J. Phys. Chem. B, 1999, 103, 8864 CrossRef CAS; (e) K. G. Thomas, V. Biju, D. M. Guldi, P. V. Kamat and M. V. George, J. Phys. Chem. A, 1999, 103, 10755 CrossRef CAS.
- (a) N. S. Sariciftci, F. Wudl, A. J. Heeger, M. Maggini, G. Scorrano, M. Prato, J. Bourassa and P. C. Ford, Chem. Phys. Lett., 1995, 247, 510 CrossRef CAS; (b) A. S. D. Sandanayaka, K. Matsukawa, T. Ishi-I, S. Mataka, Y. Araki and O. Ito, J. Phys. Chem. B, 2004, 108, 19995 CrossRef CAS; (c) A. S. D. Sandanayaka, K.-i. Ikeshita, G. A. Rajkumar, Y. Furusho, Y. Araki, T. Takata and O. Ito, J. Phys. Chem. A, 2005, 109, 8088 CrossRef CAS; (d) A. S. D. Sandanayaka, K. Ikeshita, Y. Araki, N. Kihara, Y. Furusho, T. Takata and O. Ito, J. Mater. Chem., 2005, 15, 2276 RSC.
- (a) H. Imahori, K. Hagiwara, M. Aoki, T. Akiyama, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, J. Am. Chem. Soc., 1996, 118, 11771 CrossRef CAS; (b) H. Imahori, S. Ozawa, K. Uchida, M. Takahashi, T. Azuma, A. Ajavakom, T. Akiyama, M. Hasegawa, S. Taniguchi, T. Okada and Y. Sakata, Bull. Chem. Soc. Jpn., 1999, 72, 485 CrossRef CAS; (c) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and K. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS.
- (a) A. Samanta and P. V. Kamat, Chem. Phys. Lett., 1992, 199, 635 CrossRef CAS; (b) D. M. Guldi, M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1997, 119, 974 CrossRef CAS; (c) K. G. Thomas, V. Biju, M. V. George, D. M. Guldi and P. V. Kamat, J. Phys. Chem. A, 1998, 102, 5341 CrossRef CAS; (d) D. M. Guldi, G. T. Garscia and J. Mattay, J. Phys. Chem. A, 1998, 102, 9679 CrossRef CAS; (e) M. Maggini, D. M. Guldi, S. Mondini, G. Scorrano, F. Paolucci, P. Ceroni and S. Roffia, Chem. Eur. J., 1998, 4, 1992 CrossRef CAS; (f) A. Polese, S. Mondini, A. Bianco, C. Toniolo, G. Scorrano, D. M. Guldi and M. Maggini, J. Am. Chem. Soc., 1999, 121, 3446 CrossRef CAS; (g) D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS.
- (a) J. Llacay, C. Rovira, J. Veciana, M. Mas and E. Molins, Chem. Commun., 1997, 659 RSC; (b) J. Llacay, J. Veciana, J. Vidal-Gancedo, J. L. Bourdelande, R. Gonzalez-Moreno and C. Rovira, J. Org. Chem., 1998, 63, 5201 CrossRef CAS.
- (a) C. Boulle, J. M. Rabreau, P. Hudhomme, M. Cariou, M. Jubault, A. Gorgues, J. Orduna and J. Garín, Tetrahedron Lett., 1997, 38, 3909 CrossRef CAS; (b) P. Hudhomme, C. Boulle, J. M. Rabreau, M. Cariou, M. Jubault and A. Gorgues, Synth. Met., 1998, 94, 73 CrossRef CAS; (c) D. Kreher, D. P. Hudhomme, A. Gorgues, H. Luo, Y. Araki and O. Ito, Phys. Chem. Chem. Phys., 2003, 5, 4583 RSC.
- (a) E. Allard, J. Delaunay, F. Cheng, J. Cousseau, J. Orduna and J. Garín, Org. Lett., 2001, 3, 3503 CrossRef CAS; (b) E. Allard, J. Cousseau, J. Orduna, J. Garín, H. Luo, Y. Araki and O. Ito, Phys. Chem. Chem. Phys., 2002, 4, 5944 RSC.
- N. V. Tkachenko, L. Rantala, A. Y. Tauber, J. Helaja, P. V. Hynninen and H. Lemmetyinen, J. Am. Chem. Soc., 1999, 121, 9378 CrossRef CAS.
- D. I. Schuster, P. Cheng, S. R. Wilson, V. Prokhorenko, M. Katterle, A. R. Holzwarth, S. E. Braslavsky, G. Klihm, R. M. Williams and C. Luo, J. Am. Chem. Soc., 1999, 21, 1599.
- (a) T. Yamashiro, Y. Aso, T. Otsubo, H. Tang, T. Harima and K. Yamashita, Chem. Lett., 1999, 443 CrossRef CAS; (b) M. Fujitsuka, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, J. Phys. Chem. A, 2000, 104, 4876 CrossRef CAS; (c) P. A. van Hal, J. Knol, B. M. W. Langeveld-Voss, S. C. J. Meskers, J. C. Hummelen and R. A. J. Janssen, J. Phys. Chem. A, 2000, 104, 5974 CrossRef CAS; (d) J. J. Apperloo, B. M. W. Langeveld-Voss, J. Knol, J. C. Hummelen and R. A. J. Janssen, Adv. Mater., 2000, 12, 908 CrossRef CAS; (e) M. Fujitsuka, K. Matsumoto, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, Res. Chem. Intermed., 2001, 27, 73 CrossRef CAS; (f) M. Fujitsuka, A. Masuhara, H. Kasai, H. Oikawa, H. Nakanishi, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, J. Phys. Chem. B, 2001, 105, 9930 CrossRef CAS; (g) P. A. van Hal, E. H. A. Beckers, S. C. J. Meskers, R. A. J. Janssen, B. Jousselme, P. Blanchard and J. Roncali, Chem. Eur. J., 2002, 8, 5415 CrossRef CAS; (h) E. H. A. Beckers, P. A. van Hal, A. Dhanabalan, S. C. J. Meskers, J. Knol, J. C. Hummelen and R. A. J. Janssen, J. Phys. Chem. A, 2003, 107, 6218 CrossRef CAS.
- (a) N. Martín, L. Sánchez, B. Illescas and I. Pérez, Chem. Rev., 1998, 98, 2527 CrossRef CAS; (b) N. Martín, L. Sánchez and D. M. Guldi, Chem. Commun., 2000, 113 RSC; (c) M. A. Herranz and N. Martín, Org. Lett., 1999, 1, 2005 CrossRef CAS; (d) N. Martín, L. Sanchez, M. A. Herranz and D. M. Guldi, J. Phys. Chem. A, 2000, 104, 4648 CrossRef CAS; (e) M. A. Herranz, N. Martín, L. Sánchez, C. Seoane and D. M. Guldi, J. Organomet. Chem., 2000, 599, 2 CrossRef CAS; (f) D. M. Guldi, S. González, N. Martín, A. Antón, J. Marín and J. Orduna, J. Org. Chem., 2000, 65, 1978 CrossRef CAS; (g) M. A. Herranz, B. Illescas, N. Martín, C. Luo and D. M. Guldi, J. Org. Chem., 2000, 65, 5728 CrossRef CAS; (h) S. González, N. Martín, A. Swartz and D. M. Guldi, Org. Lett., 2003, 5, 557 CrossRef CAS; (i) L. Sánchez, M. A. Herranz and N. Martín, J. Mater. Chem., 2005, 15, 1409 RSC.
- (a) J. W. Arbogast, C. S. Foote and M. Kao, J. Am. Chem. Soc., 1992, 114, 2277 CrossRef CAS; (b) C. S. Foote, Top. Curr. Chem., 1994, 169, 347 CAS.
- Y. Wang, J. Phys. Chem., 1992, 96, 764 CrossRef CAS.
- (a) R. Seshadri, C. N. R. Rao, H. Pal, T. Mukherjee and J. P. Mittal, Chem. Phys. Lett., 1993, 205, 395 CrossRef CAS; (b) H. N. Ghosh, H. Pal, A. V. Saper and J. P. Mittal, J. Am. Chem. Soc., 1993, 115, 11722 CrossRef CAS.
- E. Schaffner and H. Fischer, J. Phys. Chem., 1993, 97, 13149 CrossRef CAS.
- (a) A. Watanabe and O. Ito, J. Phys. Chem., 1994, 98, 7736 CrossRef CAS; (b) O. Ito, Y. Sasaki, Y. Yoshikawa and A. Watanabe, J. Phys. Chem., 1995, 99, 9838 CrossRef CAS; (c) Y. Sasaki, Y. Yoshikawa, A. Watanabe and O. Ito, J. Chem. Soc., Faraday Trans., 1995, 91, 2287 RSC; (d) M. Alam, A. Watanabe and O. Ito, J. Photochem. Photobiol. A, 1997, 104, 59 CrossRef CAS; (e) C. Luo, M. Fujitsuka, A. Watanabe, O. Ito, L. Gan, Y. Huang and C. H. Huang, J. Chem. Soc., Faraday Trans., 1998, 94, 527 RSC.
- (a) M. Bennati, A. Grupp, P. Bäuerle and M. Mehring, Chem. Phys., 1994, 185, 221 CrossRef CAS; (b) M. Bennati, A. Grupp, P. Bäuerle and M. Mehring, Mol. Cryst. Liq. Cryst., 1994, 256, 751 CrossRef CAS.
- (a) B. Ma, G. E. Laeson, C. E. Bunker, A. Kitaygorodskiy and Y.-P. Sun, Chem. Phys. Lett., 1995, 247, 51 CrossRef CAS; (b) Y.-P. Sun, B. Ma and G. E. Lawson, Chem. Phys. Lett., 1995, 233, 57 CrossRef CAS; (c) G. E. Lawson, A. Kitaygorodskiy, B. Ma, C. E. Bunker and Y.-P. Sun, J. Chem. Soc., Chem. Commun., 1995, 2225 RSC.
- S. Fukuzumi, T. Suenobu, M. Patz, T. Hirasaka, S. Itoh, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 1998, 120, 8060 CrossRef CAS.
- (a) H. Imahori, K. Hagiwara, T. Akiyama, M. Akoi, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, Chem. Phys. Lett., 1996, 263, 545 CrossRef CAS; (b) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and K. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS.
- J. Veciana, J. Riera, J. Castañar and N. Ferrer, J. Organomet. Chem., 1985, 297, 131 CrossRef CAS.
- (a) M. Basllester, Adv. Phys. Org. Chem., 1989, 25, 267 CAS; (b) J. Veciana, C. Rovira, N. Ventosa, M. I. Crespo and F. Palacio, J. Am. Chem. Soc., 1993, 115, 57 CrossRef CAS; (c) N. Ventosa, D. Ruiz, J. Sedó, C. Rovira, X. Tomás, J.-J. André, A. Bieber and J. Veciana, Chem. Eur. J., 1999, 5, 3533 CrossRef CAS; (d) D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190 CrossRef CAS; (e) C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266 CrossRef CAS.
- S. Chopin, J. Delaunay and J. Cousseau, Tetrahedron Lett., 2005, 46, 373 CrossRef CAS.
- M. Ballester, J. Riera, J. Castañer, C. Rovira, J. Veciana and C. Onrubia, J. Org. Chem., 1983, 48, 3716 CrossRef CAS.
- M. Ballester, J. Castañer, J. Riera, A. Ibañez and J. Pujadas, J. Org. Chem., 1982, 47, 259 CrossRef CAS.
- S. Chopin, Z. Gan, J. Cousseau, Y. Araki and O. Ito, J. Mater. Chem., 2005, 15, 2288 RSC.
- A. Weller, Z. Phys. Chem. Neue Folge, 1982, 133, 93 CAS.
- The distance was evaluated from the minimized structure of the triad 1 obtained using semiempirical AM1 calculations (see ESI† Fig. SI-3). Three resulting minimized geometries were obtained in which the distance between the PTM unit and C60 are different. The geometry used was the conformation with the lower formation enthalpy (ΔHf). Distances between the center of C60 and PTM− were evaluated to be 10 Å from this optimized structure.
- H. Luo, M. Fujitsuka, Y. Araki, O. Ito, P. Padmawar and L. Y. Chiang, J. Phys. Chem. B, 2003, 107, 9312 CrossRef CAS.
- M. A. Herranz, N. Martin, J. Ramey and M. D. Guldi, Chem. Commun., 2002, 2968 RSC.
- (a) I. Ratera, D. Ruiz-Molina, J. Vidal-Gancedo, J. J. Novoa, K. Wurst, J.-F. Letard, C. Rovira and J. Veciana, Chem. Eur. J., 2004, 10, 603 CrossRef CAS; (b) I. Ratera, D. Ruiz-Molina, F. Renz, J. Ensling, K. Wurst, C. Rovira, G. Philipp and J. Veciana, J. Am. Chem. Soc., 2003, 125, 1462 CrossRef CAS.
- (a) M. Yamazaki, Y. Araki, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 8615 CrossRef CAS; (b) F. D'Souza, G. R. Deviprasad, M. E. Zandler, V. T. Hoang, A. Klykov, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Phys. Chem. B, 2002, 106, 4952 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CV of compounds 1 and 5, minimized geometry of compound 1, time-resolved fluorescence spectra, transient spectrum and time profiles in polar solvents. See DOI: 10.1039/b514882k |
| This journal is © The Royal Society of Chemistry 2006 |