The use of enriched 111Cd as tracer to study de novo cadmium accumulation and quantitative speciation in Anguilla anguilla tissues
Andrés
Rodríguez-Cea
,
María
del Rosario Fernández de la Campa
,
José Ignacio
García Alonso
and
Alfredo
Sanz-Medel
*
Department of Physical and Analytical Chemistry, Faculty of Chemistry, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: asm@uniovi.es; Fax: 34 985103125; Tel: 34 985 103474
First published on 30th January 2006
Abstract
In order to determine de novo incorporation of cadmium into fish liver and kidney, the stable isotope 111Cd was used as tracer and the European eel (Anguilla anguilla) as model organism. The exposure of animals to 111Cd (100 ng L−1) gave rise to the in vivo dilution of the natural previously existing Cd in the selected tissues and therefore a change of Cd isotope ratios can be measured. The measurement of this new 114Cd/111Cd ratio by ICP-MS, and the subsequent application of mathematical calculations based on the isotope dilution methodology, allowed us to quantitatively discriminate between the previously accumulated natural Cd and the isotopically enriched Cd (de novo incorporated). This was observed for total cadmium in liver and kidney and also for speciated Cd (e.g., that bound to different cytosolic fractions, such as the Metallothionein fraction (MT fraction). In addition, the quantitative results observed pointed out that in liver natural Cd decreased, while kidney received an appreciable amount of endogenous cadmium, supporting the idea that a redistribution of Cd between liver and kidney in fish occurs, as has previously been described in mammals. Moreover, Cd speciation analysis provided the real cadmium balance (natural Cd versus enriched Cd) in the MT fraction, demonstrating quantitatively the Cd mobilization in hepatic and renal MT fractions. On the other hand, since MTs bind Cd, they were capable of in vivo incorporation of the enriched and stable isotope 111Cd during the period of exposure. The separation of the 111Cd labelled MTs was accomplished by anion-exchange (AE) chromatography, which was coupled on-line with ICP-MS detection to investigate the in vivo ability of individual liver MT isoforms for binding cadmium. The 114Cd/111Cd ratio, measured in three individual MT isoforms, was comparatively lower in the predominant MT-2a isoform, which seems to act in the preferential incorporation of the new incoming Cd. On the other hand, the determination of Cd in separated MT isoforms (by an on-line post-column isotope dilution technique) in conjunction with the calculated Cd isotope ratios has allowed for the first time the determination of both recently (exogenous) and previously (endogenous) Cd bound to individual MT isoforms. The results shown here could open the way to the study of other metalloproteins metals preferences, where radioactive techniques are inadequate for metal speciation analysis.
Introduction
In the last few years the use of stable isotopes and their isotope ratio measurements by mass spectrometry has gained usage for tracer experiments in biological and medical research.1,2 Also, the advantages of using stable isotopes in the field of quantitative proteomics has been demonstrated for unicellular organisms,3 nematodes,4 plants5 and even mammals6 by metabolic labelling of proteins with enriched isotopes.Inductively coupled plasma mass spectrometry (ICP-MS) is the technique of choice for the study of trace metals bound to proteins because of its extreme sensitivity and multielemental capabities.7 Nevertheless, ICP-MS potential for isotope ratio measurement and application in isotope enriched proteins is yet to be developed. Metalloproteins are ideal candidates for studying by ICP-MS detection, after in vivo labelling with enriched isotopes, because such proteins are or could be “tagged” with the metals (easily detectable by that technique even at extremely low concentration levels). However, only a few studies have used stable isotopes as tracers for in vivo labelling of metalloproteins to establish quantitatively the speciation of essential and toxic metals in biological material.8–10
On the other hand, metallothioneins (MTs) are a group of non-enzymatic metalloproteins, with a presence from bacteria to vertebrates, which exhibit high affinity for some metal ions such as Cd2+, Zn2+and Cu1+, forming characteristic metal–thiolate clusters.11 MTs involvement in transport and storage of heavy metals has been studied for decades, particularly to elucidate their exact role in the detoxification of heavy metals. In this context, metal speciation analysis of MTs from different organisms carried out by ICP-MS coupled to highly selective separation techniques has been successfully employed.12
It is known that different MT isoforms exist, consisting of minimal changes in the amino acid chain, but their biological roles in living organisms or their in vivo preferences for binding Cd (or other metals) are not fully understood yet.13,14 In any case, MTs naturally bind Cd, Cu and Zn via mercaptide bonds and therefore they offer the possibility of labelling with enriched stable isotopes for their eventual detection via ICP-MS.
One of the most harmful elements in the freshwater environment is cadmium, the long-term toxic effects of which in fish are often very similar to those described in humans and other mammals.15 It is known that after Cd absorption through the gills and the gastrointestinal tract in fish, Cd is transported primarily by the blood to liver and kidneys, forming Cd–MT complexes.15 After long-term exposure of aquatic animals to Cd several findings suggest that a redistribution of Cd occurs, in particular from liver to kidney.15–18 Indeed, in terrestrial mammals it is assumed that a proportion of liver Cd–MT complexes are redistributed by blood into different organs, particularly kidneys. Free Cd is released there, before its binding to renal MTs for final storage, and that seems to explain the chronic nephrotoxic effect of Cd.19 Classical toxicological studies are able to determine the total metal amount in the tissues at the end of the exposure, but they cannot differentiate the tissues’ existing cadmium from that incorporated de novo during the controlled experiment. The use of radioisotope tracers allows one to discriminate between both sources of Cd but their quantification requires the measurement of radioactivity in a scintillation counter and a second analytical technique (usually atomic absorption spectrometry) to determine total amounts.16 Moreover, experiments employing radioisotopes as tracers are limited when no radiotracer with a suitable half-life is available, the decrease of the tracer in the sample being not only a function of its metabolic turnover, but also of spontaneous decay.20 To avoid these limitations, the use of stable isotopes as tracers in metabolic studies has been proposed, sometimes as a realistic alternative, it being possible to determine the amounts of tracer (enriched isotope) and tracee (natural abundances) in a sample by means of mathematical calculations based on the isotope dilution approach and developed in each particular case.2,21–26 Most in vivo studies based on the tracer∶tracee ratio approach are focused on mineral metabolism, in particular study of the absorption, kinetics and excretion of total metals, but not to investigate the binding of metals to proteins, the metal speciation as, for instance, in metallothioneins.2,20 On the other hand, the limited existing studies of in vivo hetero-atom labelling of proteins (in particular selenoproteins), aiming at determining the speciation of tracer and tracee in the sample,8–10 are based on classical calibration strategies which, of course, do not offer the recognized advantages of the isotope dilution methodology.27–29 Moreover, such studies fail when assuming that the tracer (which is not 100% enriched in the isotope of interest) does not contribute to the mass measured for the tracee, leading to a possible overestimation of tracee concentration.
Here, in vivo exposure to 111Cd and measurement of Cd isotope ratios in tissues offer the possibility of discriminating between endogenous and exogenous cadmium sources by means of the application of mathematical calculations based on the isotope dilution methodology. At the same time it is possible to study the origin of Cd bound to MTs (even in individual MT isoforms) if the ICP-MS detection is carried out after on-line separation procedures. Additionally, the total amount of endogenous mobilized cadmium in those samples can be calculated by comparing the concentration of natural Cd at the end of the experiment in a given tissue (and also in a given proteins pool or individual MT isoform) with the initial values. In order to prove the validity of such in vivo isotope labelling applications, European eel was selected as a model fish because Anguilla sp. are widely distributed and long-lived fish, quite resistant to toxic substances at sublethal doses including heavy metals.30 Moreover, in a previous work we observed that eel MTs synthesis is clearly increased in response to Cd stress.31
Thus, the aim of this work is two-fold: (i) to determine de novo incorporation of Cd to eel liver and kidney and its inter-organ redistribution by means of the quantitative using of 111Cd as tracer after in vivo experiments (the needed mathematical calculations to relate the 111Cd/114Cd ratio in the exposed water with that measured experimentally in the animal are derived); (ii) to investigate the in vivo preferences of individual MT isoforms for Cd. As far as we know, stable isotope ratio measurement in metallothioneins after their in vivo isotope labelling in order to quantitatively establish the origin of metals bound to them has not been exploited so far.
Experimental
Instrumentation
A quadrupole ICP-MS Hewlett-Packard Model 4500 instrument (Yokogawa Analytical, Tokyo, Japan), equipped with a conventional Meinhard nebulizer and a double-pass spray-chamber, was utilised for ICP-MS measurements. Instrumental operating conditions for the ICP-MS detector coupled “on-line” with the exit of the HPLC column are summarised in Table 1. These conditions were tuned daily by using a multielemental aqueous standard solution containing Li, Y, Tl and Ce (at the 10 ng mL−1 level in each element).| Plasma conditions | |
| Instrument | HP4500 |
| Rf power | 1300 W |
| Plasma gas flow rate | 15.0 L min−1 |
| Carrier gas flow rate | 1.08 L min−1 |
| Nebulizer | Meinhard |
| Spray chamber | Double pass/ Peltier (4 °C) |
| Data acquisition parameters | |
| Spectrum mode: | |
| (ID analysis) | 110, 111, 112, 113, 114 |
| Monitored masses (isotopes ratios) | 106, 108, 110, 111, 112, 113, 114, 116 |
| (external calibration) | 63, 64, 65, 67, 71 |
| Points per peak | 3 |
| Integration time | 0.3 s per peak |
| Time resolved analysis mode: | |
| Monitored masses | 106, 108, 110, 111, 112, 113, 114, 116 |
| Points per peak | 1 |
| Integration time | 1 s per peak |
Liver cytosol was obtained by ultracentrifugation using an Optima L-90k Beckman ultracentrifuge (Beckman Coulter, Fullerton, CA, USA) and the fraction collection experiments were carried out with a Frac-100 Pharmacia LKB system (Bouwman, Sweden). A glass column (100 × 1 cm id) packed with Sephadex G75 (Pharmacia Fine Chemical, Uppsala, Sweden) was used to perform size-exclusion chromatography (SEC). Gradient anion-exchange chromatography was performed with two Shimadzu LC-10Avp HPLC pumps equipped with a Rheodyne Model 7125 sample injector valve (Berkeley, CA, USA) fitted with a 50 μL loop. The analytical anion-exchange column was a Mono Q HR 5/5 of 50 × 5 mm id (Pharmacia Biotech., Uppsala, Sweden). A metal scavenger column (25 × 0.5 cm id) was placed between the pumps and the injection valve in order to remove exogenous metal ions from the mobile phases used. This column was packed with the chelating material Kelex-100 impregnated on octadecylsilica, as described previously.32
Reagents and materials
Single-element standard stock solutions for ICP-MS analysis containing 1000 μg mL−1 were from Merck (Darmstadt, Germany) or J. T. Baker (Phillipsburg, NJ, USA). Stable 111Cd (96.43%) was obtained from Cambridge Isotope Laboratories (Woburn, MA, USA) as CdO and a stock solution of this metal was prepared as described in a previous work.33 The concentration of this solution was established by reverse isotope dilution analysis and it was diluted appropriately for the tracer and isotope dilution experiments.All mineral acids and metal salts used were of analytical reagent grade and Milli-Q water (Millipore) was used throughout.
Animals and experimental exposure to the stable isotope 111Cd
Fourteen specimens of Anguilla anguilla were collected by electric fishing from the Pigüeña river, which is located in a non-industrialised area in Asturias, Northern Spain. The animals were transported alive to the laboratory, divided into two groups of seven eels each and transferred to aquarium tanks (50 L dechlorinated tap water, filtered and well aerated). Before the experiments, they were acclimatised in the aquarium for a whole week, maintaining a 12 h light/12 h darkness daily cycle at a water temperature of 12 °C.Then, one group of seven eels (average weight ± SD, 52 g ± 9) was exposed to 100 μg L−1 of enriched 111Cd (96.43%) for 48 hours and then transferred to water without Cd for another 48 hours, repeating this cycle for a total period of time of three weeks. These exposure conditions were selected according to a previous experiment with natural Cd in our laboratory.31 The second group of seven eels (average weight ± SD, 50 g ± 8) was sacrificed before any exposure to 111Cd in order to determine the “background” cadmium concentration (accumulated Cd before the acute exposure experiment) in their tissues.
Eel liver and kidney cytosols preparation and MT isoforms separation
The cytosols preparation was carried out as described in a previous work.31 Livers or kidneys of the same group were excised, pooled and homogenized in four volumes (w/v) of buffer (10 mM Tris/HCl, pH 7.4; 1 mM dithiothreitol; 0.1 mM phenylmethylsulfonyl fluoride (PMSF)). The homogenates were centrifuged at 1000g for 10 min and then the decanted supernatant at 105![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000g for 60 min. All preparative steps were performed at 4 °C. The supernatant liquid was heated at 65 °C for 15 min and then re-centrifuged again for 30 min in the same way. The supernatant (cytosol) was collected and stored at −20 °C until analysis.
000g for 60 min. All preparative steps were performed at 4 °C. The supernatant liquid was heated at 65 °C for 15 min and then re-centrifuged again for 30 min in the same way. The supernatant (cytosol) was collected and stored at −20 °C until analysis.
Preparative scale size exclusion chromatography using Sephadex G75 was carried out with 10 mM Tris/HCl, pH 7.4, 5 mM 2-mercaptoethanol (2-MCE) and 25 mM NaCl (Suprapur Merck) at a flow rate of 0.2 mL min−1 with a pump Model P-500 from Pharmacia. The column was calibrated with standards of proteins (including rabbit liver MTs standards) as described in a previous paper.31 The temperature was kept constant at 4 °C. Metal impurities were removed from the mobile phase by passing it through a metal scavenger column packed with Kelex-100.32 A 1 mL aliquot of liver or kidney cytosol was applied directly to the SEC column and 30 consecutive 3 mL fractions were collected. Those fractions were measured for Cd isotope ratios and Zn and Cu concentrations by ICP-MS. The entire fraction with constant 114Cd/111Cd ratio (and high 111Cd intensity) and appreciable amounts of Zn and Cu (corresponding MT fraction) was pooled and pre-concentrated 25 times by ultrafiltration (Centricon YM-3 of 3000 MW cut-off, Millipore, Bedford, MA, USA). Aliquots of these concentrates (50 μL each) were then subjected to anion exchange separation (fast protein liquid chromatography, AE-FPLC). Mobile phases A and B and gradient elution conditions were the same as described in the previous work for metal speciation analysis in liver metallothioneins from eels exposed to natural cadmium.31 The eluate was monitored on-line for multielemental detection (Cd, Cu and Zn) by ICP-MS.
Cadmium isotope ratio measurements on cytosols, SEC fractions and separated MTs isoforms
Cadmium isotope ratios in the eel liver and kidney cytosols, in all the SEC separated fractions and in the MT fraction (pool of SEC fractions where MTs eluted) from both organs were measured by ICP-MS, using the instrumental conditions indicated in Table 1, by direct nebulisation of the samples. The measured isotope ratios were corrected for mass bias and for detector dead time as described previously.34 Additionally, the Cd isotope ratios in individual MT isoforms were measured after anion exchange separation of an eel liver MTs pool using the time resolved analysis mode. The isotope ratio on each MT peak was calculated as peak area ratio by means of the ICP-MS Chromatographic Software (Agilent Technologies). The usual corrections for mass bias and detector dead time were also applied here.Quantitative metal analysis
The total content of Cd in the liver and kidney cytosols and in their respective MT fractions isolated by SEC (from both 111Cd exposed and unexposed eels) was determined by isotope dilution analysis using a solution of enriched 111Cd as spike. Isotope ratio measurements were performed as described in the previous section after dilution with sub-boiling nitric acid and direct nebulization.On the other hand, the Cu and Zn (metals usually associated with MTs and other cytosolic proteins) content in each SEC fraction was also measured by external calibration after a 1 + 1.5 (w/w) dilution with sub-boiled 0.5% nitric acid using 71Ga as internal standard.
Mass balance for anion exchange chromatography
In order to check that all cadmium present in the chromatographed MT fractions eluted from the anion exchange column, a new experiment was performed in which the eluent from the analytical column was mixed with a solution of enriched 111Cd to perform post-column isotope dilution analysis, as explained previously.31,34,35 In brief, the mass flow of cadmium eluting from the column (ng min−1) can be calculated by measuring the 111Cd/114Cd ratio on-line in the ICP-MS. The integration of the mass flow chromatogram provides the mass of cadmium in each separated peak (each MT isoform). Moreover, the integration of the whole chromatogram indicates the total amount of cadmium eluting from the column which can be used for final mass balance purposes. These experiments (carried out in triplicate) were performed for the control eels and for the exposed to 111Cd eels.Results and discussion
Cadmium isotope ratios in cytosols and MT fractions
The Cd isotope abundances measured in the pooled liver and kidney cytosols of 111Cd exposed animals are given in Table 2. It is clear that natural cadmium isotope abundances, observed in the control animals, changed dramatically in exposed animals, reflecting the level of 111Cd incorporation (“de novo” Cd) in the tissues. As an example, data shown in Table 2 for cytosols show that liver incorporated a larger relative fraction of 111Cd than kidney, where the observed isotopic composition was closer to the natural Cd abundances.| Cytosol | MT fraction | |||||
|---|---|---|---|---|---|---|
| Isotope | Cd (natural)a | 111Cd (spike)a | Liver | Kidney | Liver | Kidney |
| a Natural Cd from ref. 46; 111Cd from ref. 33. | ||||||
| 106 | 1.25 | 0.01 | 0.69 | 0.85 | 0.72 | 1.01 |
| 108 | 0.89 | 0.01 | 0.49 | 0.63 | 0.79 | 1.30 |
| 110 | 12.49 | 0.60 | 7.06 | 8.97 | 6.94 | 9.21 |
| 111 | 12.80 | 96.43 | 51.28 | 36.52 | 49.91 | 35.53 |
| 112 | 24.13 | 1.78 | 13.90 | 17.79 | 13.82 | 17.74 |
| 113 | 12.22 | 0.42 | 6.72 | 8.84 | 6.94 | 8.91 |
| 114 | 28.73 | 0.60 | 15.80 | 21.00 | 16.16 | 20.59 |
| 116 | 7.49 | 0.14 | 4.07 | 5.40 | 4.73 | 5.70 |
| 114Cd/111Cd | 2.25 | 0.01 | 0.31 | 0.58 | 0.32 | 0.58 |
In order to evaluate the isotope ratios of cadmium present in proteins of different molecular size, liver and kidney cytosols were fractionated using a size exclusion column. The concentrations of Cu and Zn were also monitored by ICP-MS, in addition to Cd, to identify MTs elution fraction in the SEC column. The Cu and Zn concentrations found in the different fractions collected, under optimum conditions, have been plotted in Fig. 1 for both liver and kidney cytosols. As can be observed, Cu and Zn eluted in two distinct fractions, one corresponding to high-medium molecular weight proteins (fractions 10–16) and the other to low molecular weight proteins (fractions 17–24).
 | ||
| Fig. 1 Quantitative analysis of Cu and Zn in each gel-permeation fraction (n = 3) from one ml of liver or kidney cytosol from animals exposed to 111Cd. Metal detection was carried out by ICP-(Q)MS. | ||
The parallel results obtained for cadmium are illustrated in Fig. 2, showing the intensities at mass 111 for cadmium (logarithmic scale left axis) and the 114Cd/111Cd isotope ratios (linear scale right axis) in each collected fraction. As can be observed, Cd in liver and kidney cytosol eluted almost exclusively in fractions 17–24, which should correspond to the MT fraction (of course, binding of Cd, Cu and Zn by a cytosolic heat-stable protein, of about 6–7 kDa, indicates the presence of MT).36 In the kidney cytosol sample, a small fraction of cadmium eluted also in the high-medium molecular weight fractions.
 | ||
| Fig. 2 114Cd/111Cd ratios and 111Cd intensities (logarithmic scale) measured with ICP-(Q)MS detection in each gel-permeation fraction (n = 3) from one ml of liver or kidney cytosol from animals exposed to 111Cd. Fraction numbers 17–24 were pooled in a single MT fraction for further determination of total Cd and the average 114Cd/111Cd ratio. | ||
The measured 114Cd/111Cd isotope ratios in all fractions (and in both cytosol types) showed a distinct pattern: where almost no cadmium eluted from the column (fractions 1–10 and 26–30) the isotope ratio was very noisy, oscillating between the natural ratio (2.245) and that in the tracer (0.006). The liver cytosol fractions 10–15 provided an isotope ratio slightly higher than that obtained in fractions 17–24 while for kidney all fractions (10–24) provided constant isotope ratios values. Thus, it seems that in kidney the degree of incorporation of 111Cd to proteins is relatively the same in high-medium and low molecular weight proteins. Finally, the isotope ratios obtained for the liver MT fractions were closer to those of the 111Cd tracer than those found for the kidney cytosol in the same fractions, indicating different 111Cd enrichment in both organs. Fractions 17–24 (MTs containing fractions) were pooled in a single fraction (MT fraction) and the isotopic composition of cadmium (for both organs), measured by ICP-MS, is also given in Table 2. As can be observed, the isotopic Cd composition in the MT fraction is very close to that observed for the whole cytosol (i.e., most of the cadmium present eluted in this MT fraction for both organs).
Total cadmium concentrations in cytosol and MT fraction
The total concentrations of Cd in the samples under study were determined by isotope dilution analysis, both in the 111Cd exposed eels group and in the non-exposed group. For this purpose, the samples were spiked with a standard solution of 111Cd and the new isotope ratios 114/111 measured again by ICP-MS. Please note that, for the 111Cd exposed animals, the isotope composition given in Table 2 was used for the calculations, while for the non-exposed animals the natural ratio could be used directly. The results obtained, referred to the original wet tissues, are given in Table 3. As can be observed, the concentration of Cd in the non-exposed group is quite high but within the expected level for eels living in non-contaminated rivers.30,37,38 In all samples analyzed the levels of cadmium increased quite substantially after 111Cd exposure in agreement with our previous experiments using natural abundance cadmium.31| Cytosola | MT fractiona | |||
|---|---|---|---|---|
| Liver | Kidney | Liver | Kidney | |
| a Mean ± SD (n = 3). b Corresponds to the Cd concentration in the animals without exposure to 111Cd (also named Cd background in the text). | ||||
| Exposed to 111Cd group | 1219 ± 2 | 4651 ± 13 | 1173 ± 4 | 3634 ± 35 |
| Non-exposed groupb | 804 ± 5 | 2536 ± 13 | 774 ± 8 | 1852 ± 9 |
If we compare the results obtained for the cytosol and in the MT fraction we can observe that a high proportion of cadmium in the liver cytosol is present in MTs both for the exposed (96.2%) and non-exposed (96.3%) groups, respectively. However, a larger amount of cadmium seems to be bound to high-medium molecular weight proteins in kidney (as only 78.2% for the exposed and 73.0% for the non-exposed group was found in kidney MTs). In any case, it is remarkable how similar those relative figures are for a given organ in both eel groups under study.
Accumulation and redistribution of cadmium in the exposed animals
Of course, total quantitative information given in Table 3 does not, by itself, allow one to discriminate between previously accumulated cadmium and the newly incorporated 111Cd in eel tissues during the experiment, since Cd redistribution between organs occurs. However, isotope abundances in the 111Cd tracer and those measured in the fish tissues, given in Table 2, can be used to ascertain how much Cd found in the tissues actually corresponds to de novo incorporation. Mathematical reasoning is as follows.If NS represents the number of atoms of natural abundance cadmium in the tissue and NSp those of the 111Cd tracer incorporated, it is clear that the number of atoms found in the tissue, Nm, can be expressed by:
| Nm = NS + NSp | (1) |
| N114m = N114S + N114Sp | (2) |
| N111m = N111S + N111Sp | (3) |
 | (4) |
Rearranging eqn. (4) for NS/NSp we obtain:
 | (5) |
If we define as the isotope ratio (111/114) in the natural abundance cadmium and

the isotope ratio (114/111) in the tracer, eqn. (5) can be expressed as follows:
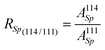
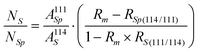 | (6) |
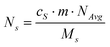 and
and  , where cS and cSp are the concentrations of natural cadmium and tracer in the tissue, m the mass of the tissue, MS and MSp the elemental atomic weights of natural cadmium and tracer, respectively, and NAvg the Avogadro Number.
, where cS and cSp are the concentrations of natural cadmium and tracer in the tissue, m the mass of the tissue, MS and MSp the elemental atomic weights of natural cadmium and tracer, respectively, and NAvg the Avogadro Number.
By the corresponding substitutions, the final equation for the 111Cd tracer experiment is:
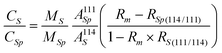 | (7) |
Thus, eqn. (7) was applied to the animals exposed to 111Cd and the data given in Table 4 were obtained. As can be observed large differences exist between liver and kidney as Cd accumulating organs. The percentage of newly incorporated cadmium in the liver reached 45–46% while only 28% of new cadmium was found in the kidney. Today it is well established that renal concentrations of Cd far exceed hepatic concentrations (which can also be appreciated in Table 3), confirming that the kidney is the most involved organ in Cd accumulation.19,39,40 On the other hand, the absolute concentrations of exogenous cadmium found in the kidney are about twice those found in the liver, both for the cytosols and the MT fraction, in agreement with the accumulating role of the kidney. However, the endogenous/exogenous Cd ratio in A. anguilla liver is lower than that observed in the kidney (Table 4), which suggests that liver is relatively more active than kidney in the sequestration of the “new” incoming Cd, at least for not so prolonged times of exposure, as tested here.
| Cytosola | MT fractiona | |||
|---|---|---|---|---|
| Liver | Kidney | Liver | Kidney | |
| a Mean ± SD (n = 3). b Concentration of natural cadmium/concentration of tracer (value of Cs/Csp in eqn. (7)). c Natural Cd concentration at the end of exposure to 111Cd minus Cd concentration before exposure (non-exposed group of Table 3). | ||||
| Relative Cd incorporationb | 1.19 ± 0.01 | 2.60 ± 0.01 | 1.26 ± 0.01 | 2.63 ± 0.01 |
| Endogenous Cd | 662 ± 1 | 3359 ± 9 | 654 ± 2 | 2632 ± 24 |
| Exogenous Cd | 557 ± 1 | 1292 ± 3 | 519 ± 2 | 1002 ± 9 |
| Mobilized Cdc | −142 | +823 | −120 | +780 |
If we assume that Cd concentrations in the non-exposed group provide the “background” Cd concentration for the exposed group before the experiment of 111Cd water exposure, then the differences between the endogenous Cd concentration found in the exposed group (Table 4) and the Cd background concentration (Table 3) will provide the amount of endogenous Cd gained or lost by the tissue during the time of exposition. Thus, this gives us the Cd “redistribution” and in our experiments it indicated that while liver cytosol accumulated the newly incoming enriched cadmium (557 ng g−1), an amount of −142 ng g−1 of endogenous cadmium was lost. On the other hand, the kidney cytosol incorporated Cd de novo (1292 ng g−1) but also received endogenous cadmium (+823 ng g−1) which, comparatively, represents a considerable proportion of the total cadmium concentration. In summary, the liver showed a negative overall balance for endogenous cadmium (it exports Cd) while the kidney showed a positive balance (it imports Cd). The same conclusion can be drawn from the MT fraction results. Most of the Cd found in liver and kidney is present in the isolated MT fraction (see also Table 3 and Fig. 2). Thus, Cd mobilization occurs basically in and out of this cytosolic fraction containing the MTs. In other words, quantitative results obtained here support the idea of Cd redistribution, between fish liver and kidney, in a similar way to that demonstrated in mammals.19
Finally, it should be pointed out that the strong binding of Cd to liver and kidney MTs explains the long half-life of Cd in fish and their extremely slow elimination.40 For this reason, Cd excretion can be considered negligible during the time of exposure in our experiment (this was demonstrated by others in several investigations16) and so its effects on our results here can be considered also negligible.
MT isoforms and their Cd associations
As pointed out before, not all functions of MTs are fully understood but they play important roles in the detoxification of heavy metals such as cadmium. For this reason, fish MTs have been investigated also as biomarkers of Cd exposure in the aquatic environment (see, for instance, ref. 41). Fig. 3a and 3b show 111Cd and 114Cd qualitative profiles obtained after further separation of eel liver and kidney MT fraction, respectively, by (AE)-FPLC-ICP-MS in the 111Cd exposed animals. As can be observed, both liver and kidney profiles are quite similar, with a predominant MT isoform eluting at a retention time of 13.1 ± 0.2 min in the liver sample and 12.8 ± 0.1 min for kidney. As can be seen, all MT isoforms turned out to be enriched in 111Cd, this being the isotope most abundant in all monitored chromatographic peaks.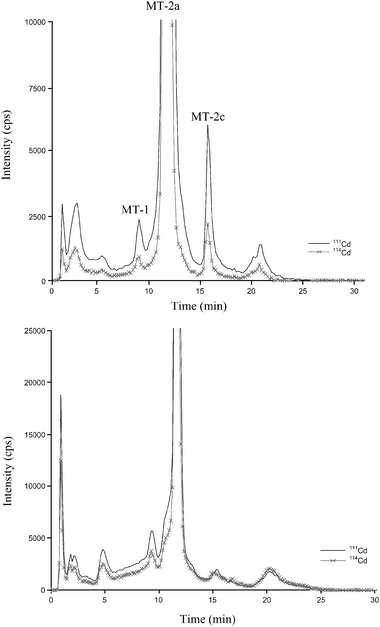 | ||
| Fig. 3 Chromatographic metal profiles of eel liver (a) and kidney (b) MTs by anion-exchange fast protein liquid chromatography coupled with on line ICP-(Q)MS detection. Hepatic MTs are numbered in line with standard rabbit liver metallothioneins.31 Total content of Cd injected in the chromatographic system was: (a) 47.7 ng of Cd; (b) 44.9 ng of Cd. | ||
In spite of years of investigations, controversial opinions still exist about the multifunctionality of MTs, the existence of several MT isoforms and sub-isoforms and their metal-specific preferences. Although it is assumed that the binding of Cd and Zn to the thioneins is kinetically labile, which negates the possibility of the co-existence of Zn and Cd specific forms of the protein,42 some studies showed (at least in invertebrates) the existence of distinct MT isoforms dedicated to Cd detoxification or homeostasis regulation.43–45 The use of stable isotopes of Cd and the measurement of adequate isotope ratios (i.e., 114Cd/111Cd ratio) in individual isoforms could help to elucidate their in vivo individual preferences for the newly absorbed Cd in the organism. In this vein, Cd isotopes ratios in the individual chromatographic peaks under study (named in Fig. 3a as MT-1, MT-2a and MT-2c for comparison with standard MT31) were calculated here for liver sample. The results are illustrated in Table 5. As can be observed, the 114Cd/111Cd ratios measured in the three peaks under study are quite similar and differ substantially from the natural ratio. It is worth noting that the isotope ratio was comparatively lower in the predominant peak (MT-2a). It could be thought that the differences between the three peaks under study are not enough to demonstrate clearly the presence of a MT isoform more involved than others in de novo Cd accumulation. However, the lowest 114Cd/111Cd ratio of the MT-2a isoform is in agreement with the idea of a preferential induction of this eel liver MT isoform by Cd exposure, as demonstrated by sulfur analysis in a previous work when animals were exposed to natural Cd.31 Both results tend to indicate that the new biosynthesis of MT-2a isoform, promoted by Cd in the eel liver, is followed by preferential incorporation in that isoform of the new incoming Cd.
| MT isoform | Retention time/min | 114Cd/111Cd ratio |
|---|---|---|
| MT-1 | 9.4 ± 0.0 | 0.413 ± 0.007 |
| MT-2a | 13.1 ± 0.2 | 0.318 ± 0.001 |
| MT-2c | 17.3 ± 0.3 | 0.359 ± 0.008 |
| Natural Cd | 2.24 |
Mass balances for the anion exchange separation were checked for the control group and for the exposed to 111Cd animals, using post-column isotope dilution analysis, with 111Cd as spike. It is necessary to clarify that this spike (96.43%) could be used for the exposed to 111Cd group because the isotope abundances of Cd in the liver MT fraction were substantially different from those observed in this 111Cd spike (see Table 2). The detailed experimental conditions for such post-column isotope dilution analysis have been described elsewere.31 Basically, the eluent from the anion exchange column is mixed with a known flow of enriched 111Cd and the mass flow of cadmium present in the sample can be calculated from the measured isotope ratios.34,35Fig. 4 shows the mass flow chromatogram obtained for the liver MT fraction of the control animals. The mass flow chromatogram corresponding to the exposed to 111Cd animals is not showed here since it is identical to the chromatographic profile showed in Fig. 3a except for the units (ng min−1 of Cd instead of intensity in cps). As can be observed in Fig. 4, the profile shown here is very similar to the qualitative profile shown in Fig. 3a for the 111Cd exposed animals. The integration of the different MT peaks indicated quantitative recoveries (98 ± 2% and 97 ± 1% for the control and for the exposed to 111Cd group samples, respectively) of cadmium with reference to the values given in Table 3. Therefore, no losses of cadmium during anion exchange separation were apparent. In other words, all cadmium species present (injected) eluted from the chromatographic column.
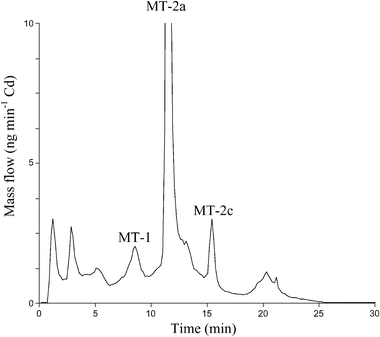 | ||
| Fig. 4 Mass flow chromatogram obtained for the separation of eel liver MTs (control group) by AE-FPLC-ICP-MS. Total content of Cd injected in the chromatographic system was 16 ng. | ||
The integration of the MT-2a peak in the mass flow chromatograms provided the amount of Cd bound to this MT isoform in the control group (599 ± 23 ng g−1) and in the exposed to 111Cd group (883 ± 32 ng g−1). This predominant peak represents 77% and 75% of the total Cd found in the MT fraction (see Table 3) in the control and in the exposed eels, respectively. Once the cadmium concentration in the MT-2a isoform is known (obtained here by post-column isotope dilution analysis), the 114Cd/111Cd ratio measured in that peak (see Table 5) can be used (as described before for the cytosols and the MT fraction samples) to ascertain the exact amounts of exogenous and endogenous cadmium bound to this MT isoform if we apply here eqn. (7). Thus, total Cd bound to MT-2a in 111Cd exposed animals could be “dissected” to 489 ng g−1 and 394 ng g−1 of endogenous and exogenous Cd, respectively. If we compare this endogenous Cd concentration with that found in the animals before the exposure to 111Cd (concentration of Cd bound to MT-2a in the control group) it indicates that this MT-2a isoform has lost 110 ng g−1 of Cd, which represents the 91% of the endogenous Cd mobilized from the whole MT fraction (see Table 4). These quantitative results unequivocally demonstrate that liver MT-2a isoform is the more implicated protein in the detoxification, accumulation and mobilization of Cd in A. anguilla.
Conclusions
Many previous papers have studied toxic metals accumulation in tissues of animals to which radioisotopes of this element had been administered. However, apart from safety considerations, the half-life of the radioisotope can affect the length of study and the quantitative determination is influenced by spontaneous decay. Here, the use of a stable isotope of Cd (111Cd) administered chronically, followed by ICP-MS determinations of isotope ratios in liver and kidney, has allowed us to distinguish between the naturally occurring Cd (previously accumulated in the tissues) and the isotopically enriched one (cadmium incorporated by the animal from the spiked water). The isotope dilution analysis methodology extended to in vivo conditions allowed us to ascertain the origin of Cd (endogenous or exogenous) in the whole cytosol and in metallothioneins. Likewise, we have been able to determine the amount of natural cadmium mobilized in the organs under study. In this way, Cd redistribution in liver and kidney has been proved for fish for the first time.Kidney accumulated more Cd than liver. However, the latter organ seems to receive more new incoming Cd and mobilise more efficiently endogenous cadmium. As a result, the final balance for endogenous cadmium was negative in liver and positive in kidney (this finding could be explained in a similar way to that demonstrated in mammals, where a proportion of liver Cd–MT complexes are transported to the kidneys for final Cd storage and future excretion19).
Finally, the use of stable isotopes in combination with hyphenated techniques (e.g., AE-FPLC-ICP-MS) seems adequate for studying the binding patterns of metals to metallothionein isoforms. In particular, the coupling of HPLC-ICP-MS to measure on-line isotope ratios in metallothioneins (in conjunction with the post-column isotope dilution technique) has proved most successful to determine both recently (exogenous) and previously (endogenous) Cd bound to individual MT isoforms. These results could open the way to using other enriched isotopes of essential elements (e.g., Fe, Cu, Zn, Se, etc.) in a similar way. In vivo enriched-isotope experiments to investigate the origin, mobilization and quantitative redistribution of such essential elements in animal tissues are now straightforward and could be extended to many metalloproteins in human beings.
Acknowledgements
The authors are grateful to Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología (FICYT) and Ministerio de Ciencia y Tecnología de España for the financial support through projects FC-02-PC-REC01-12 and BQU-2003-04671, respectively. The staff of the Consejería de Medio Rural y Pesca (Asturian Regional Government, Spain) is also acknowledged for allowing the necessary fishing licenses and collaborating in field tasks.References
- J. S. Becker, J. Anal. At. Spectrom., 2002, 17, 1172–1185 RSC.
- I. J. Griffin, J. Anal. At. Spectrom., 2002, 17, 1186–1193 RSC.
- Y. Oda, K. Huang, F. R. Cross, D. Cowburn and B. T. Chait, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6591–6596 CrossRef CAS.
- J. Krijgsveld, R. F. Ketting, T. Mahmoudi, J. Johansen, M. Artal-Sanz, C. P. Verrijzer, R. H. Plasterk and A. J. Heck, Nat. Biotechnol., 2003, 21, 927–931 CrossRef CAS.
- J. H. Ippel, L. Pouvreau, T. Kroef, H. Gruppen, G. Versteeg, P. van den Putten, P. C. Struik and C. P. M. van Mierlo, Proteomics, 2004, 4, 226–234 CrossRef CAS.
- C. C. Wu, M. J. MacLoss, K. E. Howell, D. E. Matthews and J. R. Yates, Anal. Chem., 2004, 76, 4951–4959 CrossRef CAS.
- A. Sanz-Medel, M. Montes-Bayón and M. L. Fernández Sánchez, Anal. Bioanal. Chem., 2003, 377, 236–247 CrossRef CAS.
- K. T. Suzuki and M. Itoh, J. Chromatogr. B, 1997, 692, 15–22 CrossRef CAS.
- Y. Kobayashi, Y. Ogra and K. T. Suzuki, J. Chromatogr. B, 2001, 760, 73–81 CrossRef CAS.
- K. T. Suzuki and Y. Ogra, Food Addit. Contam., 2002, 19, 974–983 CrossRef CAS.
- J. H. R. Kägi, in Metallobiochemistry. Part B Metallothionein and Related Molecules, Methods in Enzymology 205, eds. J. F. Riordan and B. L. Vallee, Academic Press, San Diego, California, 1991, pp. 613–626 Search PubMed.
- A. Prange and D. Schaumlöffel, Anal. Bioanal. Chem., 2002, 373, 441–453 CrossRef CAS.
- G. Roesijadi, Cell Mol. Biol., 2000, 46, 393–405 Search PubMed.
- P. Hunziker and J. H. R. Kägi, Nature, 1997, 388, 237–238 CrossRef.
- P. E. Olsson, P. Kling and C. Hogstrand, in Metal Metabolism in Aquatic Environments, eds. W. J. Langston and M. J. Bebianno, Chapman & Hall, London, 1998, pp. 321–350 Search PubMed.
- M. J. Chowdhury, M. Grosell, D. G. McDonald and C. M. Wood, Aquat. Toxicol., 2003, 64, 259–275 CrossRef CAS.
- C. De Conto Cinier, M. Petit-Ramel, R. Faure and D. Garin, Ecotox. Environ. Saf., 1997, 38, 137–143 Search PubMed.
- M. T. Rie, K. A. Lendas and I. P. Callard, Comp. Biochem. Physiol., 2001, 130C, 41–51 Search PubMed.
- G. Nordberg, T. Jin, P. Leffler, M. Svensson, T. Zhou and M. Nordberg, Analusis, 2000, 28, 396–400 CrossRef CAS.
- K. Y. Patterson and C. Veillon, Exp. Biol. Med., 2001, 226, 271–282 Search PubMed.
- J. R. Turnlund, W. R. Keyes and G. L. Peiffer, Anal. Chem., 1993, 65, 1717–1722 CrossRef CAS.
- M. Janghorbani, R. F. Martin, L. J. Kasper, X. F. Sun and V. R. Young, Am. J. Clin. Nutr., 1990, 51, 670–677 CAS.
- M. Janghorbani, Y. Xia, P. Ha, P. D. Whanger, J. A. Butler, J. W. Olesik and L. Daniels, Br. J. Nutr., 1999, 82, 291–297 CAS.
- C. Cobelli, G. Toffolo and D. M. Foster, Am. J. Physiol. Endocrinol. Metab., 1992, 262, E968–E975 Search PubMed.
- N. M. Lowe, D. M. Shames, L. R. Woodhouse, J. S. Matel, R. Roehl, M. P. Saccomani, G. Toffolo, C. Cobelli and J. C. King, Am. J. Clin. Nutr., 1997, 65, 1810–1819 CAS.
- S. Sturup, Anal. Bioanal. Chem., 2004, 378, 273–282 CrossRef CAS.
- P. Rodríguez-González, J. M. Marchante-Gayón, J. I. García Alonso and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef.
- L. Rottman and K. G. Heumann, Fresenius’ J. Anal. Chem., 1994, 350, 221–227 CrossRef CAS.
- K. G. Heumann, L. Rottman and J. Vogl, J. Anal. At. Spectrom., 1994, 9, 1351–1355 RSC.
- J. Bruslé, Aquat. Living Resour., 1990, 3, 131–141 Search PubMed.
- A. Rodríguez-Cea, M. R. Fernández de la Campa, E. Blanco González, B. Andón Fernández and A. Sanz-Medel, J. Anal. At. Spectrom., 2003, 18, 1357–1364 RSC.
- A. Lopez García, E. Blanco González and A. Sanz-Medel, Mikrochim. Acta, 1993, 1–2, 19–29.
- J. P. Valles Mota, J. Ruiz Encinar, M. R. Fernández de la Campa, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 1999, 14, 1467–1473 RSC.
- C. Sariego Muñiz, J. M. Marchante Gayón, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2001, 16, 587–592 RSC.
- L. Hinojosa Reyes, J. M. Marchante Gayón, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2003, 18, 1210–1216 RSC.
- K. T. Suzuki, in Metallobiochemistry. Part B Metallothionein and Related Molecules, Methods in Enzymology, eds. J. F. Riordan and B. L. Vallee, Academic Press, San Diego, California, 1991, pp. 252–263 Search PubMed.
- A. R. Linde, P. Arribas, S. Sánchez-Galán and E. García-Vázquez, Arch. Environ. Contam. Toxicol., 1996, 31, 297–302 CrossRef CAS.
- W. J. Langston, B. S. Chesman, G. R. Burt, N. D. Pope and J. McEvoy, Mar. Environ. Res., 2002, 53, 263–293 CrossRef CAS.
- H. De Smet, B. De Wachter, R. Lobinski and R. Blust, Aquat. Toxicol., 2001, 52, 269–281 CrossRef CAS.
- C. Hogstrand and C. Haux, Comp. Biochem. Physiol., 1991, 100C, 137–141 Search PubMed.
- D. R. Livingstone, J. Chem. Technol. Biotechnol., 1993, 57, 195–211 CAS.
- J. D. Otvos, X. Lui, H. Li, G. Shen and M. Basti, in Metallothionein III, eds. K. T. Suzuki, N. Imura and M. Kimura, Birkhäuser, Basel, 1993, pp. 57–74 Search PubMed.
- P. Hunziker and J. H. R. Kägi, Nature, 1997, 388, 237–238 CrossRef.
- F. Geret and R. P. Cosson, Arch. Environ. Contam. Toxicol., 2002, 42, 36–42 CrossRef CAS.
- S. R. Stürzenbaum, C. Winters, M. Galay, A. J. Morgan and P. Kille, J. Biol. Chem., 2001, 276, 34013–34018 CrossRef CAS.
- K. J. R. Rosman and P. D. P. Taylor, Pure Appl. Chem., 1998, 70, 217–235 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |