Spin-glass behaviour in the double perovskite “Sr2FeTeO6” due to mis-site disorder and cation vacancy formation
Luis
Ortega-San Martin
a,
Jon P.
Chapman
a,
Luis
Lezama
a,
José J.
Saiz Garitaonandia
b,
Jorge
Sánchez Marcos
d,
Jesús
Rodríguez-Fernández
d,
María I.
Arriortua
c and
Teófilo
Rojo
*a
aDepartamento de Química Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco (UPV/EHU), Apdo. 644, E-48080 Bilbao, Spain. E-mail: teo.rojo@ehu.es
bDepartamento de Física Aplicada II, Facultad de Ciencia y Tecnología, Universidad del País Vasco (UPV/EHU), Apdo. 644, E-48080 Bilbao, Spain
cDepartamento de Mineralogía y Petrología, Facultad de Ciencia y Tecnología, Universidad del País Vasco (UPV/EHU), Apdo. 644, E-48080 Bilbao, Spain
dCITIMAC, Facultad de Ciencias, Universidad de Cantabria, 39005 Santander, Spain
First published on 16th November 2005
Abstract
The double perovskite of nominal composition Sr2FeTeO6 has been prepared by the freeze-drying method. Simultaneous Rietveld refinement of X-ray and high resolution D2B neutron diffraction data has shown it to have space group I12/m1 but with a significant degree of pseudo-tetragonal symmetry with a = 5.6080(3) Å, b = 5.5952(3) Å, c = 7.9081(5) Å and β = 90°, and a far more complex cation distribution over the B- and B′-sites. Refinement of Fe and Te fractions over these sites yielded the composition (Fe0.84□0.16)B(Te0.87Fe0.13)B′. This, and the absence of any significant formation of oxygen vacancies, yields an oxidation state for Fe close to +3, confirmed by EPR measurements (g ∼ 2.00). The disorder found over the B- and B′-sites implies that each Fe3+O6 octahedron is coordinated to six species, each of which can be Te, □ or another Fe, producing a statistical distribution of exchange pathways. The presence of several signals in the room temperature 57Fe Mössbauer spectrum and the broadening of the ESR signals can be related to this statistical distribution of the coordination environments around Fe3+. Magnetic DC susceptibility measurements indicate that the most important interactions are of antiferromagnetic nature. Absence of magnetic peaks in the 4 K neutron diffraction data, divergence below 40 K of field-cooled and zero-field-cooled DC susceptibilities, the frequency dependence of χ′ and χ″ magnetic AC susceptibilities and the lack of a three-dimensional λ-type peak in the specific heat measurements are all consistent with spin-glass type behaviour, as expected from the variety of frustrated magnetic interactions arising from the observed mis-site disorder and vacancies.
Introduction
Mixed metal oxides with the perovskite and double perovskite structures attract intense interest in many applied and fundamental areas of solid state chemistry, physics, advanced materials and catalysis due to the diverse physical properties that they present.1 Among them, perovskites with the general formula SrFe0.5B0.5O3 show a wide variety of interesting magnetic properties, which vary from low temperature antiferromagnetic insulators such as SrFe0.5Ru0.5O3,2 to room temperature ferrimagnets showing the magnetoresistive effect, as in the case of the technologically important Sr2FeMoO6 oxide.3 These properties are known to be highly dependent on the nature of the magnetic cations present (both Fe and B) and on structural features such as the degree of order between Fe and B, as explained below.It has been observed experimentally that in systems consisting of Fe3+ and a B transition element which can only be in a +5 oxidation state, a disordered arrangement of the two cations over the B and B′ sites of the perovskite structure is favoured. Examples of this type are SrFe0.5Ru0.5O3,2 SrFe0.5Ta0.5O34 and SrFe0.5Nb0.5O3.5 In all of these cases, magnetic behaviour has been explained considering the phases as spin-glass systems which result from competing ferromagnetic and antiferromagnetic interactions either between paramagnetic iron ions (in Ta and Nb compounds) or between iron and ruthenium ions statistically distributed over the octahedral B sites. If the B5+ cation is a p-group element such as Sb or Bi, a high degree of ordering is often achieved between iron and B′ and the magnetic behaviour is explained considering an antiferromagnetic ordering (usually of Type I) with moderately low Néel temperatures (TN < 100 K).6,7 The magnetic behaviour changes again when the most stable oxidation state of the B′ ion is +6, as in Sr2FeWO6, which shows complete structural ordering and is an antiferromagnet with type II three-dimensional ordering below ∼30–40 K.8
When the B′ cation is an element that is stable in both the +5 and +6 oxidation states, such as the paramagnetic cations Mo5+,6+ (S = ½, 0) or Re5+,6+ (S = 1, ½), the Sr2FeB′O6 phases obtained can be ordered or disordered depending on the synthetic conditions.9,10 These are also the most important phases from the magnetic and applications points of view as they present room temperature ferrimagnetism resulting from the incomplete cancellation of the localised Fe moments antiferromagnetically aligned with delocalised Mo/Re moments. This delocalisation leads to a half metallic conductivity that can be modified by an external applied field giving rise to the magnetoresistive effect.11
In the present study we have selected tellurium(VI) as the B′ cation in order to check the effects that can be produced on the magnetic properties of these phases when a p-group element (with a d10 shell) substitutes a transition element with an empty d shell. Previously, we have observed that the introduction of Te6+ in the series A2CoWO6 (A = Ba, Sr) induces a change in the magnetic structure from the typical Type II (that favours next-nearest neighbour antiferromagnetic interactions) to Type I (in which antiferromagnetic nearest-neighbour interactions are dominant) antiferromagnetic structure.12 In this paper, the physical properties of the “highly ordered perovskite, Sr2FeTeO6” have been correlated to significant vacancy formation and B-cation mis-site disorder.
Experimental
A polycrystalline sample of nominal composition Sr2FeTeO6 was synthesised by the freeze-drying method. Stoichiometric quantities of strontium carbonate, iron(II) acetylacetonate and tellurium dioxide were dissolved in dilute aqueous nitric acid in order to obtain 8 g of the final product. The solution was drop-by-drop frozen under liquid nitrogen. The frozen solution was subsequently freeze-dried and the powder obtained was ground and calcined at 800 °C for 6 hours. The sample was ground, pelleted and calcined several times for 8 hours at 900 °C until phase purity was confirmed by X-ray powder diffraction. Contents of Sr, Fe and Te were determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) performed with an ARL Fisons 3410 spectrometer. The morphology of the obtained powder was analysed using a JEOL JSM-6400 scanning electron microscope with an accelerating voltage of 20 kV.High resolution neutron powder diffraction data were collected at 4, 60 and 298 K on Instrument D2B at the Institut Laue Langevin (I.L.L.), Grenoble, France. Thermal neutrons of wavelength 1.594 Å were incident on 8 g of polycrystalline “Sr2FeTeO6”, packed in an 8 mm diameter vanadium can contained in a liquid helium cryostat. Data were collected in the angular range 5 ≤ 2θ ≤ 162° in steps of 0.05° with an integration time of 50000 monitor counts per step and with each point in the diffraction pattern being recorded by 6 independent detectors for subsequent normalisation and summation, giving an overall collection time of approximately 3 hours for the entire data set. Room temperature X-ray powder diffraction data were collected in the range 17 ≤ 2θ ≤ 105° with an integration time of 17 seconds per 0.02° step using a Philips X'Pert-MPD X-ray diffractometer with secondary beam graphite monochromated Cu-Kα radiation. The crystal structure was refined simultaneously from neutron and X-ray data by the Rietveld method13 using the GSAS software package.14
Small Angle Neutron Scattering (SANS) data were collected on the low-to-middle-angle diffractometer D16 at the I.L.L. Data were first corrected for differences in detector efficiency, then for background, multiple scattering and self-absorption and normalised to absolute scale using the CORRECT software.15
X-Band EPR spectra of “Sr2FeTeO6” were recorded between 0 and 7 kOe from 300 to 5 K on a Bruker ESP 300 spectrometer. The temperature was controlled by an Oxford Instruments (ITC4) regulator. The magnetic field was measured with a Bruker BNM 200 Gaussmeter and the frequency inside the cavity was determined using a Hewlett Packard 5352B microwave frequency counter. The 57Fe Mössbauer spectrum was recorded at room temperature in transmission geometry using a conventional constant-acceleration spectrometer with a 57Co ∶ Rh source. Measured isotopic shifts were then corrected relative to metallic Fe. DC magnetic susceptibility measurements were performed using a Quantum Design MPMS-7 SQUID magnetometer whilst heating from 5 to 300 K in an applied field of 1 kOe, after cooling either in the presence (field cooling, FC) or absence (zero field cooling, ZFC) of the applied field. Magnetisation as a function of applied field (H) was measured using the same MPMS-7 SQUID magnetometer at 5 K after cooling the sample in zero field. During the measurement, the field was swept between 70 and −70 kOe.
AC magnetic susceptibility measurements were made using a standard QD PPMS system with an alternate excitation field (Hac) of 1 Oe. Data were recorded from 4 to 100 K as a function of both frequency and applied DC magnetic field. Frequencies from 100 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz were measured in the absence of an applied magnetic field and field dependent data were collected at a fixed frequency of 1000 Hz with applied fields from 0 to 40 kOe. Heat capacity measurements were carried out by a relaxation method using the same PPMS system. The sample was an 11 mg plate of 0.3 mm thickness obtained by compressing the polycrystalline powder. Data were collected with zero field and under an applied field of 90 kOe from 1.8 to 300 K. The phonon contribution, modelled by two independent Debye16 spectra, was subtracted to give the magnetic specific heat, Cp,mag, from which the magnetic entropy Smag was calculated.
000 Hz were measured in the absence of an applied magnetic field and field dependent data were collected at a fixed frequency of 1000 Hz with applied fields from 0 to 40 kOe. Heat capacity measurements were carried out by a relaxation method using the same PPMS system. The sample was an 11 mg plate of 0.3 mm thickness obtained by compressing the polycrystalline powder. Data were collected with zero field and under an applied field of 90 kOe from 1.8 to 300 K. The phonon contribution, modelled by two independent Debye16 spectra, was subtracted to give the magnetic specific heat, Cp,mag, from which the magnetic entropy Smag was calculated.
Results
The crystal structure determination of the ordered double perovskite “Sr2FeTeO6” was performed on 8 g of a homogeneous orange–brown sample with a mean grain size of 0.5 µm determined by Scanning Electron Microscopy (SEM) measurements, as shown in Fig. 1. | ||
| Fig. 1 SEM image of polycrystalline “Sr2FeTeO6” recorded at 20 kV and ×5000 magnification showing a homogeneous powder of 0.5 µm mean grain size. | ||
The high resolution neutron and X-ray powder diffraction patterns were indexed within a unit cell of approximate dimensions √2ap
×
√2ap
× 2ap, where ap is the primitive cubic perovskite parameter. The poor resolution of the ‘characteristic’ (404) X-ray diffraction maximum (which corresponds to the unique (444) in the 2ap
× 2ap
× 2ap cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m lattice) and the absence of reflections breaking the I centring condition on hkl (h + k + l = 2n), compared to those observed in other tellurium double perovskites recently studied by our group (Sr2BTeO6 with B = Mn, Ni and Co12,17) show the symmetry to be I12/m1 monoclinic (#12; b3). Despite the monoclinic symmetry required to describe the rotational distortion of the BO6 and B′O6 octahedra, the structure shows a high degree of pseudo-tetragonal symmetry with reduced refined lattice parameters (see Table 1), especially b and c/√2, showing little difference. The monoclinic β angle refined to 90° within its standard deviation and free refinement of the fractional coordinates for Sr yielded values corresponding to the
m lattice) and the absence of reflections breaking the I centring condition on hkl (h + k + l = 2n), compared to those observed in other tellurium double perovskites recently studied by our group (Sr2BTeO6 with B = Mn, Ni and Co12,17) show the symmetry to be I12/m1 monoclinic (#12; b3). Despite the monoclinic symmetry required to describe the rotational distortion of the BO6 and B′O6 octahedra, the structure shows a high degree of pseudo-tetragonal symmetry with reduced refined lattice parameters (see Table 1), especially b and c/√2, showing little difference. The monoclinic β angle refined to 90° within its standard deviation and free refinement of the fractional coordinates for Sr yielded values corresponding to the ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) position in the tetragonal I4/m space group. In the final model, these variables were therefore fixed to these pseudo-tetragonal values. The crystal structure is characterised by the tilting of the BO6 and B′O6 octahedra along the [100]p and [010]p axes of the primitive perovskite (Fig. 2), the rotations being anti-phase (−) around both axes.18
position in the tetragonal I4/m space group. In the final model, these variables were therefore fixed to these pseudo-tetragonal values. The crystal structure is characterised by the tilting of the BO6 and B′O6 octahedra along the [100]p and [010]p axes of the primitive perovskite (Fig. 2), the rotations being anti-phase (−) around both axes.18
 | ||
| Fig. 2 Projections of the room temperature structure of Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6 along the three primitive axes of the cubic perovskite showing the absence (○) or presence (–) of out-of-phase rotations. Dark octahedra represent BO6, light octahedra B′O6 and spheres Sr atoms. | ||
| 298 K (XPD/NPD) | 60 K (NPD) | 4 K (NPD) | ||
|---|---|---|---|---|
| a/Å | 5.6080(3) | 5.5997(2) | 5.5995(2) | |
| b/Å | 5.5952(3) | 5.5805(2) | 5.5802(2) | |
| c/Å | 7.9081(4) | 7.8854(4) | 7.8847(3) | |
| (c/√2)/Å | 5.5919(3) | 5.5758(3) | 5.5753(2) | |
| β/° | 90 | 90.03(1) | 90.03(1) | |
| Volume/Å3 | 248.14(2) | 246.41(2) | 246.37(1) | |
| Sr | x | 0.5 | 0.5 | 0.5 |
| y | 0 | 0 | 0 | |
| z | 0.75 | 0.75 | 0.75 | |
| U iso (×100)/Å2 | 1.91(2) | 1.61(2) | 1.59(2) | |
| B x = y | 0.5 | 0.5 | 0.5 | |
| z | 0 | 0 | 0 | |
| U iso (×100)/Å2 | 1.02(5) | 1.3(1) | 1.4 (2) | |
| B′ x = y = z | 0 | 0 | 0 | |
| U iso (×100)/Å2 | 0.61(4) | 0.5(2) | 0.4(2) | |
| O1 | x | 0.0395(9) | 0.040(1) | 0.043(1) |
| y | 0 | 0 | 0 | |
| z | 0.755(1) | 0.751(2) | 0.754(2) | |
| U iso (×100)/Å2 | 1.4(1) | 1.4(1) | 1.3(1) | |
| O2 | x | 0.239(1) | 0.241(1) | 0.239(1) |
| y | 0.254(1) | 0.254(1) | 0.257(2) | |
| z | 0.0157(4) | 0.0190(4) | 0.0201(4) | |
| U iso (×100)/Å2 | 2.03(6) | 1.70(6) | 1.69(6) | |
| R wp (%) (XRD/NPD) | 13.7/4.0 | 4.8 | 4.8 | |
| R p (%) (XRD/NPD) | 8.6/3.1 | 3.8 | 3.8 | |
| χ 2 | 1.5 | 1.5 | 1.5 | |
| R F (XRD/NPD) | 3.0/1.4 | 1.8 | 1.8 | |
Peak profiles and background were fitted as previously reported for related Sr2BTeO6 (B = Mn, Ni and Co) oxides. Isotropic thermal parameters of all atoms were allowed to refine freely. The contrast provided between Fe and Te when refining X-ray and neutron diffraction data simultaneously (the ratio of scattering factors for X-rays and neutrons, Te ∶ Fe, being 2.00 ∶ 1 and 0.37 ∶ 1,19 respectively) allowed the free refinement of Fe and Te fractions over the B and B′ sites. The refined B/B′-site composition of (Fe0.841(4)□0.159)B(Te0.847(4)Fe0.126(6))B′ showed evidence for significant mis-site disorder and B-site vacancy (□) formation. As no evidence for vacancy formation on the B′-site was found, this site was returned to full occupancy giving a B′-site cation distribution of (Te0.87Fe0.13). Three independent oxygen fractions were also allowed to refine but all remained close to unity. In the final model, all oxygen fractions were therefore fixed at unity yielding a refined composition Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6. Refined parameters and statistics are given in Table 1 and selected bond distances and angles in Table 2. Rietveld fits to the room temperature X-ray and neutron diffraction data are shown in Fig. 3.
 | ||
| Fig. 3 Rietveld fit of Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6 in space group I12/m1, showing observed, calculated and difference curves, to (a) the room temperature X-ray diffraction data, (b) the room-temperature D2B neutron data and (c) the 4 K D2B neutron data. Lower and upper reflection markers correspond, in neutron diffraction data, to Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6 and ca. 0.6% Fe2O3, respectively. Fits to the high angle data are inset. | ||
| 298 K | 60 K | 4 K | |
|---|---|---|---|
| a Value calculated considering the following ionic radii (in Å) and coordination numbers, CN: Fe3+CN=6 = 0.645, Te6+CN=6 = 0.56 and O2−CN=2 = 1.35.20 | |||
| B–O1 ×2 | 2.03(1) | 2.00(1) | 2.02(1) |
| B–O2 ×4 | 2.011(6) | 2.006(3) | 2.003(8) |
| Mean | 2.017 | 2.002 | 2.002 |
| Calculated Fe–O a | 1.995 | ||
| O1–B–O2 | 91.0(2) | 90.8(1) | 90.5(1) |
| O2–B–O2 | 93.7(3) | 93.6(5) | 94.5(5) |
| B′–O1 ×2 | 1.95(1) | 1.98(1) | 1.96(1) |
| B′–O2 ×4 | 1.960(6) | 1.964(6) | 1.964(8) |
| Mean | 1.957 | 1.969 | 1.973 |
| Calculated Te–O a | 1.910 | ||
| O1–B′–O2 | 90.9(2) | 90.6(2) | 90.2(2) |
| O2–B′–O2 | 93.0(3) | 92.6(5) | 93.4(5) |
| B–O1–B′ | 167.2(3) | 165.8(2) | 166.0(2) |
| B–O2–B′ | 172.0(2) | 170.8(2) | 170.2(2) |
Observed values from atomic emission spectroscopy of Sr 1.92(2), Fe 1.07(1) and Te 0.90(2) were more consistent with those calculated for the refined cation distribution, Sr2(Fe0.84)(Te0.87Fe0.13)O6: Sr 2.00, Fe 0.97 and Te 0.87 than for the nominal Sr2FeTeO6 stoichiometry. The oxidation state of +3 for Fe, calculated from this refined cation distribution, will be discussed later in relation to the spectroscopic measurements. A small fraction, 0.6(1)% by mass, of Fe2O3, only visible in the neutron diffraction data, was refined as a secondary phase.
The room temperature EPR data (see Fig. 4a) showed a single, broad resonance which was fitted by a Lorentzian function with a peak-to-peak linewidth, ΔHpp, of 1200(5) Oe and a resonance field of 3375(2) Oe, the latter corresponding to a g value of 1.995(3). These results are consistent with an oxidation state of +3 for the iron cations.21
 | ||
| Fig. 4 (a) Normalised EPR spectra between 290 and 40 K. The inset shows the EPR spectrum at room temperature fitted with a single Lorentzian function. (b) Evolution with temperature of the signal area (AEPR) and the peak-to-peak linewidth (ΔHpp). | ||
The thermal evolution of the integrated area (AEPR) and the peak-to-peak linewidth (ΔHpp) of the EPR signal from room temperature to 40 K are shown in Fig. 4b. The peak-to-peak linewidth can be related to the Fe3+ spins' relaxation time, τ, in a way that ΔHpp is proportional to the inverse of τ.22 Usually, τ increases with decreasing temperature reducing the signal broadening. This effect is not always observed in ΔHpp as the exchange and dipolar interactions between iron ions act in the opposite sense. Fig. 4b shows that ΔHpp increases monotonically on lowering the temperature, indicating that the exchange and dipolar interactions between paramagnetic cations, and not the spin–lattice relaxation time, are dominating the resonance process. This is a consequence of the mis-site disorder producing pairs of Fe ions that are less separated than they would be in a fully ordered perovskite, giving rise to stronger exchange interactions. Below 40 K, the signal diminishes, which is usually an indication of a magnetically ordered state below this temperature. In the present case no magnetic peaks were observed in the neutron diffraction data (discussed later), indicating that this low temperature state is not three-dimensionally ordered.
On the other hand, AEPR is related to the number of paramagnetic spins and is generally considered as the “EPR susceptibility”. In order to avoid intensity losses due to broadening of the signal into magnetic field regions not covered by the measurements, AEPR was calculated as Y·Γ·π by double integration of the derivative equation that defines the resonance process,21 where Y is the height of the signal and Γ the half-width at half-height, both of which can be directly obtained by fits to the experimental data. As observed, AEPR increases slightly with decreasing temperature down to 70 K. Below this temperature there is significant broadening of the signal together with a reduction in intensity. These effects are consistent with the existence of strong magnetic correlations between Fe3+ spins below this temperature but which, in this case, do not lead to a three dimensionally ordered state.
The fit to the 57Fe Mössbauer spectrum at room temperature is represented in Fig. 5. The spectrum is well fitted by a pair of doublets with equal half-width of 0.186(2) mm s−1. Isomer shifts, quadrupolar splittings and integrated areas were independently refined for each doublet. Two doublets with equal isomer shifts, δ, of 0.435(2) and 0.434(1) mm s−1 relative to metallic Fe with quadrupolar splittings of 0.23 and 1.21 mm s−1, respectively, are both consistent with high-spin Fe in the +3 oxidation state with S = ![[/]](https://www.rsc.org/images/entities/char_e11f.gif) .23 The mean Fe–O bond distance of 2.02 Å obtained from the diffraction data agrees well with the expected δ value of 0.43 mm s−1 as indicated by McCammon for iron containing perovskites.24
.23 The mean Fe–O bond distance of 2.02 Å obtained from the diffraction data agrees well with the expected δ value of 0.43 mm s−1 as indicated by McCammon for iron containing perovskites.24
 | ||
| Fig. 5 Room temperature 57Fe Mössbauer spectrum of “Sr2FeTeO6” showing the two fitted doublets (D1 and D2) of equal half-width, their sum and the difference curve on the same scale. Isomer shift is relative to metallic Fe. | ||
The temperature dependence of the magnetic and reciprocal susceptibilities measured at 1 kOe are shown in Fig. 6. The molar magnetic susceptibility, χm, increases with decreasing temperature and reaches a broad maximum at ca. 29 K. The continuous decrease of the product χmTvs. temperature, inset in Fig. 6, indicates that the predominant interactions are of antiferromagnetic nature. As the high temperature molar magnetic susceptibility does not follow the classical Curie–Weiss law, it was not possible to fit the data over the whole high-temperature range, even with the introduction of a temperature-independent factor, χo, meaning that an effective paramagnetic moment (μeff) could not be estimated. However, from the slope of the reciprocal susceptibility at 300 K, a magnetic moment, μ300K, of 5.4 μB could be calculated, in excellent agreement with that expected for Fe3+.
 | ||
| Fig. 6 (a) Temperature dependence of the magnetic and reciprocal susceptibilities measured at 1 kOe. (b) Low temperature field cooled (FC) and zero field cooled (ZFC) susceptibilities. The inset in (b) shows the thermal evolution of the product χmT. | ||
The low-temperature magnetic susceptibility, measured whilst warming from 5 K under 1 kOe after cooling from room temperature firstly without applied field (ZFC) and subsequently under applied field (FC), is characterised by a clear divergence of the ZFC and FC signals below the maximum observed at 29 K, indicating the possible presence of weak ferromagnetic interactions due to an incomplete antiferromagnetic ordering of the sample or, more probably, to a spin-glass-like behaviour.25
Field dependent magnetisation measurements, performed at temperatures slightly above (50 K) and below (5 K) the observed maximum in the χm curves, are shown in Fig. 7. A small hysteresis loop develops at low temperature with Hc and Mr values of 1970 Oe and 110 emu mol−1, respectively. Saturation is not reached, even at an applied field of 70 kOe. The hysteresis loop is no longer observed at temperatures above the observed maximum in the χm curves, indicating that the observed ferromagnetic moment is only associated with the magnetic state adopted below ∼30–40 K. The evolution with temperature of the low temperature magnetisation, obtained at HDC = 0 after cooling from T > TN under a magnetic field of 100 Oe (see Fig. 7a) confirms this point, as no remnant magnetisation is observed above 40 K.
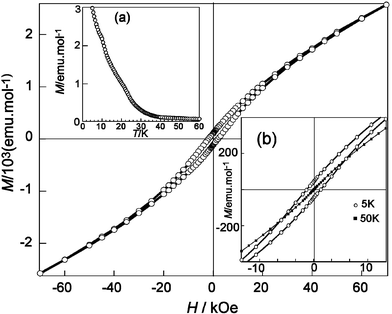 | ||
| Fig. 7 Magnetisation vs. applied field at 5 K. Inset are (a) remnant magnetisation vs. temperature after cooling the sample under an applied field of 100 Oe and (b) detail of the −10 ≤ H/kOe ≤ 10 region showing magnetic irreversibility at 5 K compared to that at 50 K. | ||
Low temperature neutron diffraction data collected at 60 and 4 K are presented in Fig. 8, together with the room temperature data for comparison. Additional maxima in the 4 K neutron diffraction data, expected for a three-dimensionally ordered antiferromagnetic state below 30 K, were not present, indicating that a long-range magnetically ordered state is not achieved. This feature can not be explained by a canted antiferromagnetic state and points directly to a freezing of the spins below a temperature close to 30 K, i.e. spin glass behaviour. Small angle neutron scattering (SANS) measurements were performed in order to obtain information about the possible existence of a magnetic cluster structure of the scattering system. Data at 60, 35 and 5 K (represented in the inset of Fig. 8) showed no change in cross section as a function of temperature, indicating that no magnetic contribution to the SANS exists, even well below the observed maximum in χm.
 | ||
| Fig. 8 Neutron diffraction data of “Sr2FeTeO6” at 4, 60 and 298 K showing the absence of magnetic peaks at low temperatures. The inset shows the differential cross section vs.Q-space at 5, 35 and 60 K. | ||
To ascertain the possible existence of spin-glass behaviour in this phase, AC magnetic susceptibility measurements were carried out. The real (or in-phase, χ′) and imaginary (or out-of-phase, χ″) components of the AC magnetic susceptibility at different frequencies are given in Fig. 9a and b. In apparent contrast with the unique and broad maximum observed at the intermediate temperature of ∼30 K in χm, the χ′ curve showed a broad maximum around 40 K followed by a change of slope around 20 K, indicating that the magnetic interactions in the sample are complex and proceed in more than one step. This complex behaviour is also evidenced by the two maxima in the out-of-phase susceptibility, which is associated to energy losses due to the dynamical process.
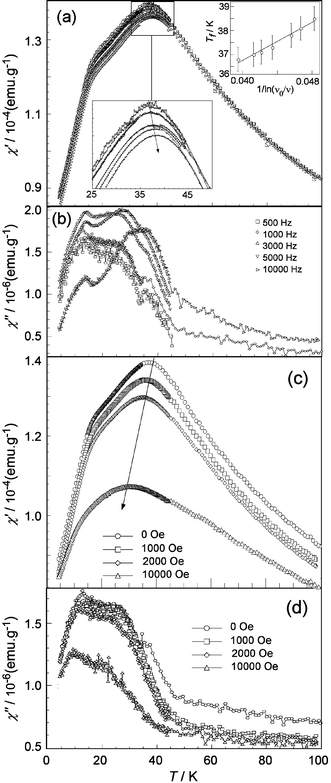 | ||
| Fig. 9 Real (χ′) and imaginary (χ″) components of the AC magnetic susceptibility as a function of (a, b) frequency and (c, d) applied DC field. In (a) details of the maximum susceptibility and its thermal evolution (1/ln(ν0/ν) vs.Tf) are inset. | ||
The effect on χ′ of the increasing frequency is a shift of the first maximum to higher temperatures. If we denote the temperature at which this maximum appears as Tf, it can be said that, depending on the intensity of the magnetic interactions Tf can be strongly (weak interactions) or weakly (strong interactions) dependent on the measuring time. This frequency dependence of Tf is usually characterised by the term ΔTf/[Tf·Δ(logν)]. In the present case, the obtained value of 0.022 falls within the range 0.004–0.080 reported for spin glasses.25 Another way to characterise this type of behaviour is to monitor the dependence of Tf as if it were a real glass. This dependence usually obeys the empirical Vogel–Fulcher law:25

 | ||
| Fig. 10 Specific heat vs. temperature of “Sr2FeTeO6” from 1.8 to 80 K showing total specific heat, Cp, the calculated phonon contribution, Cp,pho, and the resultant magnetic contribution, Cp,mag. The inset shows the calculated magnetic entropy, Smag. | ||
The influence of a superimposed DC field in χ′ and χ″ is also shown in Fig. 9c. The AC susceptibility shows a strong dependence on magnetic field, although lower than that expected for canonical spin glasses. The two χ′ features evolve towards a rounded peak of significantly smaller amplitude when the intensity of the DC field increases and in addition this peak is displaced to lower temperatures. The χ″ curve shows a similar displacement with magnetic field. The fact that the χ′ and χ″ maxima do not disappear, even under applied fields up to 40 kOe, indicates the existence of stronger magnetic interactions than in canonical spin glasses. Furthermore, the displacement of the maximum to lower temperatures with increasing magnetic field could be associated to a preponderant AF character of the magnetic interactions.
The specific heat data between 1.8 and 80 K, shown in Fig. 10, do not exhibit a maximum due to any magnetic ordering. The contribution of the lattice vibrations to the specific heat capacity was fitted to the high temperature data (above 50 K) with a modified Debye model16 considering the existence of two phonon spectra. For the unit cell containing N atoms, n1 were assigned a Debye temperature θ1 and n2, constrained such that n2 = N − n1, a Debye temperature θ2, yielding three independent variables to be refined, namely θ1, θ2 and n1. Refined values indicate that, of the 10 atoms in the unit cell, 5.7 (n1) have a Debye temperature of 260.9 K and 4.3 (n2 = N − n1) have 767.6 K. This result not only produces an excellent fit to the experimental data (see Fig. 10) but is also in good agreement with the unit cell contents of 4 heavy atoms and 6 lighter atoms with a higher Debye temperature. The magnetic contribution to the heat capacity was calculated as Cp,mag = Cp − Cp,pho. A small and very broad hump with no resemblance to a λ-type peak was observed around 30 K confirming that the magnetic transition observed around this temperature is far from a three dimensional one. The thermal evolution of the magnetic entropy, Smag, for the ordering process in this phase is shown in the inset of Fig. 10. The total magnetic entropy, calculated as Smag(T) = ∫T0Cmag(T)/TdT, was found to be ≈2.9 J mol−1 K−1 above 40 K. This value is less than 20% of the theoretical value, Rln(2S + 1) = 14.9 J mol−1 K−1, expected for the S = 5/2 state of the Fe3+ cation at low temperatures. As observed by Wiebe et al.27 in the related ordered double perovskite, Sr2MgReO6, these low values of entropy removal seem to be a common feature of spin-glasses. In particular, in “Sr2FeTeO6” this result indicates that, at 1.8 K, the magnetic moments still have a high degree of disorder, close to that of the paramagnetic state.
Discussion and conclusions
Ordered double perovskites are known to display a varied structural chemistry with a wide range of physical properties.28 From the structural point of view these perovskites are a classic example of compounds showing a high degree of pseudo-symmetry.29 The present “Sr2FeTeO6” oxide is a good example of this behaviour in which, despite a highly pseudo-tetragonal lattice, the true symmetry is monoclinic, space group I12/m1. The resulting crystal structure is characterised by the tilting of the BO6 and B′O6 octahedra along the [100]p and [010]p axes of the primitive perovskite, the rotations being anti-phase (−) around both axes.X-Ray and neutron diffraction data have shown that this compound presents a high degree (up to 15%) of tellurium vacancies that are concentrated in the iron site and no deviations from nominal stoichiometry for iron, strontium and oxygen, giving the following chemical formula: Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6. These tellurium vacancies result in an increase of the nominal iron oxidation state from +2 to +3 in order to maintain the electroneutrality of the sample. This iron oxidation state has been verified using EPR (with a gyromagnetic factor of g ≈ 2.00) and 57Fe Mössbauer (which consist of two signals both centred at isomer shifts of δ ≈ 0.43 mm s−1) spectroscopic measurements.
The presence of iron in the +3 oxidation state may also be the reason for the observed 15% of mis-site disorder between tellurium and iron sites. Considering the results proposed by Galasso30 that the degree of mis-site disorder increases when the differences between ionic radii and oxidation state between B and B′ ions decrease, the disorder found in the oxide “Sr2FeTeO6” is justified due to a minimisation of these parameters compared to other fully ordered tellurium(VI) double perovskites listed in Table 3.
The presence of B-cation vacancies is, however, the most important structural feature of this sample. A thorough search of the available literature on double perovskites has shown that this feature is an uncommon characteristic in these perovskites but some examples are known.33 This ability of the perovskite structure to allow such a high level of cation defects is a confirmation of the varied structural chemistry of perovskites and a clear proof of the robustness of the perovskite structural framework. In “Sr2FeTeO6”, these vacancies, together with the presence of disorder, result in a complex distribution of B-cations over both the B and B′ octahedral sites.
In an Sr2BB′O6 double perovskite with no mis-site disorder, all BO6 octahedra are coordinated to six B′ cations and, conversely, all B′O6 octahedra to six B cations. In the current case, mis-site disorder leads to compositions of B = Fe0.84□0.16 (where □ represents a B-cation vacancy) and B′ = Te0.87Fe0.13.
In order to understand the physical properties of Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6, it is necessary to first consider the statistical distribution of the coordination environments of the paramagnetic cations. Firstly, the majority of Fe3+ (the only paramagnetic cation present) is located on the B-site, FeB, and is coordinated to either Te or FeB′ with fractional occupancies 0.87 and 0.13 respectively. The remaining Fe3+ on the B′-site, FeB′, is coordinated to either FeB or vacancy □ with fractional occupancies 0.84 and 0.16.
For m independent events, each of which has two possible outcomes A and B with probabilities PA and PB respectively, the probability P of A occurring n times (and B therefore occurring m
−
n times) is given by the expression:34
| P = mCn.PAn.PB(m−n) |
| mCn = m!/(m − n)! n! |
Values of mCn and the calculated statistical probability distribution of all possible coordination environments for FeB and FeB′ are given in Table 4 and represented graphically in Fig. 11. Of particular importance when discussing the physical properties of Sr2(Fe0.84□0.16)(Te0.87Fe0.13)O6, will be the relative fractions of the highly symmetrical FeX6 and the remaining less symmetrical coordination environments. The sum of the calculated fractions of Fe⋯Te6 and Fe⋯Fe6 (with a negligible fraction for Fe⋯□6) is 0.425, leaving a total fraction of 0.575 for those of lower symmetry.
| FeB… | B′6 | mCn | P a | FeB′… | B6 | mCn | P a | |
|---|---|---|---|---|---|---|---|---|
a
Probability, normalised over all Fe on B and B′ sites, with fractional occupancies, f(FeB) and f(FeB′) such that:
|
||||||||
| Fe6 | 1 | 0.000 | Fe6 | 1 | 0.046 | |||
| Fe5Te | 6 | 0.000 | Fe5□ | 6 | 0.052 | |||
| Fe4Te2 | 15 | 0.003 | Fe4□2 | 15 | 0.025 | |||
| Fe3Te3 | 20 | 0.025 | Fe3□3 | 20 | 0.006 | |||
| Fe2Te4 | 15 | 0.126 | Fe2□4 | 15 | 0.001 | |||
| FeTe5 | 6 | 0.338 | Fe□5 | 6 | 0.000 | |||
| Te6 | 1 | 0.379 | □6 | 1 | 0.000 | |||
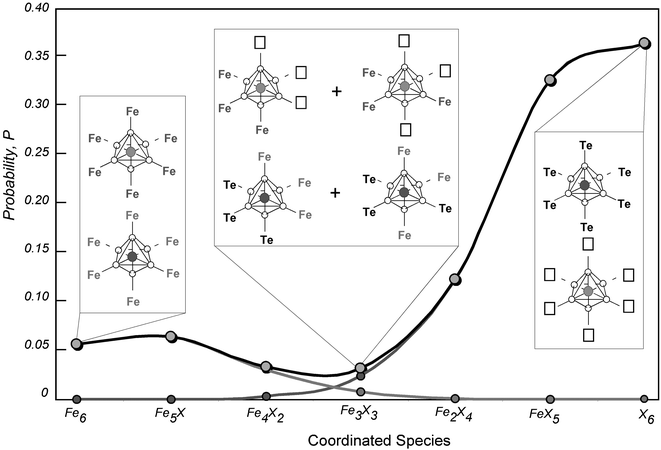 | ||
| Fig. 11 Probability distribution of species coordinated to the FeO6 octahedra, where X is either Te or a cation vacancy (□). Normalised probability curves correspond to the Fe0.84 on the B-site, the Fe0.13 on the B′-site and the sum over all Fe within the double perovskite phase. Selected coordination geometries of FeB and FeB′ are depicted. | ||
The two observed signals in the Mössbauer spectra can therefore be assigned to this cation distribution in the sample. Doublet 1, with relative area of 0.554(1) and a quadrupolar splitting, Δ, of 1.214(1) mm s−1, has been assigned to the lower symmetry coordination environments around the FeO6 octahedra and the relative area is in good agreement with the 0.575 expected from the calculated statistical distribution. Doublet 2, with a lower quadrupolar splitting of 0.230(2) mm s−1 and relative area of 0.425(3) can be assigned to the highly symmetrical coordination to either six Fe or six Te ions. It may be expected that these two symmetrical species would be distinguishable by their different isomer shifts due to the different electron density on the central Fe when coordinated to Fe3+ or Te6+. However, attempts to resolve this doublet into two signals, even by fixing the relative areas to those values expected from the statistical distribution of 0.379 and 0.046, were unsuccessful. Likewise, doublet 1, corresponding to the more asymmetric coordination environments, could not be further resolved despite the variety of symmetries and electron densities expected around the Fe3+ centre.
The observed transition temperature of ∼70 K, taken to be the maximum in the integrated area of the EPR signal, AEPR, is significantly higher than the 30–40 K from DC and AC susceptibility measurements. This is consistent with the existence of a variety of magnetic interactions of different strengths resulting from the anisotropic distribution of iron atoms in the sample. Taking into account the greater sensitivity of EPR spectroscopy to the local environment of the paramagnetic atoms, compared to DC and AC magnetic susceptibilities, the shift of the maximum in AEPR to higher temperature is a direct consequence of the strongest exchange and dipolar interactions between nearest-neighbour iron atoms in the disordered regions.
The evolution of the EPR signal with temperature indicates that exchange and dipolar interactions between iron cations dominate the resonance process down to 35–40 K. Below this temperature the disappearance of the EPR resonance means that strong magnetic correlations between iron spins are established. These results are in good agreement with the antiferromagnetic behaviour observed from DC magnetic susceptibility measurements which showed a broad maximum at a temperature of approximately 30 K.
Magnetic susceptibility measurements did not follow the Curie-Weiss Law in the temperature range covered. This behaviour has also been observed in similar complex perovskites such as SrLaFeSnO6, SrEuFeSnO6, BaLaFeSnO6, Ba2FeNbO6 and ALaFeVO6 (A = Ca and Sr)35 which show the simultaneous presence of locally ordered and disordered regions. Consequently, this behaviour could be associated to the complex distribution of Fe exchange paths arising from the disorder and vacancies in the present “Sr2FeTeO6” oxide.
The strong irreversibility in the ZFC and FC curves and the presence of a small hysteresis loop below the ordering temperature indicate that weak ferromagnetic interactions exists. These interactions usually exist when there is an incomplete antiferromagnetic ordering of the compound or, if some degree of disorder exists, in systems with a spin-glass-like behaviour. Low temperature neutron powder diffraction has shown that no magnetic peaks appear even at 4 K, precluding the possibility that the divergence of the ZFC and FC curves at ≈29 K is due to long range three-dimensional antiferromagnetic ordering. The observed frequency dependence of the χ′ and χ″ AC magnetic susceptibilities has demonstrated that this oxide behaves as an spin-glass. We can approximate the freezing temperature to be around 40 K although the deviation of the magnetic susceptibility from the Curie–Weiss law, expected at around T ∼ 5Tf in spin glasses,36 seems to begin at temperatures higher than the range covered in this experiment. The presence of multiple peaks in the χ″ curves could also be related to the presence of distinct ordered and disordered regions in the sample with different, and competing, magnetic interactions.
This behaviour has been summarised in Fig. 12, where a two-dimensional projection of the “Sr2FeTeO6” B-site cation distribution, including the disorder and vacancies observed in the sample, is presented. From this representation, it can be deduced that the exchange interactions inside both regions must be different. In the disordered region, the behaviour must be the same as in a simple ABO3 perovskite and it is expected that the nearest-neighbour interaction (NNd) between Fe3+ ions, separated by a distance of ∼4 Å through a 180° exchange path, be antiferromagnetic and that the resulting diagonal next-nearest-neighbour interaction (NNNd), at a distance of ∼5.5 Å through a 90° exchange path, be ferromagnetic.37 In these cases the NNd interactions are stronger than the NNNd interactions.
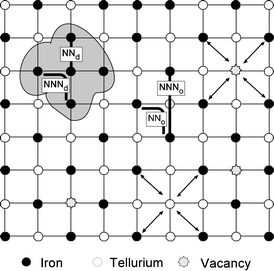 | ||
| Fig. 12 Schematic representation of the B-site cation distribution showing nearest-neighbour (NN) and next-nearest neighbour (NNN) interactions. The shaded zone indicates a disordered region in which the Fe–Fe exchange path lengths are significantly reduced with respect to those in an ordered perovskite. Diagonal arrows represent interrupted exchange paths between paramagnetic cations arising from mis-site disorder and vacancy formation (see text). | ||
In an ordered double perovskite the nearest-neighbour interactions, NNo, occur through a 90° and ∼5.5 Å exchange path and are equivalent to the NNNd. These NNo interactions are mostly ferromagnetic if the NNNo interactions (through a 180°, ∼7.9 Å exchange path) are antiferromagnetic yielding the common Type II antiferromagnetic structure. If the strength of the NNNo interaction approaches zero, however, the NNo interactions are dominant and antiferromagnetic giving the Type I structure.38 In this scenario, it is easily inferred that the exchange interactions inside both regions can be of opposite sense and result in magnetic frustration.39
Occasionally, it has been argued that the presence of small degrees of disorder, as in the case of the ordered double perovskites Sr2FeSbO6 or Sr2FeBiO6, characterised by high irreversibilities in the ZFC and FC susceptibility curves, may cause a spin-glass-like behaviour. The observation of a Type I, three-dimensional antiferromagnetic structure at low temperatures in the former case, however, indicates that the origin of this irreversibility may be due to a canting in the antiferromagnetic structure rather than from a spin-glass behaviour.
In consequence, the lack of a measurable magnetic structure in “Sr2FeTeO6” and the complex behaviour observed in the AC susceptibility curves must be the result of a more complicated frustration mechanism. The presence of iron site vacancies could be another source of frustration as these vacancies interrupt some of the exchange paths between iron ions in the ordered zones, further complicating the magnetic interactions.
Recently, Blasco et al.40 observed that only the simultaneous presence of ordered and disordered regions, together with a small amount of vacancies in the oxygen positions, can be the origin of spin-glass behaviour in the ordered perovskite La2MgMnO6−δ, and that in antiferromagnetically ordered double perovskites it is not possible to observe spin-glass behaviour considering only mis-site defects. The presence of vacancies, either in the B cation site or in the oxygen content, must exist to consider this type of behaviour. We therefore consider that the coexistence of ordered and disordered zones, plus B-site site vacancies, must be the origin of the observed spin glass behaviour in “Sr2FeTeO6”.
Finally, we must distinguish the spin-glass behaviour observed in these oxides from the spin-glass behaviour of other double perovskites in which the only paramagnetic element is an S = ½ cation. In these cases, spin glass behaviour has been associated with the so called spin-liquid state that arises from quantum effects of frustrated ½ spin systems. The ordered double perovskites Sr2MgReO6, Sr2CaReO6 and Ba2CuWO6 are considered examples of this behaviour as the perovskite structure can be viewed as a frustrated fcc lattice. As mentioned previously, however, in the oxide “Sr2FeTeO6” the driving force that leads to spin-glass behaviour is the coexistence of disorder and vacancies in the B and B′ sites of the ordered double perovskite structure.
In summary, the double perovskite of nominal composition Sr2FeTeO6 has been prepared by the freeze-drying method. The structure has been refined from X-ray and high resolution D2B neutron diffraction data which indicate that this oxide can be described with the space group I12/m1. Refinement of Fe and Te fractions over B cation sites yielded the composition Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6. This, and the absence of any significant formation of oxygen vacancies, yields a nominal oxidation state for Fe close to +3, that has been confirmed by EPR and Mössbauer measurements. Magnetic DC and AC susceptibility measurements indicate that, although the most important interactions are of antiferromagnetic nature, the sample shows spin-glass-like behaviour below ∼40 K. The lack of a three-dimensional peak in the specific heat measurements and the absence of magnetic peaks in 4 K neutron diffraction data are consistent with the proposed spin-glass behaviour.
The presence of 15% B-cation mis-site disorder and tellurium vacancies in Sr2(Fe0.84□0.16)B(Te0.87Fe0.13)B′O6 results in a variety of exchange pathways between the paramagnetic species and is the origin of a spin-glass state in which no long range magnetic order is established.
Acknowledgements
This work has been funded by the Spanish ‘Ministerio de Ciencia y Tecnología’ (MCyT, MAT-2004-02425). L. Ortega-San Martin acknowledges MCyT for a Doctoral Fellowship. Dr J. P. Chapman thanks MCyT for funding (MAT 2001-0064). Dr M. Insausti is acknowledged for her help with the SEM microscope. The authors gratefully acknowledge Drs G. Cuello, Jon Gutiérrez, Javier Bermejo and Nestor Veglio for assistance with neutron data collection and the I.L.L., Grenoble, France for provision of beam-time on Instruments D2B and D16.References
- R. H. Mitchell, Perovskites modern and ancient, Almaz Press, Ontario, 2002 Search PubMed; Properties and applications of perovskite type oxides, ed. L. G. Tejuca and J. L. G. Fierro, Marcel Decker, New York, 1993 Search PubMed.
- P. D. Battle, T. C. Gibb, C. W. Jones and F. Studer, J. Solid State Chem., 1989, 78, 281 CrossRef CAS.
- K.-I. Kobayashi, T. Kimura, H. Sawada, K. Terakura and Y. Tokura, Nature, 1998, 395, 667 CrossRef.
- E. J. Cussen and P. D. Battle, J. Mater. Chem., 2003, 13, 1210 RSC.
- (a) R. Rodríguez, A. Fernández, A. Isalgué, J. Rodríguez, A. Labarta, J. Tejada and X. Obradors, J. Phys. C, 1985, 18, L401 CrossRef CAS; (b) T. C. Gibb, J. Mater. Chem., 1993, 3, 441 RSC; (c) K. Tezuka, K. Hemni, Y. Hinatsu and N. M. Masaki, J. Solid State Chem., 2000, 154, 591 CrossRef CAS.
- E. Cussen, J. F. Vente, P. D. Battle and T. C. Gibb, J. Mater. Chem., 1997, 7, 459 RSC.
- S.-O. Lee, T. Y. Cho and S. H. Byeon, Bull. Korean Chem. Soc., 1997, 18, 91 CAS.
- (a) A. K. Azad, S.-G. Eriksson, A. Mellergard, S. A. Ivanov, J. Eriksen and H. Rundlöf, Mater. Res. Bull., 2002, 37, 1797 CrossRef CAS; (b) K.-I. Kobayashi, T. Okuda, Y. Tomioka, T. Kimura and Y. Tokura, J. Magn. Magn. Mater., 2000, 218, 17 CrossRef CAS.
- (a) D. Sánchez, J. A. Alonso, M. García-Hernández, M. J. Martínez-Lope, J. L. Martínez and A. Mellegård, Phys. Rev. B: Condens. Matter, 2002, 65, 104426 CrossRef; (b) M. García-Hernández, J. L. Martínez, M. J. Martínez-Lope, M. T. Casais and J. A. Alonso, Phys. Rev. Lett., 2001, 86, 2443 CrossRef CAS.
- (a) H. Kato, T. Okuda, Y. Okimoto, T. Tomioka, K. Oikawa, T. Kamiyama and Y. Tokura, Phys. Rev. B: Condens. Matter, 2004, 69, 184412 CrossRef; (b) J. Gopalakrishnan, A. Chattopadhyay, S. B. Ogale, J. Venkatesan, R. L. Greene, A. J. Millis, K. Ramesha, B. Hannoyer and G. Marest, Phys. Rev. B: Condens. Matter, 2000, 62, 9538 CrossRef CAS.
- D. D. Sarma, Curr. Opin. Solid State Mater. Sci., 2001, 5, 261 CrossRef CAS.
- L. Ortega-San Martin, J. P. Chapman, L. Lezama, J. Sánchez Marcos, J. Rodríguez-Fernández, M. I. Arriortua and T. Rojo, J. Mater. Chem., 2005, 15, 183 RSC.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, GSAS: General Structure Analysis System, LAUR 86-748, 1994.
- M. A. Howe, R. L. McGreevy and P. Zetterström, CORRECT (computer code), NFL Studsvik, 1996.
- (a) P. Debye, Ann. Phys., 1912, 39, 789 Search PubMed; (b) B. Bazán, J. L. Mesa, J. L. Pizarro, J. Rodríguez-Fernández, J. Sánchez-Marcos, A. Roig, E. Molins, M. I. Arriortua and T. Rojo, Chem. Mater., 2004, 16, 5249 CrossRef CAS.
- (a) L. Ortega-San Martín, J. P. Chapman, E. Hernández-Bocanegra, M. Insausti, M. I. Arriortua and T. Rojo, J. Phys.: Condens. Matter, 2004, 16, 3879 CrossRef; (b) L. Ortega-San Martín, J. P. Chapman, G. Cuello, J. M. González-Calbet, M. I. Arriortua and T. Rojo, Z. Anorg. Allg. Chem., 2005, 631(11), 2127 CrossRef CAS.
- C. J. Howard, B. Kennedy and P. M. Woodward, Acta Crystallogr., Sect. B, 2003, 59, 463 CrossRef.
- V. F. Sears, Neutron News, 1992, 3, 29 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef.
- R. L. Carlin, Magnetochemistry, Springer, Berlin, 1986 Search PubMed.
- (a) F. E. Mabbs and D. Collison, Electron paramagnetic resonance of d transition metal compounds, Elsevier, Amsterdam, 1992 Search PubMed; (b) J. A. Weil, J. R Bolton and J. E. Wertz, Electron Paramagnetic Resonance – Elementary Theory and Practical Applications, Wiley & Sons, New York, 1994 Search PubMed.
- N. N. Greenwood and T. C. Gibb, Mössbauer spectroscopy, Chapman and Hall, London, 1992 Search PubMed.
- C. McCammon, Phase Transitions, 1996, 58, 1 CrossRef CAS.
- J. A. Mydosh, Spin glasses: An Experimental Introduction, Taylor & Francis, London, 1993 Search PubMed.
- J. L. Tholence, Solid State Commun., 1993, 88, 917 CrossRef.
- C. R. Wiebe, J. E. Greedan, P. P. Kyriakou, G. M. Luke, J. S. Gardner, A. Fukaya, I. M. Gat-Malureanu, P. L. Russo, A. T. Savici and Y. J. Uemura, Phys. Rev. B: Condens. Matter, 2003, 68, 134410 CrossRef.
- M. T. Anderson, K. B. Greenwood, G. A. Taylor and K. R. Poeppelmeier, Prog. Solid State Chem., 1993, 22, 197 CrossRef CAS.
- M. Gateshki, J. M. Igartua and E. Hernández-Bocanegra, J. Phys.: Condens. Matter, 2003, 15, 6199 CrossRef CAS.
- F. Galasso, Perovskites and High Tc superconductors, Gordon & Breach, New York, 1990 Search PubMed.
- M. S. Augsburger, M. C. Viola, J. C. Pedregosa, A. Muñoz, J. A. Alonso and R. E Carbonio, J. Mater. Chem., 2005, 15, 993 RSC.
- D. Iwanaga, Y. Inaguma and M. Itoh, J. Solid State Chem., 1999, 147, 291 CrossRef CAS.
- (a) J.-H. Park and P. M. Woodward, Int. J. Inorg. Mater., 2000, 2, 153–166 CrossRef CAS; (b) W. Wischert, D. Oelkrug, H. J. Schittenhelm and S. Kemmler-Sack, Z. Anorg. Allg. Chem., 1982, 495, 219 CrossRef CAS (this is the last paper of a long series devoted to the subject “ordered perovskites with cationic defects/vacancies”).
- J. H. Conway and R. K. Guy, The Book of Numbers, Springer-Verlag, New York, 1996 Search PubMed.
- (a) T. C. Gibb, J. Roger and J. Whitehead, J. Mater. Chem., 1993, 3, 591 RSC; (b) P. D. Battle, T. C. Gibb, A. J. Herod and J. P. Hodges, J. Mater. Chem., 1995, 5, 75 RSC.
- K. Binder and A. P. Young, Rev. Mod. Phys., 1986, 58, 801 CrossRef CAS.
- (a) J. B. Goodenough, Magnetism and the chemical bond, Wiley, New York, 1966 Search PubMed; (b) L. F. De Jongh and R. Block, Physica, B + C, 1975, 79, 568 CrossRef.
- P. D. Battle and C. W. Jones, J. Solid State Chem., 1989, 78, 108 CrossRef CAS.
- T. C. Gibb, J. Mater. Chem., 2001, 11, 456 RSC.
- J. Blasco, J. García, G. Subías and M. C. Sánchez, Phys. Rev. B: Condens. Matter, 2004, 70, 094426 CrossRef.
| This journal is © The Royal Society of Chemistry 2006 |