Airborne exposure and biological monitoring of bar and restaurant workers before and after the introduction of a smoking ban†
Dag G.
Ellingsen
*,
Geir
Fladseth
,
Hanne L.
Daae
,
Merete
Gjølstad
,
Kristina
Kjærheim‡
,
Marit
Skogstad
,
Raymond
Olsen
,
Syvert
Thorud
and
Paal
Molander
National Institute of Occupational Health, P.O. Box 8149 Dep, N-0033, Oslo, Norway. E-mail: dag.ellingsen@stami.no; Fax: +47/23195205; Tel: +47/23195205
First published on 20th February 2006
Abstract
The aims were to assess the impact of a total smoking ban on the level of airborne contaminants and the urinary cotinine levels in the employees in bars and restaurants. In a follow up design, 13 bars and restaurants were visited before and after the implementation of a smoking ban. Ninety-three employees in the establishments were initially included into the study. The arithmetic mean concentration of nicotine and total dust declined from 28.3 μg m−3 (range, 0.4–88.0) and 262 μg m−3 (range, 52–662), respectively, to 0.6 μg m−3 (range, not detected–3.7) and 77 μg m−3 (range, not detected–261) after the smoking ban. The Pearson correlation coefficient between airborne nicotine and total dust was 0.86 (p < 0.001; n = 48). The post-shift geometric mean urinary cotinine concentration declined from 9.5 μg g−1 creatinine (cr) (95% CI 6.5–13.7) to 1.4 μg g−1 cr (95% CI 0.8–2.5) after the ban (p < 0.001) in 25 non-snuffing non-smokers. A reduction from 1444 μg g−1 cr (95% CI 957–2180) to 688 μg g−1 cr (95% CI 324–1458) was found (p < 0.05) in 29 non-snuffing smokers. The urinary cotinine levels increased from 11.7 μg g−1 cr (95% CI 7.0–19.6) post-shift to 21.9 μg g−1 cr (95% CI 13.3–36.3) (p < 0.01) in the next morning in 24 non-snuffing non-smokers before the smoking ban. A substantial reduction of airborne nicotine and total dust was observed after the introduction of a smoking ban in bars and restaurants. The urinary cotinine levels were reduced in non-smokers. The decline found in smokers may suggest a reduction in the amount of smoking after intervention. In non-smokers cotinine concentrations were higher based on urine sampled the morning after a shift than based on urine sampled immediately post-shift.
Introduction
Exposure to environmental tobacco smoke (ETS) is associated with a wide range of health effects. The increased risk of acquiring severe diseases such as lung cancer and cardiovascular diseases has been associated with such exposure.1–4 An increased prevalence of upper airway or respiratory symptoms, or impaired lung function parameters, are some other health effects.5In many countries smoking has been banned from most workplaces, but employees in the hospitality industry such as nightclubs, bars and restaurants are still occupationally exposed to ETS. Airborne gaseous nicotine is a frequently used marker of exposure because of its specificity as one of the components in ETS. However, other less used markers such as solanesol in the particulate matter or gaseous 3-ethenylpyridine, may be used as well.6 The reported exposure levels of nicotine differ substantially between restaurants, bars, nightclubs and other workplaces, ranging up to nearly 100 μg m−3 in the most contaminated bars/taverns.7
Smoking tobacco is basically a combustion process of tobacco leaves, generating particles in addition to the more specific markers of ETS. However, airborne particulate matter has less often been measured in bars and restaurants, perhaps because these particles are not specific to ETS. On the other hand, such particles may contribute to upper airways discomfort. Weighted mean averages of 348 μg m−3 total dust, based on measurements from 10 bars and 117 μg m−3 in 12 restaurants, have been reported.8 Experimental studies have shown that particles from freshly smoked cigarettes may be in the respirable range, and even ultrafine particles have been demonstrated.9–11
Cigarette smoking is the most commonly used delivery system of nicotine to the body, but the habit of snuffing tobacco is also prevalent in the Scandinavian countries. The blood nicotine concentration peaks less than ½ an hour after administration. It is somewhat faster after cigarette smoking than after snuffing.12 Nicotine is distributed to most tissues after absorption, mainly to the skeletal muscles but also to the brain.12 Nicotine is mainly metabolised in the liver to cotinine and further to trans-3′-hydroxycotinine.13 Although the latter compound is the main urinary metabolite, cotinine is regarded as the better predictor for the total nicotine intake.13 The elimination half-life of cotinine in urine (U-cotinine) is about 18 hours.14 The concentration of U-cotinine is regarded as the better predictor of nicotine intake when compared to cotinine in saliva, both in smokers and snuffers.13
Norway enacted comprehensive legislation on smoking in public places in 1988 as a reaction to the increased awareness of the dangers of passive smoking. Restaurants and bars were, however, exempted from these regulations. Due to the increasing evidence linking passive smoking to a range of diseases, the Norwegian government proposed a ban on smoking in restaurants, pubs and bars. This total smoking ban in bars, nightclubs and restaurants was introduced on 1st June 2004.
This study is part of an investigation to assess some of the effects of this intervention. We describe the airborne exposure to nicotine and dust before the new regulation banning the smoking of tobacco in bars and restaurants was implemented. The measurements were repeated after the smoking ban was introduced. Further, U-cotinine levels were determined in identical subjects before and after the new regulations were enforced. Thus the study is one of very few assessing the impact of a smoking ban on exposure in the same establishments and with the same individuals in a follow-up design.
Material and methods
Study design
The first round of sampling took place during the last month before the smoking ban in bars and restaurants was introduced in Norway on 1st June 2004. The second round of sampling took place at least three months after the implementation of the new regulations, in the period from September 2004 to February 2005. The sampling took place on busy days for the establishments, on evening shifts between Thursday and Saturday. The second round of sampling was carried out on the same weekdays in the respective bars and restaurants as the first sampling.Selection of establishments and air sampling strategy
Fifteen bars and restaurants in Oslo were approached, based on geographical location, differences in size and whether they were restaurants or pubs. They were asked for their willingness to participate in the study. Two establishments refused, leaving 13 for the study.Depending on the size of the establishment, between four and six pairs of stationary samplers were placed in parallel approximately at the height of the breathing zone, usually about two metres above the floor. One sampler collected nicotine in the gaseous phase. The second sampler of the pair collected total dust. As the establishments could be overcrowded, and the employees could be very busy, only a limited number of personal samplers were employed in order to assess any association between the mean concentration of nicotine in the respective establishments and the nicotine concentration in personally collected samples. Three of the establishments were restaurants only. The remaining establishments were a mixture of other public venues such as pubs and nightclubs.
Air sampling equipment
Airborne nicotine was trapped on XAD-4 sampling tubes (SKC cat. no. 226-93, SKC Ltd, Dorset, UK) using active sampling SKC Pocket Pumps or in-house made pumps operated at 200 ml min−1 or 1.0 l min−1, respectively, according to an established method.15 The pumps were routinely calibrated using flow-meters before and after sampling. Total airborne dust was collected by use of 25 mm blank cassettes (cat. no. M000025A0) from Millipore (Billerica, MA, USA) and Millipore 25 mm Teflon filters with 1.0 μm pore size (cat no. FAL02500) equipped with Millipore Adsorbant pads (cat. no. AP1002500). The air sampling flow rate was 2.0 l min−1. In-house made pumps were used. The pumps were routinely calibrated using flow-meters before and after sampling.Selection of subjects and sampling of urine
All workers on the selected shift in the 13 included establishments were invited to participate in the study. In all, 112 subjects were identified and asked to participate. Ninety-three subjects agreed, yielding a participation rate of 83%. The background characteristics of the subjects participating in the first sampling round are shown (Table 1).| All (n = 93) | Men (n = 46) | Women (n = 47) | ||||
|---|---|---|---|---|---|---|
| Mean | Range | Mean | Range | Mean | Range | |
| Age/years | 30.7 | 19–55 | 31.3 | 19–54 | 30.1 | 19–55 |
| Height/cm | 172.5 | 155–194 | 179.1 | 165–194 | 166.0 | 155–181 |
| Smokers only (%) | 52.7 | — | 41.3 | — | 63.8 | — |
| Snuffers only (%) | 3.2 | — | 4.3 | — | 2.1 | — |
| Smoker and snuffer (%) | 11.8 | — | 23.9 | — | 0 | — |
| Non-smoker/non-snuffer (%) | 32.3 | — | 30.4 | — | 34.0 | — |
The participants voided a post-shift urine sample that was immediately brought back to the laboratory and frozen. They were further instructed to deliver a first voided urine sample the next morning, and send it by mail immediately to the laboratory. When received, usually one day later, the samples were frozen at −20 °C until analysis. Urines were collected in Urine-Monovette® 10 ml tubes (Sarstedt, Numbrecht, Germany).
Re-examination
Between four and eight months later, the same establishments were re-visited in the second round of sampling. Of those original 93 examined individuals, 74 subjects participated. None of the original participants refused to participate, but most of the non-participants had quit their jobs and were thus not available for the study. On some occasions, the subjects were not present at work on the second visit.The samplers for the collection of dust and nicotine were placed in exactly the same positions in the second round of sampling as in the first. The same routine for collecting urine samples was also employed. To ensure a high participation, some establishments had to be visited more than once, resulting in more air measurements than in the first sampling round.
The study protocol was approved by the Regional Ethics Committee for Medical Research, and an informed written consent was obtained from the participants.
Laboratory analysis
Dichloromethane (99.5%), sodium hydroxide (16.75 M) and hydrochloric acid (37%) were obtained from VWR (Darmstadt, Germany), and nitrogen was obtained from a Whatman nitrogen generator (Haverhill, MA, USA). Argon (99.99%) and helium (99.99%) were obtained from Yara (Oslo, Norway).
A Waters CapLC™ System (Milford, MA, USA) with a binary gradient pump was used to separate cotinine and 2-phenylimidazole, using an analytical column (1.0 × 150 mm, 3.5 μm Kromasil C18 (G&T Septech, Kolbotn, Norway) and a mobile phase gradient starting isocratically at composition A ∶ B (90 ∶ 10, v/v) for 2 min, prior to a linear increment to A ∶ B (40 ∶ 60, v/v) in 10 min, and finally to 100% B in 5 min (5 min hold time), where A and B are acetonitrile ∶ methanol ∶ 30 mM ammonium formate (2 ∶ 5 ∶ 93, v/v) and neat methanol, respectively. The injection volume was 2.0 μl.
A Quattro LC tandem quadrupole MS with positive electrospray ionization (ESI) (Micromass, Manchester, UK) was used for mass spectrometric (MS) detection of the compounds of interest in the multiple reaction monitoring (MRM) mode. Cotinine and 2-phenylimidazole were monitored as product ions of their respective [M + H]+ molecular ions with m/z transitions of 177 → 80 and 145 → 104, respectively. The ion source capillary voltage, sample cone and extraction cone voltages were 4200 V, 35 V and 2 V, respectively. The nitrogen nebulizer gas flow was 90 l h−1, while the desolvation gas flow and temperature were 360 l h−1 and 300 °C, respectively. The source temperature was 100 °C, and the argon collision cell pressure was set to 2.8 × 10−3 mbar with collision and RF lens voltages set to 15 V and 0.4 V, respectively. The LC system and the tandem quadrupole MS instrument were controlled and data were acquired using MassLynx v. 3.5 software.
Cotinine was quantified by use of the addition of internal standard and relative comparisons to spiked urine blank samples prepared identically. The method were validated over the concentration range 0.5–1.0 × 103 ng cotinine ml−1 urine, displaying a coefficient of correlation >0.998, and within-assay (n = 6) and between-assay (n = 6) precisions <10%. The detection limit (DL) was 0.2 μg cotinine l−1 urine.
Urine creatinine was determined with a customized FIA-2000 flow injection analyser equipped with an autosampler (Burkard Scientific, UK). Creatinine was measured spectrophotometrically with the Jaffé reaction in accordance with FIA-2000 method sheet 0319-02.
Statistics
Concentrations below the DL were substituted with values of the half of the DL. Distributions of measures with skewness exceeding 2.0 were log-transformed to achieve normalisation. Thus, the U-cotinine concentrations were log-transformed and the geometric mean (GM) is presented. The arithmetic mean (AM) is presented for the other variables. Student’s t-test for paired samples was used for comparison of measurements carried out in identical individuals, whereas Student’s t-test for unpaired samples was used otherwise for the comparison of continuous variables. Least square regression analysis was employed to assess relationships between variables, and Pearson’s correlation coefficients calculated to assess the strength of the associations. The level of significance was set at 0.05 (two-tailed). The statistics were calculated on a PC with the data package SPSS 11.5.Results
Table 2 shows the results of all measurements of airborne total dust and nicotine for all establishments before the introduction of the smoking ban. The AM total dust and nicotine concentrations were 262 μg m−3 and 28.3 μg m−3, respectively. The total dust and nicotine concentrations were lower in the restaurants than in the other establishments. The measured mean concentrations were substantially lower for the measurements collected after the introduction of the smoking ban, being 77 μg m−3 and 0.6 μg m−3 for total dust and nicotine, respectively. The differences in the measured concentrations of nicotine and total dust before and after the smoking ban was introduced were highly significant (p < 0.001).| N | AM/μg m−3 | Range/μg m−3 | 90th perc./μg m−3 | |
|---|---|---|---|---|
| a Nd: not detected. | ||||
| Before intervention | ||||
| Nicotine | 58 | 28.3 | 0.4–88.0 | 54.5 |
| Restaurants | 14 | 7.7 | 0.4–18.2 | 18.0 |
| Others | 44 | 34.9 | 8.1–88.0 | 62.9 |
| Total dust | 71 | 262 | 52–662 | 511 |
| Restaurants | 14 | 115 | 52–218 | 210 |
| Others | 57 | 298 | 89–662 | 531 |
| After intervention | ||||
| Nicotine | 96 | 0.6 | Nda–3.7 | 1.5 |
| Total dust | 93 | 77 | Nd–261 | 152 |
In all, 48 stationary samples of dust and 48 stationary samples of nicotine were sampled in parallel. The Pearson’s correlation coefficient between the two variables was high (r = 0.86; p < 0.001). The relationship between the total dust concentrations and the nicotine concentrations is presented (Fig. 1). The regression equation (Fig. 1) predicts that when the nicotine concentration in air is 0.6 μg m−3, as it was measured after the smoking ban was introduced, the point estimate of the total dust concentration in the workroom atmosphere should be 68 μg m−3. This is quite close to the actual measured AM concentration of 77 μg m−3 in the second round of sampling. Seventeen dust samples were analysed also for their content of nicotine. Very small quantities of nicotine were detected in these samples (from 0.03 μg m−3–0.45 μg m−3). This corresponds to 0.02% (range 0.01%–0.07%) nicotine in relation to total dust in these samples. The concentration of nicotine in dust in relation to nicotine in air in samples collected in parallel was 0.16% (range 0.06%–0.68%).
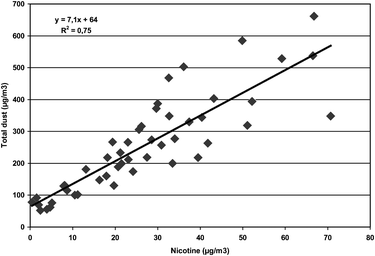 | ||
| Fig. 1 The association between total dust and nicotine in the workroom air assessed by stationary sampling of workroom air collected in parallel. | ||
Fourteen nicotine samples were collected by personal sampling. Fig. 2 shows the association between personally sampled airborne nicotine and the mean nicotine concentrations calculated from the stationary samples in the respective establishments. A high correlation between the two variables was observed (Pearson’s r = 0.81; p < 0.001).
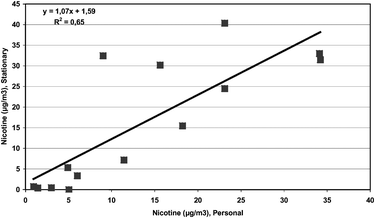 | ||
| Fig. 2 The association between personally sampled airborne nicotine and the mean nicotine concentrations assessed by stationary sampling. | ||
The concentrations of U-cotinine in those 84 subjects who delivered urine samples directly after the shift before the smoking ban was introduced are presented in Table 3. The GM concentration of U-cotinine was 11.8 μg g−1 creatinine in those subjects who were non-smokers and non-snuffers. The GM concentration was much higher, 1305 μg g−1 creatinine, in the 41 current smokers (who were also non-snuffers). Having the habit of both smoking and snuffing did not result in a statistically significantly higher mean U-cotinine concentration than being a smoker only.
| Smoking | Snuffing | N | GM/μg g−1 | 95% CI/μg g−1 | Range/μg g−1 |
|---|---|---|---|---|---|
| Yes | No | 41 | 1305 | 926–1840 | 54–8950 |
| Yes | Yes | 11 | 1635 | 729–3668 | 221–10812 |
| No | No | 29 | 11.8 | 7.6–18.3 | 2.4–230 |
| No | Yes | 3 | 888 | 122–6457 | 375–1815 |
Table 4 and Table 5 show the U-cotinine concentrations in those subjects who delivered urine samples at both sampling occasions that are compared. All subjects who reported that they used snuff were excluded from the analysis because of the substantial contribution of this habit to the levels of cotinine. The GM U-cotinine concentration increased statistically significantly from the evening after a shift until the next morning before the smoking ban was introduced, from 11.7 to 21.9 μg g−1 creatinine in those 24 subjects who were non-smokers and who delivered urine samples on these two occasions (Table 4). No similar increase was observed after the smoking ban was introduced. A statistically significant reduction, from 9.5 μg g−1 creatinine to 1.4 μg g−1 creatinine, in the non-smokers’ U-cotinine levels was found in the samples collected directly after a shift before and after the smoking ban was introduced (Table 5). The difference was even larger when urine samples from the subsequent morning before and after the smoking ban were compared. A substantial reduction in U-cotinine was also found in the smokers after the smoking ban was introduced. For samples collected after a shift before and after the smoking ban, the reduction was of statistical significance (GM 1444 μg g−1 creatinine vs. 688 μg g−1 creatinine; p = 0.03). The difference did not attain statistical significance when samples collected across intervention on the morning after a shift were compared.
| Intervention | N | Evening GM (95% CI)/μg g−1 | Morning GM (95% CI)/μg g−1 | Ratio (95% CI) | p value | |
|---|---|---|---|---|---|---|
| a The ratio is used for expressing the difference in concentrations because the variables were log-transformed. | ||||||
| Smokers | Before | 30 | 1462 (1009–2117) | 1597 (1137–2242) | 0.92 (0.71–1.17) | 0.47 |
| After | 20 | 556 (247–1254) | 956 (553–1652) | 0.58 (0.35–0.98) | 0.04 | |
| Non-smokers | Before | 24 | 11.7 (7.0–19.6) | 21.9 (13.3–36.3) | 0.53 (0.38–0.75) | 0.001 |
| After | 23 | 1.3 (0.7–2.6) | 1.8 (1.1–3.0) | 0.75 (0.42–1.34) | 0.32 | |
| Time of sampling | N | Before intervention GM (95% CI)/μg g−1 | After intervention GM (95% CI)/μg g−1 | Ratio (95% CI) | p value | |
|---|---|---|---|---|---|---|
| a The ratio is used for expressing the difference in concentrations because the variables were log-transformed. | ||||||
| Smokers | Evening | 29 | 1444 (957–2180) | 688 (324–1458) | 2.10 (1.09–4.04) | 0.03 |
| Morning | 17 | 1664 (971–2852) | 1095 (620–1934) | 1.52 (0.84–2.76) | 0.16 | |
| Non-smokers | Evening | 25 | 9.5 (6.5–13.7) | 1.4 (0.8–2.5) | 6.76 (3.57–12.81) | <0.001 |
| Morning | 20 | 15.3 (10.3–22.7) | 1.6 (0.9–3.0) | 9.27 (4.72–18.22) | <0.001 | |
Discussion
This study has assessed exposure to airborne nicotine and total dust in the same bars and restaurants before and after a total smoking ban was introduced in Norway. At the same time U-cotinine levels were determined in the identical subjects working in these establishments. To our knowledge, other studies that have used a follow-up design to assess the effects of such an intervention are not described in the literature. This study may be termed as an intervention follow-up study. This type of observational study most closely resembles an experiment, thus having major advantages with respect to the interpretation of the results when compared to cross-sectional studies carried out in two different populations before and after an intervention.The results showed substantially lower airborne nicotine and total dust levels after the smoking ban was introduced compared to the levels when smoking was allowed. A high correlation was calculated between airborne dust and nicotine before the smoking ban. The U-cotinine levels were substantially lower in the non-smokers after the introduction of the smoking ban. However, the magnitude of the reduction was dependent on whether after-shift or next morning urine samples were compared. A reduction in U-cotinine concentrations was found in the smokers as well. Finally, the U-cotinine levels measured in samples collected the morning after the shift were much higher than levels found in samples collected directly after the shift.
The level of airborne nicotine in the studied establishments before the smoking ban is comparable to what has been reported previously, being in the range from 2.3–65.5 μg m−3.18 The levels in the restaurants were substantially lower than in the other establishments. After the smoking ban was introduced, some nicotine was still measured in all except one of the establishments. The detection of nicotine in air after the smoking ban is not necessarily related to smoking inside the establishments. This was only observed once by our staff who performed the air sampling. Other more likely explanations could be that smokers going outside for a cigarette may bring the contamination back inside with them or when smoking occurs outside close to the entrance or to the air intake, small amounts of nicotine may diffuse into the establishment. No decreasing trend in the nicotine concentrations in air was obvious during the follow-up period, suggesting that the decrease occurred immediately after the introduction of the smoking ban.
In contrast to the abundance of reports on nicotine levels in bars and restaurants, fewer reports are available on dust concentrations. We measured on average 262 μg m−3 total dust in air before the introduction of the smoking ban, which is much higher than what has been measured, for instance, in office buildings in Oslo.19 Based on 12 studies, a weighted mean of 117 μg m−3 of dust was reported from restaurants, which is close to the 115 μg m−3 measured in the restaurants in our study.8 Also the total dust concentration of 298 μg m−3 in the bars in this study is close to the weighted average of 348 μg m−3 from 10 bars reported previously.8 The strong association between gaseous airborne nicotine and total dust measured in samples collected in parallel (Fig. 1) suggests a common source of the two contaminants. Similar associations have been proposed previously.20 Based on the presented regression equation (Fig. 1) a total dust concentration of 64 μg m−3 should be expected in the complete absence of airborne nicotine. This is close to the levels found in office buildings in Oslo, and also close to the mean level of 77 μg m−3 that we measured on average after the smoking ban.19 This suggests that after a total smoking ban, the total dust concentration will be substantially reduced to levels that are comparable to measured in smoke free office buildings in Oslo where the study took place.
The sampling equipment used gives an estimate of the total dust concentration. However, a similar association between airborne nicotine and particles in the PM2.5 fraction has been shown, thus suggesting that particles measured in air contaminated with ETS may be in the respirable range.21 The regression line calculated between the particle concentration in the PM2.5 fraction and nicotine in the air indicated that an increase of 1 μg nicotine m−3 would result in an increase of 9.05 μg dust m−3 (after exclusion of outliers). Other authors have found a coefficient of about 10.22 We found a corresponding increase of 7.1 μg dust m−3 for each 1 μg nicotine m−3. An increased concentration of particles in the size PM2.5 was measured in a coffee room where smoking took place.23 Experimental studies have also shown that particles from freshly smoked cigarettes are in the respirable range or even ultrafines, supporting the view that the reduction in total dust observed in this study is mainly attributed to a reduction of smaller particles in the respirable aerosol fraction which may penetrate below the larynx into the deeper parts of the lungs.9–11
Airborne nicotine is almost exclusively determined in the gaseous phase, and in this study only traces of nicotine were apparent in the particulate mass. One may assume that nicotine in the gaseous phase will distribute fairly homogeneously throughout the workroom air due to high diffusion. This assumption is supported by the high correlation observed between personally sampled nicotine and the mean level of airborne nicotine in the respective establishments. This suggests that, on the average, stationary sampled nicotine levels are quite representative for an individual’s personal exposure level in bars and restaurants. The U-cotinine levels measured before the smoking ban was introduced are in agreement with levels previously reported.13 A substantial reduction was observed in the non-smokers, which was compatible with the a priori hypothesis. However, the magnitude of the reduction is dependent on the timing of the sampling of urine in relation to ETS exposure. Nearly a doubling of the U-cotinine concentrations was found in the non-smokers in the morning urine samples as compared to the post-shift urines collected the night before. Taking into account the metabolism of nicotine and the urinary cotinine half-life of around 18 hours, this finding could be expected.14 However, this point raises the question of what would be the appropriate time to sample urines for cotinine determination of non-smokers exposed to ETS.
A substantially larger decrease in U-cotinine levels was found in the smokers compared to the non-smokers after the smoking ban was introduced. This reduction cannot be explained by the much lower exposure to ETS in the smokers. Of the 74 subjects attending the examinations after the smoking ban, only three had changed their smoking habits, of whom one had started and two had quit smoking. They were, however, excluded from the analysis. One explanation for the finding could be that the smokers actually smoke less than before the smoking ban was introduced. Because this reduction in U-cotinine was found both post-shift and the next morning, it could be that their reduction in smoking is not related to their working hours only. We have not found any literature showing reduced concentrations of U-cotinine in smokers after the introduction of a smoking ban in the hospitality industry.
In conclusion, a large reduction of airborne nicotine and total dust was found after the introduction of a smoking ban in bars and restaurants. Also the U-cotinine concentrations were reduced, both in the non-smokers and also in the current smokers.
Acknowledgements
The workers and staffs of the restaurants and bars are gratefully acknowledged for participation in the study.References
- A. K. Hackshaw, M. R. Law and N. J. Wald, Br. Med. J., 1997, 315, 980 CAS.
- M. R. Law, J. K. Morris and N. J. Wald, Br. Med. J., 1997, 315, 973 CAS.
- P. Boffetta, Scand. J. Work Environ. Health, 2002, 28, 30 Search PubMed.
- P. Jousilahti, K. Paja and V. Salomaa, Scand. J. Work Environ. Health, 2002, 28, 41 Search PubMed.
- M. S. Jaakkola and J. J. K. Jaakkola, Scand. J. Work Environ. Health, 2002, 28, 52 Search PubMed.
- T. Johnsson, T. Tuomi, M. Hyvärinen, J. Svinhufvud, M. Rothberg and K. Reijula, Am. J. Ind. Med., 2003, 43, 523 CrossRef.
- S. K. Hammond, Environ. Health Perspect., 1999, 107, 329 CrossRef.
- M. Siegel, J. Am. Med. Assoc., 1993, 270, 490 Search PubMed.
- P. J. Anderson, J. D. Wilson and F. C. Hiller, Am. Rev. Respir. Dis., 1989, 140, 202 Search PubMed.
- D. M. Broday and R. Robinson, Aerosol Sci. Technol., 2003, 37, 510 CAS.
- A. Afshari, U. Matson and L. E. Ekberg, Indoor Air, 2005, 15, 141 CrossRef CAS.
- J. Le Houezec and N. L. Benowitz, Clin. Chest Med., 1991, 12, 681 Search PubMed.
- C. Boswell, M. Curvall, R. K. Elswick Jr and D. Leyden, Biomarkers, 2000, 5, 341 CrossRef CAS.
- M. J. Jarvis, M. A. H. Russell, N. L. Benowitz and C. Feyerabend, Am. J. Public Health, 1988, 78, 696 Search PubMed.
- Method 2551, Manual of Analytical Methods, National Institute of Occupational Safety and Health, Cincinatti, OH, 4th edn, 1998 Search PubMed.
- T. Tuomi, T. Johnsson and K. Reiula, Clin. Chem., 1999, 45, 2164 CAS.
- M. W. Ogden, C. W. Eudy, D. L. Heavner, F. W. Conrad jr and C. R. Green, Analyst, 1989, 114, 1005 RSC.
- D. Trout, J. Decker, C. Mueller, J. T. Bernert and J. Pirkle, J. Occup. Environ. Med., 1998, 40, 270 CAS.
- K. R. Skulberg, K. Skyberg, K. Kruse, W. Eduard, P. Djupesland, F. Levy and H. Kjuus, Epidemiology, 2004, 15, 71 CrossRef.
- J. L. Repace and A. H. Lowrey, Risk Anal., 1993, 13, 463 CAS.
- E. A. Miesner, S. N. Rudnick, F.-C. Fu, J. D. Spengler, L. Preller, H. Özkaynak and W. Nelson, J. Air Pollut. Control Assoc., 1989, 39, 1577 CAS.
- B. P. Leaderer, Environ. Sci. Technol., 1991, 25, 770 CAS.
- S. Nardini, R. Cagnin, G. Invernizzi, A. Ruprecht, R. Boffi and S. Formentini, Monaldi Arch. Chest Dis., 2004, 61, 183 Search PubMed.
Footnotes |
| † The study was carried out with financial support from the Directorate for Health and Social Affairs, Norway. |
| ‡ Current address: The Cancer Registry, N-0369 Oslo, Norway. |
| This journal is © The Royal Society of Chemistry 2006 |