Detection and quantification of Cladosporium in aerosols by real-time PCR
Qing-Yin Zengab, Sven-Olof Westermarka, Åsa Rasmuson-Lestanderb and Xiao-Ru Wang*a
aNational Institute for Working Life, SE-90713, Umeå, Sweden. E-mail: Xiao-Ru.Wang@niwl.se; Fax: +46-90-176 123; Tel: +46-90-176 115
bDepartment of Molecular Biology, Umeå University, SE-90187, Umeå, Sweden
First published on 9th November 2005
Abstract
Cladosporium is one of the most common airborne molds found in indoor and outdoor environments. Cladosporium spores are important aeroallergens, and prolonged exposure to elevated spore concentrations can provoke chronic allergy and asthma. To accurately quantify the levels of Cladosporium in indoor and outdoor environments, two real-time PCR systems were developed in this study. The two real-time PCR systems are highly specific and sensitive for Cladosporium detection even in a high background of other fungal DNAs. These methods were employed to quantify Cladosporium in aerosols of five different indoor environments. The investigation revealed a high spore concentration of Cladosporium (107 m−3) in a cow barn that accounted for 28–44% of the airborne fungal propagules. In a countryside house that uses firewood for heating and in a paper and pulp factory, Cladosporium was detected at 104 spores m−3, which accounted for 2–6% of the fungal propagules in the aerosols. The concentrations of Cladosporium in these three indoor environments far exceeded the medical borderline level (3000 spores m−3). In a power station and a fruit and vegetable storage, Cladosporium was found to be a minor component in the aerosols, accounted for 0.01–0.1% of the total fungal propagules. These results showed that monitoring Cladosporium in indoor environments is more important than in outdoor environments from the public health point of view. Cladosporium may not be the dominant fungi in some indoor environments, but its concentration could still be exceeding the threshold value for clinical significance. The methods developed in this study could facilitate accurate detection and quantification of Cladosporium for public health related risk assessment.
1. Introduction
Cladosporium is one of the most common molds found in indoor and outdoor environments. The abundance of the airborne conidia is due in a large part to their ability to grow on a wide array of substrates, e.g. plants, wood and paper products, leather goods and foods.1–3Cladosporium spores are found throughout the year in outdoor aerosols,1 but high concentrations of spores occur in the warmer summer months.2 The peak levels of up to 105 spores m−3 are observed in outdoor agriculture environment.4 The genus Cladosporium consists of ca. 60 species.5 Several species are plant pathogens,6 and some others are allergenic to human.1,7–11 Prolonged exposure to high concentrations of Cladosporium spores can weaken the immune system allowing opportunistic bacteria and viruses to infect the host.12,13Cladosporium sensitization is associated with severe or life-threatening asthma. Upper respiratory symptoms occur with exposure to Cladosporium, but asthma symptoms are more prevalent.1 Up to now, the investigations on Cladosporium’s distribution have mainly focused on outdoor environments.2,3,14,15 Information on the prevalence of Cladosporium in different indoor environments is limited. Occupational health risk associated with exposure to Cladosporium has not been thoroughly assessed. Accurate detection and quantification methods are needed to better clarify the distribution of Cladosporium in working and living environments. Although it is known that Cladosporium spores are important aeroallergen, the majority of Cladosporium species are still not characterized. Developing species-specific detection and quantification systems for each Cladosporium species are not feasible and time consuming in applications. Generic group-specific detection and quantification methods are desirable and would facilitate the environmental monitoring of Cladosporium for exposure risk assessment.Up to date, the detection and quantification of Cladosporium in aerosols have relied on microscopic and culture techniques.3,12,14,16 Culture-based examinations are time consuming and laborious and not all airborne spores can be cultivated due to variations in viability. Thus, quantification of airborne fungi based on cultivation may not accurately reflect the true concentrations.17–20 Molecular techniques are promising approaches complementary to the conventional detection methods. Polymerase chain reaction (PCR) based methods have the advantage of detecting the presence of microorganisms in a sample regardless of their culturability at the time of analysis. However, conventional PCR does not allow for accurate quantification. Recently, the introduction of real-time PCR has offered the ability of simultaneous detection and quantification of DNA of a specific microbe in one reaction. The main advantages of real-time PCR are its quantitative property, high sensitivity, high specificity and no post-PCR steps. Several approaches have been developed to detect the amplification products in real-time PCR. The most popular approaches are TaqMan probe and SYBR Green I based assays. The TaqMan real-time PCR assay is based on the specific hybridization of a double-dye oligonucleotide probe to the target DNA molecule. SYBR Green real-time PCR assay is based upon the binding of the fluorescent dye SYBR Green I into the double-stranded PCR product. Based on these two approaches, several medically important fungi are detected and quantified in environmental and medical samples.18,21–24
In this study, we aimed at the development of rapid and sensitive generic methods for the detection and quantification of Cladosporium. Based on the patterns of mitochondrial small subunit rDNA sequence variation, we developed a TaqMan probe and a SYBR Green I based real-time PCR system specific for Cladosporium. The two systems were applied to the detection of Cladosporium in indoor aerosols of different environments. The concentrations of Cladosporium derived from the real-time PCR were compared to microscopic counting and cultural-based colony forming unit (CFU) counting. The real-time PCR systems proved to be specific and sensitive and could facilitate the environmental monitoring for Cladosporium in indoor and outdoor aerosols.
2. Materials and methods
2.1 Fungal strains and genomic DNA extraction
Fourteen strains of Cladosporium representing ten different species (Table 1) were obtained from Uppsala University Culture Collection of Fungi (UPSC, Uppsala, Sweden), Centraalbureau voor Schimmelcultures (CBS, Holland) and China General Microbiological Culture Collection Center (CGMCC, Beijing, China). Twenty-four strains of Cladosporium cladosporioides and seven strains of Cladosporium spp. were isolated from outdoor air in northern Sweden. These strains were identified through cultivation on malt extract agar (MEA) medium containing 5% purified agar (OXOID L28), 2% malt extract (OXOID L39), 30 mg l−1 streptomycin sulfate and 30 mg l−1 penicillin G, followed by morphological examinations. Another 45 fungal strains representing 45 species from 16 genera of common airborne fungi were also included in this study (Table 1). Most of these fungi were obtained from UPSC, CBS and our laboratory culture collection (ALI, Arbetslivsinstitutet, Umeå, Sweden). All the fungal strains were cultivated on MEA or dichloran-18% glycerol (DG18) medium at room temperature (22 °C) for two weeks. Genomic DNA was isolated from pure culture of each fungal strain using the procedure described by Wu et al.20| Fungal strains | GenBank accession | Cladosporium | GenBank accession |
|---|---|---|---|
| Alternaria solani CBS 109157 | Cladosporium cucumerinum CBS 177.54 | DQ089645 | |
| Aspergillus niger UPSC 1769 | AY291253 | Cladosporium effusum CBS 172.52 | DQ089648 |
| Aspergillus ochraceus UPSC 1983 | AY291267 | Cladosporium elatum UPSC 2558 | DQ089640 |
| Aspergillus fumigatus UPSC 1771 | AY291258 | Cladosporium herbarum UPSC 846 | DQ089642 |
| Aspergillus flavus UPSC 1768 | AY291268 | Cladosporium herbarum CBS 673.69 | |
| Aspergillus penicilloides ALI 231 | AY291264 | Cladosporium herbarum ALI 466 | |
| Aspergillus versicolor UPSC 2027 | AY291275 | Cladosporium macrocarpum CBS 181.54 | DQ089649 |
| Aspergillus silvaticus ALI 234 | AY291266 | Cladosporium macrocarpum CBS 420.92 | |
| Aspergillus clavatus CBS 470.91 | Cladosporium oxysporum CBS 125.80 | DQ089646 | |
| Aspergillus oryzae var. oryzae CBS 819.72 | Cladosporium sphaerospermum UPSC 957 | DQ089641 | |
| Botrytis aclada CBS 101961 | Cladosporium sphaerospermum CGMCC 3.3583 | DQ089644 | |
| Botrytis cinerea CBS 676.89 | Cladosporium sphaerospermum ALI 465 | ||
| Chrysonilia sitophila ALI 346 | AY291272 | Cladosporium sphaerospermum ALI 467 | |
| Eurotium herbariorum ALI 216 | AY291259 | Cladosporium variabile CGMCC 3.4011 | DQ089643 |
| Fusarium culmorum UPSC 1981 | AY291276 | Cladosporium variabile CBS 195.54 | DQ089650 |
| Fusarium oxysporum CBS 744.97 | Cladosporium cladosporides f. pisicola CBS 144.35 | DQ089647 | |
| Fusarium cerealis CBS 100101 | Cladosporium cladosporioides ALI 50 | AY291273 | |
| Microdochium nivale UPSC 3273 | AY291254 | Cladosporium cladosporioides UPSC 1657 | |
| Mucor plumbeus UPSC 1492 | AY291277 | Cladosporium cladosporioides ALI 2 | |
| Mucor piriformis CBS 255.85 | Cladosporium cladosporioides ALI 3 | ||
| Mucor mucedo CBS 109.16 | Cladosporium cladosporioides ALI 5 | ||
| Penicillium commune CBS 343.51 | AY291261 | Cladosporium cladosporioides ALI 6 | |
| Penicillium italicum UPSC 1577 | AY291256 | Cladosporium cladosporioides ALI 9 | |
| Penicillium chrysogenum UPSC 2020 | AY291284 | Cladosporium cladosporioides ALI 10 | |
| Penicillium brevicompactum ALI 319 | AY291282 | Cladosporium cladosporioides ALI 12 | |
| Penicillium frequentans ALI 218 | AY291260 | Cladosporium cladosporioides ALI 13 | |
| Paecilomyces lilacinus UPSC 1722 | AY291280 | Cladosporium cladosporioides ALI 14 | |
| Paecilomyces variotii UPSC 1651 | AY291281 | Cladosporium cladosporioides ALI 23 | |
| Paecilomyces inflatus CBS 288.90 | Cladosporium cladosporioides ALI 24 | ||
| Rhizopus microsporus UPSC 1758 | AY291255 | Cladosporium cladosporioides ALI 25 | |
| Rhizopus microsporus var. rhizopodiformis CBS 258.79 | AY291263 | Cladosporium cladosporioides ALI 26 | |
| Rhizopus stolonifer var. stolonifer CBS 819.96 | Cladosporium cladosporioides ALI 28 | ||
| Stachybotrys dichroa CBS 182.80 | AY291269 | Cladosporium cladosporioides ALI 29 | |
| Stachybotrys oenanthes CBS 252.76 | AY291271 | Cladosporium cladosporioides ALI 30 | |
| Stachybotrys kampalensis CBS 388.73 | AY291270 | Cladosporium cladosporioides ALI 31 | |
| Stachybotrys chartarum CBS 330.37 | AY291283 | Cladosporium cladosporioides ALI 37 | |
| Stachybotrys bisbyi CBS 317.72 | Cladosporium cladosporioides ALI 48 | ||
| Stachybotrys cylindrospora CBS 878.68 | Cladosporium cladosporioides ALI 468 | ||
| Stachybotrys parvispora CBS 253.75 | Cladosporium sp. | ||
| Stachybotrys microspora CBS 186.79 | Cladosporium sp. | ||
| Saccharomyces cerevisiae ALI 308 | Cladosporium sp. | ||
| Trichoderma harzianum ALI 232 | AY291265 | Cladosporium sp. | |
| Trichoderma viride ALI 210 | AY291257 | Cladosporium sp. | |
| Ulocladium botrytis CBS 173.82 | AY291262 | Cladosporium sp. | |
| Wallemia sebi UPSC 2502 | AY291274 | Cladosporium sp. |
2.2 Amplification of mt SSU rDNA and sequencing
Mitochondrial (mt) small subunit rDNA (SSU rDNA) of Cladosporium was amplified by PCR using the universal fungal mitochondrial primers MS1 (5′-CAGCAGTCAAGAATATTAGTCAATG-3′) and MS2 (5′-GCGGATTATCGAATTAAATAAC-3′).25 PCR mixture contained 10 pmol of each primer, 0.75 U of Taq DNA polymerase (Invitrogen Life Technologies, Carlsbad, CA,. USA), 200 μM of each dNTP (Invitrogen Life Technologies), 1.5 mM MgCl2 and 1–5 ng of template DNA in a volume of 25 μl. PCR conditions were optimized to consist of an initial denaturation of 3 min at 95 °C, followed by 36 cycles of 30 s at 94 °C, 45 s at 45 °C and 90 s at 72 °C, and a final extension of 10 min at 72 °C. Negative control was included in all PCR runs. PCR products (3 μl) were analyzed by electrophoresis on 1.4% agarose gels in 1 × TAE buffer to verify the successful amplification. The gels were stained with ethidium bromide and visualized under UV light using a Gel Doc 2000 fluorescent gel documentation system (Bio-Rad, Hercules, CA, USA). Subsequently, the PCR products were purified with a GFX™ PCR DNA and Gel Band Purification Kit (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, England) and sequenced in both directions using the same PCR amplification primers and BigDye Terminator Cycle Sequencing Ready Reaction Kit v3.0 (Applied Biosystems, Foster City, CA, USA). The obtained sequences have been deposited in the GenBank database (Table 1).2.3 Primers and TaqMan probe for Cladosporium
The mt SSU rDNA sequences of ten Cladosporium species and 24 other fungal species26 were aligned using Clustal X.27 A partial alignment is shown in Fig. 1. The sequence alignment showed all species of Cladosporium shared the same sequence pattern in the region between alignment position 81 and 110, which differed from the other fungi by a 6 bp insertion. Another discriminative sequence pattern was observed between alignment position 172 and 197. These regions were utilized to design PCR primers and probes specific for Cladosporium using the software Primer Premier v.5.0 (PREMIER Biosoftware International, Palo Alto, CA, USA) and Beacon Designer (PREMIER Biosoftware International). Two specific primers (Clado-SYBRG-PF: 5′-TACTCCAATGGTTCTAATATTTTCCTCTC-3′ and Clado-SYBRG-PR: 5′-GGGTACCTAGACAGTATTTCTAGCCT-3′) were designed for the SYBR Green I real-time PCR assay (Fig. 1). The expected amplicon size for primer pair Clado-SYBRG-PF/R is 87 bp. Another two primers (Clado-TaqMan-PF: 5′-CCTCAACGTCAGTTATTACAT-3′ and Clado-TaqMan-PR: 5′-ACCTAGACAGTATTTCTAGCCT-3′) and a TaqMan probe (Clado-TaqMan-PB: 5′-CTACTCCAATGGTTCTAATATTTTCCTCTC-3′) were designed for a TaqMan real-time PCR system. The TaqMan probe was labeled with the reporter dye 6-carboxyfuorescein (FAM) at the 5′ end and a DarkQuencher at the 3′ end (MedProbe, Oslo, Norway).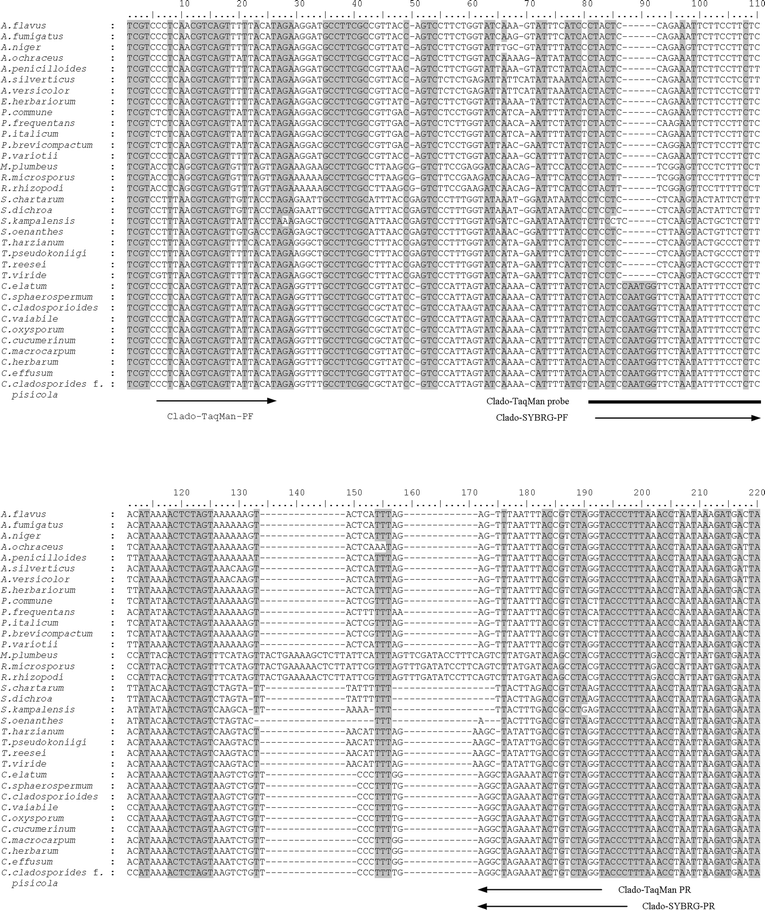 | ||
| Fig. 1 Alignment of partial mt SSU rDNA sequences from 34 fungi. The real-time PCR primers and probe sequences are indicated by arrows and line, respectively. Dash indicates alignment gap. Shadowed sites indicate conserved regions. | ||
2.4 Real-time PCR systems specific for Cladosporium
The real-time PCR was performed on an iCycler iQ™ Real-Time PCR Detection System (Bio-Rad). For the SYBR Green I based assay, iQ™ SYBR Green Supermix kit (Bio-Rad) was used in all reactions consisting of template DNA, 10 pmol of each primer (Clado-SYBRG-PF/R) and 12.5 μl of iQ™ SYBR Green Supermix in a volume of 25 μl. The real-time PCR conditions were optimized as initial denaturation of 3 min at 95 °C, followed by 40 cycles of 95 °C for 10 s and 68 °C for 30 s. For the TaqMan probe based assay, iQ™ Supermix kit (Bio-Rad) was used in reactions consisting of template DNA, 10 pmol of each primer (Clado-TaqMan-PF/R), 10 pmol TaqMan probe and 12.5 μl of iQ™ Supermix in a volume of 25 μl. The real-time PCR conditions were optimized as initial denaturation of 5 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 45 s. Each DNA sample, including negative control, was analyzed in three replicates.Standard curves based on threshold cycles (Ct, at which the fluorescence signal exceeds the background during the exponential phase of amplification) were constructed using a 10-fold dilution series of C. cladosporioides DNA (Test 1, Table 2). A 3 μl aliquot of each dilution (equivalent to 8.1 ∼ 8.1 × 10−6 ng DNA), in three replicates, was used in SYBR Green and TaqMan real-time PCR assays. After amplification, a standard curve was automatically generated by the iCycler software v. 3.0a (Bio-Rad).
| Test 1 | Test 2 | |
|---|---|---|
| Sample | C. cladosporioides | C. cladosporioides + fungal mix |
| 1 | 2.7 ng μl−1 | 2.7 ng μl−1 + 2.8 ng μl−1 |
| 2 | 2.7 × 10−1 ng μl−1 | 2.7 × 10−1 ng μl−1 + 2.8 ng μl−1 |
| 3 | 2.7 × 10−2 ng μl−1 | 2.7 × 10−2 ng μl−1 + 2.8 ng μl−1 |
| 4 | 2.7 × 10−3 ng μl−1 | 2.7 × 10−3 ng μl−1 + 2.8 ng μl−1 |
| 5 | 2.7 × 10−4 ng μl−1 | 2.7 × 10−4 ng μl−1 + 2.8 ng μl−1 |
| 6 | 2.7 × 10−5 ng μl−1 | 2.7 × 10−5 ng μl−1 + 2.8 ng μl−1 |
| 7 | 2.7 × 10−6 ng μl−1 | 2.7 × 10−6 ng μl−1 + 2.8 ng μl−1 |
| 3 μl in PCR | 1 ∶ 1 vol. mix, 3 μl in PCR |
2.5 Sensitivity evaluation of real-time PCR
To determine the detection limit of the SYBR Green I and TaqMan real-time PCR assays, two DNA dilution series were created and subjected to real-time PCR analyses (Table 2). The first test was on a 10-fold dilution series of C. cladosporioides genomic DNA with concentration ranging from 2.7 ng μl−1 to 2.7 × 10−6 ng μl−1 (Test 1, Table 2). The initial DNA concentration of 2.7 ng μl−1 was quantified using a GeneQuant Pro RNA/DNA Calculator spectrophotometer (Amersham Pharmacia Biotech, Uppsala, Sweden). A 3 μl aliquot of each dilution (equivalent to 8.1–8.1 × 10−6 ng DNA) was used in real-time PCR. To simulate the detection of Cladosporium in a mixed fungal background, genomic DNAs of ten common airborne molds detected world-wide, Alternaria solani, Aspergillus clavatus, Wallemia sebi, Eurotium herbariorum, Chrysonilia sitophila, Fusarium culmorum, Paecilomyces varioti, Penicillium commune, Stachybotrys chartarum and Ulocladium botrytis, each had a concentration of 1–5 ng μl−1, were mixed in equal volume and formed a composite fungal DNA of 2.8 ng μl−1. The 10-fold dilution series of C. cladosporioides DNA was mixed with this composite fungal DNA in equal volumes for real-time PCR analyses (Test 2, Table 2). A 3 μl aliquot of this mixture at each dilution (equivalent to 4–4 × 10−6 ng C. cladosporioides DNA plus 4.2 ng composite fungal DNA) was used in each PCR (Table 2).2.6 Bioaerosol sampling and analysis
Cladosporium spp. are often associated with plant materials and wood and paper products.1–3 Accordingly, we selected five different indoor environments for sampling: a cow barn where hay and feed are handled, a paper and pulp factory where timbers are debarked and wood chips produced, a power station that uses woodchips and garbage as biofuel, a fruit and vegetable storage facility, and a countryside family house that uses firewood for heating. Air samples were taken in the active working hours. Air sample was also taken from our DNA laboratory, where DNA isolation and PCR were performed, as background control.The airborne particles were collected and placed onto 25 mm-diameter polycarbonate filters with a pore size of 0.4 μm (Isopore; Millipore, County Cork, Ireland). The filter was mounted in a 25 mm carbon-filled polypropylene cassette (Millipore, Molsheim, France). Air was drawn through the filter with an Aircheck Sampler model 224-PCXR7 (SKC Inc., Dorset, UK). The airflow rate was 1.5 litres min−1. The sampling time was 100–150 min and 150–225 litres of air were collected in each sampler.
After sampling, 2 ml of suspension buffer (50 mM Tris-HCl, pH 7.5; 50 mM EDTA; 2% SDS; 1% Triton-100) was added into each sampling cassette. The cassettes were shaken on a shaker for 10 min to suspend the particles. From each suspension samples were taken for determination of total fungal spores (based on particle shape and size) under microscope using a Bürker chamber. A 500 μl aliquot of the suspension was serially diluted in 0.05% Tween 80. Colony counting was performed by spreading 100 μl of each dilution on MEA medium plates, in duplicates. Cultivation on MEA medium is commonly used for isolation and detection of airborne fungi.28–30 The plates were incubated at room temperature (22 °C) for 14 days before the Cladosporium CFUs were determined. Another 600 μl of the particle suspension was used for DNA extraction according to the method described by Wu et al.31 To ensure maximum DNA recovery from each aerosol sample, DNA was eluted from the binding membrane column (DNeasy Plant Mini Kit, Qiagen, Hilden, Germany) three times, each with 100 μl of elution buffer (Buffer AE, DNeasy Plant Mini Kit, Qiagen). Each DNA elution was kept separately. These DNAs were analyzed by the SYBR Green I and TaqMan based real-time PCR assay. The quantity of Cladosporium DNA used as original template in each reaction was calculated from the standard curve. From which, the concentration of Cladosporium in the aerosol samples was deduced. Each of the aerosol sample and their dilutions were repeated three times in the real-time PCR analysis.
3. Results and discussion
3.1 Specificity and detection sensitivity of the designed real-time PCR systems
In both SYBR Green I and TaqMan real-time PCR assays, all isolates of Cladosporium gave strong positive fluorescent signals after 13 to 22 cycles, while the other 45 fungal strains produced very faint signals only after 36 cycles (Fig. 2A and B). The suitability of these DNAs for PCR was tested using universal fungal mitochondrial primers MS1/2 to exclude the possibility of the negative PCR due to the absence of amplifiable DNA. In this test all the fungal DNAs gave positive amplification (result not shown). Thus, the two real-time PCR systems are highly specific for Cladosporium.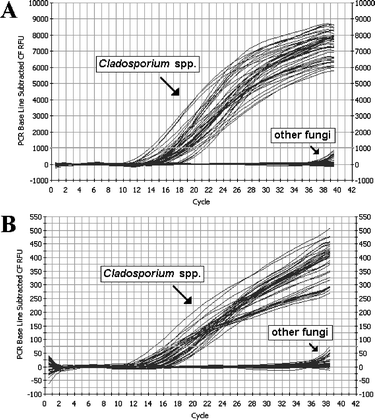 | ||
| Fig. 2 Specificity examination of SYBR Green I (A) and TaqMan probe (B) based real-time PCR assay. | ||
Two dilution series of C. cladosporioides genomic DNA, with and without background fungal DNA, were created to determine the detection sensitivity of the two real-time PCR systems. Without other background DNA (Test 1, Table 2), 8.1 × 10−5 ng of C. cladosporioides genomic DNA can be detected by the two real-time PCR methods without ambiguity (lane 6 in Fig. 3A and B). When C. cladosporioides genomic DNA was mixed with DNAs from ten other fungi (Test 2, Table 2), 4 × 10−5 ng of C. cladosporioides DNA still could be detected against a background of 4.2 ng unrelated fungal DNA (PCR amplification profile identical to Test 1, Fig. 3A and B, thus not shown), which indicates that the presence of other fungal background did not affect the detection of target DNA. The average ascomycetous fungal genome size is 36 Mb,32 corresponding to 40 fg (i.e. 4.0 × 10−5 ng) genomic DNA. This conversion of 40 fg DNA per fungal cell (spore) is frequently used in other reports.33–35 Thus, the two real-time PCR systems developed in this study could potentially detect one fungal cell or spore in a reaction.
 | ||
| Fig. 3 Detection sensitivity of SYBR Green I (A) and TaqMan probe (B) based real-time PCR assay. Samples 1–7, a 10-fold dilution series of C. cladosporioides DNA used as the template, corresponding to Test 1 in Table 2. | ||
The TaqMan real-time PCR assay is based on measuring the fluorescence released during primer extension as the 5′-nuclease activity of Taq DNA polymerase cleaves a dual-labeled fluorescent hybridization probe, designed to bind inside the amplified region.36 SYBR Green I is an intercalating dye that gives fluorescent signal when bound to double-stranded DNA. If not designed properly, the SYBR Green I based assay could have poor specificity due to the disturbance of nonspecific PCR products, which would further affect the detection sensitivity. It is critical that the primers used in SYBR Green real-time PCR are optimized with high stringency. With primers highly specific to the target DNA SYBR Green I based real-time PCR can have the same detection efficiency as the TaqMan system. The two real-time PCR systems developed in this study showed very comparable detection sensitivity both in the pure DNA samples and in aerosol samples (Fig. 3, Table 3).
| Cladosporium spores m−3 by real-time PCR with | |||||
|---|---|---|---|---|---|
| Sampling site | Number of samples | Cladosporium CFU m−3 | Total fungal spores m−3 | SYBR Green I | TaqMan probe |
| ND: not detected; NT: not tested. | |||||
| Cow barn | 12 | ND | 3 × 107–5 × 107 (4.3 × 107) | 1.0 × 106–1.5 × 107 (1.2 × 107) | 1.6 × 107–2.1 × 107 (1.9 × 107) |
| Countryside house | 5 | ND | 6 × 105–2 × 106 (1.4 × 106) | 1.5 × 104–6.3 × 104 (3.1 × 104) | 5.1 × 104–9.9 × 104 (6.8 × 104) |
| Fruit and vegetable storage | 4 | NT | 1 × 107–2 × 107 (1.8 × 107) | 4.7 × 102–6.9 × 103 (2.5 × 103) | 5.0 × 102–2.4 × 103 (1.2 × 103) |
| Power station | 12 | NT | 1 × 106–1 × 107 (4.7 × 106) | 1.0 × 103–3.2 × 103 (2.3 × 103) | 1.1 × 103–9.4 × 103 (4.8 × 103) |
| Paper and pulp factory | 6 | 2 × 103–5 × 103 | 4 × 105–8 × 105 (5.8 × 105) | 7.3 × 103–6.5 × 104 (2.5 × 104) | 1.5 × 104–5.8 × 104 (3.7 × 104) |
| Lab background | 1 | NT | NT | ND | ND |
Cladosporium consists of about 60 different species.5 This study tested 10 species including the most common ones in aerosols, such as C. herbarum, C. cladosporioides and C. sphaerospermum. These three species are considered to constitute nearly all spores of Cladosporium in aerosols.4 It is impossible for us to include all the Cladosporium species in this experiment. Thus, although the two real-time PCR systems showed high specificity for all Cladosporium species listed in Table 1, they may not be applicable to some of the untested species. Further sequence analysis of the whole genus would validate the applicability of the methods to other Cladosporium spp. Nevertheless, the most common Cladosporium species were included in this study which validates the application value of our methods for generic detection of Cladosporium for environmental monitoring.
3.2 Detection and quantification of Cladosporium in aerosols from different environments
All the aerosol samples were analyzed by SYBR Green I and TaqMan real-time PCR, cultivation for CFU counting and microscopic examination of total spore count. Cladosporium was detected in all air samples by real-time PCR except for the sample from the DNA lab (Table 3). The two real-time PCR systems gave similar estimation of Cladosporium concentration in each air sample. Among all the 40 air samples, the highest Cladosporium concentration was observed in the cow barn. Assuming one fungal genome is ca. 4 × 10−5 ng of DNA,33–35 the two real-time PCR methods generated an estimate of 107Cladosporium cells m−3 in the aerosols from the cow house (Table 3). In the countryside house and the paper and pulp factory the concentrations of Cladosporium were measured at 104 cells m−3. In the power station and the fruit and vegetable storage the concentrations were at levels of 103 cells m−3 (Table 3). The DNA method measures aerosolized all fungal propagules including spores and hyphal fragments. Microscopic examination of the 40 aerosol samples included in this study indicated that spores were the major component of fungal particles and hyphal fragments were in minute amount (data not shown). Thus, we roughly regard the DNA quantification of cells m−3 as spore concentration in the aerosols (Table 3). The total spore counts of the 40 aerosols are listed in Table 3. The samples from the cow barn and the fruit and vegetable storage showed the highest fungal spores concentration (4.3 × 107 and 1.8 × 107 m−3, respectively). The countryside house and the power station were measured at 1.4 × 106 and 4.7 × 106 m−3, respectively, and the paper and pulp factory at a level of 5.8 × 105 m−3. Detection of Cladosporium through cultivation on MEA medium was performed for aerosols from the cow barn, the countryside house and the paper and pulp factory. For the samples from the cow barn and the family house, the cultivation failed to detect Cladosporium. For the samples from the paper and pulp factory, the cultivation revealed a Cladosporium concentration of 2 × 103–5 × 103 CFU m−3 (Table 3).Compared to the cultivation result from the paper and pulp factory, the PCR quantification was about 10-fold higher than the CFU counting. Similar trend is reported in the quantification of Wallemia sebi in aerosols from farms using real-time PCR.18 This difference can be explained by the presence of unviable spores and hyphal fragments in the aerosols that originated from old fungal colonies and multiple-spore aggregates that form single colonies. In DNA-based detections, all the collected bioparticles are analyzed regardless of their culturability. Thus, the real-time PCR gave estimates that better reflect the true concentration of Cladosporium propagules in aerosols. The lack of detectable Cladosporium by cultivation in aerosols from the cow barn and the family house reflects the detection uncertainty using cultivation. Contaminants, spore viability, growth medium and the abundance of other fast growing fungi could all have affected the detection of Cladosporium colonies.
Comparison of the Cladosporium quantity to the total fungal spore concentration revealed the relative prevalence of Cladosporium in different environments. In the cow barn, Cladosporium accounted for 28–44% of the airborne fungal propagules. A study on poultry houses revealed a total fungal spore concentration of 2 × 107 m−3, but Cladosporium is not among the prevalent groups.37 The high composition of Cladosporium detected in the cow barn could be associated with hay handling. In the fruit and vegetable storage and the power station, high concentrations of fungal spores were also detected (106–107 m−3). The Cladosporium composition, however, was very low in these two environments representing 0.01% and 0.1% of the total airborne fungal flora, respectively (Table 3). A separate study using probe hybridization technique revealed that Penicillium and Aspergillus are the predominant groups in biofuel power stations.31 In the paper and pulp factory and the countryside house that uses firewood for heating, Cladosporium accounted for 2–6% of the fungal propagules in the aerosols. Cladosporium are often associated with wood products.3,38 During the long period of storage and transportation, timbers and firewood can be colonized by Cladosporium spp.39 Thus, indoor environments handling hay, timber, wood chips and firewood carry the potential risk of increased exposure to Cladosporium.40
Cladosporium spores are important aeroallergens. The concentration of 3000 Cladosporium spores m−3 in the air is suggested as threshold value for clinical significance.3,41Cladosporium spore concentration in outdoor environments are often below this threshold value.2,3,14,15 Certain indoor environments, like the cow barn, countryside house and paper and pulp factory investigated in this study, however, may host high Cladosporium concentrations far exceeding the medical borderline level. To date, information on the accurate quantification of Cladosporium in different indoor environments is limited. Our results showed that monitoring Cladosporium in indoor environments is more important than in outdoor environments from the public health point of view. Cladosporium may not be the dominant fungi in some indoor environments, but its concentration could still be exceeding the threshold value for clinical significance.
In conclusion, the present study developed two real-time PCR systems for rapid detection and quantification Cladosporium in aerosols. The two real-time PCR methods are highly specific for Cladosporium, and can potentially detect one spore in a reaction. Application of these methods in the quantification of Cladosporium in the aerosols of different indoor environments revealed concentrations of Cladosporium in cow barn, paper and pulp factory and countryside house far exceeded the threshold value for clinical significance. Prolonged exposure in these environments would impose certain health risk. Further accurate monitoring of the distribution and concentrations of Cladosporium in various environments would advance our understanding on the risk factors associated with this group of mold. The methods developed in the study could facilitate the rapid and accurate measurement of Cladosporium in environmental samples.
Acknowledgements
We thank Mr Hans Thorén, SCA, for assistance in air sampling. This study was supported by grant from the Swedish Council for Working Life and Social Research (FAS).References
- R. W. Weber, Ann. Allergy Asthma Immunol., 2002, 89, A6 Search PubMed.
- P. D. Hollins, P. S. Kettlewell, M. D. Atkinson, D. B. Stephenson, J. M. Corden, W. M. Millington and J. Mullins, Int. J. Biometeorol., 2004, 48, 137 Search PubMed.
- R. Peternel, J. Culig and I. Hrga, Ann. Agric. Environ. Med., 2004, 11, 303 Search PubMed.
- D. Kahane, D. Sime, M. McGinnis, S. Vesper and R. A. Haugland, in Proceedings of Indoor Air 2002 Conference, Indoor Air 2002, Santa Cruz, CA, ed. H. Levin, G. Bendy and J. Cordell, The Printing House, Inc., Stoughton, Wisconsin, 2002, pp. 69–73 Search PubMed.
- P. M. Kirk, P. F. Cannon, J. C. David and J. A. Stalpers, in Dictionary of the fungi, CAB International, Wallingford, 9th edn, 2001 Search PubMed.
- R. A. Samson, E. S. Hoekstra, J. C. Frisvad and O. Filtenborg, in Introduction to food- and airborne fungi, Centraalbureau voor Schimmelcultures, Utrecht, 6th edn, 2002, p. 107 Search PubMed.
- K. Okada, K. Takizawa, Y. Maebayashi, L. Xi, G. M. de Campos-Takaki, K. Nishimura, M. Miyaji and K. Fukushima, FEMS Immunol. Med. Microbiol., 1996, 16, 39 CrossRef CAS.
- E. Piecková and Z. Jesenská, Ann. Agric. Environ. Med., 1999, 6, 1 Search PubMed.
- J. E. Moore, J. Xu, B. C. Millar and S. Elshibly, Mycopathologia, 2002, 154, 25 CrossRef.
- S. Singh, P. Singh, C. Sarkar, V. Goel, T. Srivastava, M. C. Sharma and M. Behari, J. Neurol. Sci., 2005, 228, 109 CrossRef.
- A. P. Verhoeff and H. A. Burge, Ann. Allergy Asthma Immunol., 1997, 78, 544 Search PubMed.
- S. M. Hasnain, A. S. Al-Frayh, A. Al-Suwaine, M. O. Gad-El-Rab, K. Fatima and S. Al-Sedairy, Mycopathologia, 2004, 157, 171 CrossRef.
- X. Havaux, A. Zeine, A. Dits and O. Denis, Clin. Exp. Immunol., 2005, 139, 179 CrossRef CAS.
- A. Konopinska, Ann. Agric. Environ. Med., 2004, 11, 347 Search PubMed.
- F. J. Rodriguez-Rajo, I. Iglesias and V. Jato, Mycol. Res., 2005, 109, 497 Search PubMed.
- S. G. R. Wirsel, C. Runge-Frobose, D. G. Ahren, E. Kemen, R. P. Oliver and K. W. Mendgen, Fungal Genet. Biol., 2002, 35, 99 CrossRef CAS.
- J. Borneman and R. J. Hartin, Appl. Environ. Microbiol., 2000, 66, 4356 CrossRef CAS.
- Q. Y. Zeng, S. O. Westermark, A. Rasmuson-Lestander and X. R. Wang, Appl. Environ. Microbiol., 2004, 70, 7295 CrossRef CAS.
- D. Polzehl, M. Weschta, A. Podbielski, H. Riechelmann and D. Rimek, J. Med. Microbiol., 2005, 54, 31 CrossRef.
- Z. Wu, X. R. Wang and G. Blomquist, J. Environ. Monit., 2002, 4, 377 RSC.
- R. A. Haugland, M. Varma, L. J. Wymer and S. J. Vesper, Syst. Appl. Microbiol., 2004, 27, 198 Search PubMed.
- J. Morrison, C. Yang, K. T. Lin, R. A. Haugland, A. N. Neely and S. J. Vesper, J. Hosp. Infect., 2004, 57, 85 CrossRef CAS.
- P. L. White, A. Shetty and R. A. Barnes, J. Med. Microbiol., 2003, 52, 229 CrossRef.
- M. C. Hsu, K. W. Chen, H. J. Lo, Y. C. Chen, M. H. Liao, Y. H. Lin and S. Y. Li, J. Med. Microbiol., 2003, 52, 1071 CrossRef CAS.
- T. J. White, T. Bruns, S. Lee and J. Taylor, in Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, ed. M. A. Innis, D. H. Gelfand, J. J. Sninsky and T. J. White, Academic Press, San Diego, CA, 1990, pp. 315–322 Search PubMed.
- Q. Y. Zeng, X.-R. Wang and G. Blomquist, Mol. Cell. Probes, 2003, 17, 281 CrossRef CAS.
- J. D. Thompson, T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, Nucleic Acids Res., 1997, 24, 4876 CrossRef CAS.
- P. C. Wu, H. J. Su and H. M. Ho, Environ. Res., 2000, 82, 253 CrossRef CAS.
- P. Ren, T. M. Jankun and B. P. Leaderer, J. Expo. Anal. Environ. Epidemiol., 1999, 9, 560 CrossRef CAS.
- H. A. Burge, M. Chatigny, J. Feeley, K. Kreiss, P. Morey, J. Otten and K. Peterson, Appl. Ind. Hyg., 1987, 2, R10 Search PubMed.
- Z. Wu, G. Blomquist, S. O. Westermark and X. R. Wang, J. Environ. Monit., 2002, 4, 673 RSC.
- D. M. Kupfer, C. A. Reece, S. W. Clifton, B. A. Roe and R. A. Prade, Fungal Genet. Biol., 1997, 21, 364 CrossRef CAS.
- R. Wahyuningsih, H. J. Freisleben, H. G. Sonntag and P. Schnitzler, J. Clin. Microbiol., 2000, 38, 3016 CAS.
- A. S. Pham, J. J. Tarrand, G. S. May, M. S. Lee, D. P. Kontoyiannis and X. Y. Han, Am. J. Clin. Pathol., 2003, 119, 38 Search PubMed.
- R. Bu, R. K. Sathiapalan, M. M. Ibrahim, I. Al-Mohsen, E. Almodavar, M. I. Gutierrez and K. Bhatia, J. Med. Microbiol., 2005, 54, 243 CrossRef CAS.
- C. Heid, J. Stevens, K. Livak and P. Williams, Genome Res., 1996, 6, 986 CrossRef CAS.
- K. Radon, B. Danuser, M. Iversen, E. Monso, C. Weber, J. Hartung, K. J. Donham, U. Palmgren and D. Nowak, Ann. Agric. Environ. Med., 2002, 9, 41 Search PubMed.
- T. Meklin, T. Husman, A. Vepsalainen, M. Vahteristo, J. Koivisto, J. Halla-Aho, A. Hyvarinen, D. Moschandreas and A. Nevalainen, Indoor Air, 2002, 12, 175 CrossRef CAS.
- J. M. Thwaites, R. L. Farrell, K. Hata, P. Carter and M. Lausberg, J. Wood Sci., 2004, 50, 459 Search PubMed.
- J. Dutkiewicz, S. Olenchock, E. Krysinska-Traczyk, C. Skorska, J. Sitkowska and Z. Prazmo, Ann. Agric. Environ. Med., 2001, 8, 191 Search PubMed.
- S. Gravesen, Grana, 1981, 20, 225 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |