Synthesis, characterization and reactivity of tetranuclear ruthenium hydrido clusters containing chiral phosphine ligands
Viktor
Moberg
a,
Pertti
Homanen
a,
Simona
Selva
b,
Roger
Persson
a,
Matti
Haukka
c,
Tapani A.
Pakkanen
c,
Magda
Monari
*b and
Ebbe
Nordlander
*a
aInorganic Chemistry, Center for Chemistry and Chemical Engineering, Lund University, Box 124, SE-221 00, Lund, Sweden. E-mail: Ebbe.Nordlander@inorg.lu.se; Fax: +46 46 222 4439
bDipartimento di Chimica “G. Ciamician”, Università di Bologna, via Selmi 2, I-40126, Bologna, Italy
cDepartment of Chemistry, University of Joensuu, BOX 111, FIN-80101, Joensuu, Finland
First published on 21st November 2005
Abstract
The chiral clusters [H4Ru4(CO)12−n(L)n] (n = 1, 2; L = NMDPP), 1,1-[H4Ru4(CO)10(L–L)] (L–L = DUPHOS, DIPAMP), 1,2-[H4Ru4(CO)10(DIOP)] and [{H4Ru4(CO)10(DIOP)}2] have been synthesized by derivatizing the parent carbonyl cluster [H4Ru4(CO)12] with the appropriate mono- or didentate chiral phosphine ligand. The phosphine-substituted clusters were found to be able to catalyze the (asymmetric) hydrogenation of tiglic acid albeit with relatively low selectivity (enantiomeric excesses varying from 0 to 23%). It was found that the stability of the chiral ruthenium hydride clusters and the product distribution obtained in the catalytic reactions are dependent on the nature of the chiral phosphine. The crystal structures of [H4Ru4(CO)12−n(L)n] (n = 1, 2; L = NMDPP), 1,1-[H4Ru4(CO)10(L–L)] (L–L = DUPHOS, O-DUPHOS (partially oxygenated ligand), DIPAMP), 1,2-[H4Ru4(CO)10(DIOP)] and [{H4Ru4(CO)10(DIOP)}2] are presented.
Introduction
Asymmetric hydrogenation reactions are predominantly catalysed by Rh- and Ru-based catalysts. In the case of Rh(I)-containing catalysts,1 enantioselectivities have been found to be dependent on the substrate used,1 and the most frequently reported results for asymmetric hydrogenations affording high enantiomeric excesses (ee) involve unsaturated carboxylic acids with α-acylamido groups.1,2 Systematic studies of Ru(II) complexes derivatized with chiral diphosphines have led to the discovery of highly active catalysts of the type [Ru(O2CR)2(L2)],3 (L2 = BINAP derivative; R = CH3 or C(CH3)3), which are much less sensitive to the double bond geometry and the substituents present in the substrate. Despite the excellent results obtained with mononuclear ruthenium complexes, relatively few studies have been carried out on asymmetric catalysis based on phosphine-derivatized ruthenium clusters. Notable exceptions are the tetranuclear ruthenium hydrido clusters of the general formulas [H4Ru4(CO)10(L–L)] or [H4Ru4(CO)8(L–L)2] (L–L = chiral bidentate phosphine).4 These clusters have been used as catalysts for several asymmetric reactions, including isomerization,5 hydroformylation,6 and homogeneous hydrogenation5 of various carboxylic acids. In the application of such clusters to asymmetric hydrogenation of tiglic acid (trans-2,3-dimethylacrylic acid; trans-2-methyl-2-butenoic acid) the enantiomeric discrimination and the catalyst activity have been reported to depend on the chiral phosphines coordinated to parent clusters, and hydrogenation under relatively harsh reaction conditions result in the formation of S- and R-forms of 2-methylbutyric acid with enantiomeric excesses (ee) that vary from 6–39%.7 In an earlier study, we have shown that higher ee's and high conversion rates can be obtained under relatively mild reaction conditions when [H4Ru4(CO)10(x-BDPP)] (x = R,R- or S,S-) is used as a catalyst.8 Furthermore, the predominating enantiomeric form of the hydrogenated acid was shown to strongly depend on the chiral conformation of the cluster-coordinated bis-2,4-(diphenylphosphino)pentane (BDPP) ligand. In order to study the effect of cluster-coordinated chiral phosphines on the stability of modified tetranuclear ruthenium species, as well as the product distribution(s) in asymmetric hydrogenation reactions, we are currently studying the reactivity of a number of ruthenium clusters containing chiral ligands. Here, we describe the synthesis and characterization of new tetranuclear ruthenium hydrido clusters containing the chiral mono- and/or didentate phosphine ligands (S)-(+)-neomenthyldiphenylphosphine (NMDPP), 1.2-bis[(2R,5R)-2,5-dimethylphospholano]benzene (DUPHOS), (4R,5R)-(−)-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane (DIOP), and (1R,2R)-bis{(2-methoxyphenyl)phenylphosphino}ethane (DIPAMP, Fig. 1), and we report the performance of the clusters in catalytic asymmetric hydrogenation of tiglic acid.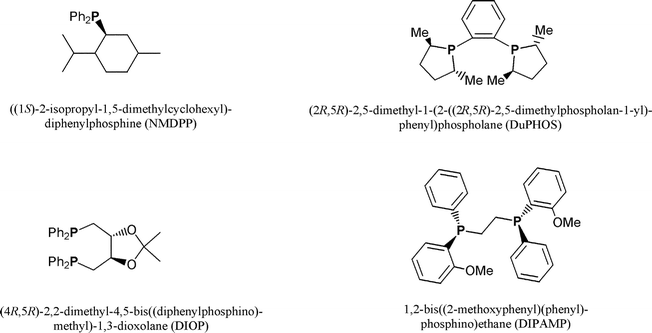
Experimental
General experimental procedures
All reactions were carried out under nitrogen atmosphere and manipulations of the products were performed in air. All solvents used in syntheses and catalysis tests were distilled under nitrogen and dried prior to use. Infrared spectra were measured using a Nicolet Avatar 360 FT-IR spectrometer with the appropriate solvent as background. 1H and 31P NMR spectra were recorded at 298 K on a Varian Unity 300 MHz spectrometer with CDCl3 used as solvent and 31P NMR shifts were referenced to external H3PO4 (85%). Thin-layer chromatography (TLC) was performed on commercial 20 × 20 cm plates covered with Merck Kiselgel 60 F254 to 0.25 mm thickness. HPLC purifications were carried out on a Rainin Dynamax chromatograph equipped with a reverse phase Dynamax C18 100 Å column, with acetonitrile used as the mobile phase. In the catalysis experiments, a Parr autoclave with PTFE lining (V = 20 mL) was used as the reaction vessel. The reagents were of analytical grade and were used as received. The chiral phosphines were enantiomerically pure samples that were purchased from STREM Chemicals (DUPHOS and DIOP) and from Larodan Fine Chemicals AB (NMDPP and DIPAMP). Tiglic acid, 2-methylbutyric acid (in pure S-form as well as a racemic mixture), and (S)-methyl mandelate were purchased from Sigma-Aldrich. The parent ruthenium cluster [H4Ru4(CO)12] was synthesized following the method of Kaesz and co-workers9 and its purity was assessed using TLC and IR spectroscopy.Preparation of [H4Ru4(CO)11(NMDPP)] (1) and [H4Ru4(CO)10(NMDPP)2] (2)
Preparation of 1,1-[H4Ru4(CO)10(DUPHOS)] (3) and 1,1-[H4Ru4(CO)10(O-DUPHOS)] (4)
Cluster 3 was also formed when the ligand substitution reaction was tested in refluxing THF. After TLC separation, the red–orange product (spectral data similar to those of 3) was recrystallized from dichloromethane. In addition to an orange microcrystalline powder of 3, a few red crystals were formed, which were analyzed by single-crystal X-ray diffraction (vide infra) and identified as [H4Ru4(CO)10(O-DUPHOS)] 4, containing partially oxygenated chelating diphosphine ligand. Because of the very poor yield, no spectroscopic data for this compound are available (attempts to resynthesize compound 4 have failed).
Preparation of 1,2-[H4Ru4(CO)10(DIOP)] (5) and [{H4Ru4(CO)10(µ-DIOP)}2] (6)
A total of 40 mg (54 µmol) of [H4Ru4(CO)12] and 32.2 mg (65 µmol) of DIOP [= (4R,5R)-(−)-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane] were dissolved in 10 ml of CH2Cl2 and 7 ml of CH3CN and the solution was stirred at room temperature. A slight excess of Me3NO (9.75 mg, 0.13 mmol) dissolved in 3 ml of CH3CN was added dropwise and very slowly to the stirred solution. The initial deep yellow solution darkened and changed colour to deep red during the addition of Me3NO. After 2 h the solvent was evaporated under vacuum. The resultant residue was subjected to preparative TLC, using n-hexane–CH2Cl2 (1 : 1 v/v) as eluent, to give two main bands which were extracted with CH2Cl2. A dark orange band (Rf = 0.75), identified as 1,2-[H4Ru4(CO)10(DIOP)] 5 (33 mg, 51%) νCO(CH2Cl2)/cm−1: 2071s, 2051s, 2029vs, 2009s, 1964w, 1945w; 1H NMR (hydride resonances): −16.8 (s, br), −16.9 (d), −17.4 (s) ,−17.7 (m); 31P{1H} NMR: 16.9 (s), 23.4 (s) 24.7 (s), 26.7 (s) (JP–P not resolved), and a dark yellow band (Rf = 0.8), identified as [{H4Ru4(CO)10(µ-DIOP)}2] 6 (10 mg, 8%) νCO(CH2Cl2)/cm−1: 2074s, 2055vs, 2016s, br, 1992m, br, 1951w, 1934w; 1H NMR (hydride resonances): −17 ppm (m); 31P{1H} NMR: 29.6 (s). Red crystals of 5 were obtained by slow evaporation of a CHCl3 solution and the yellow product 6 was crystallized from n-hexane–CH2Cl2 (2 : 1).Preparation of 1,1-[H4Ru4(CO)10(DIPAMP)] (7) and 1,2-[H4Ru4(CO)10(DIPAMP)] (8)
Catalysis experiments
In a typical reaction, an autoclave was loaded with catalyst and substrate under N2, and the degassed solvent mixture was added (2.5 mL EtOH/2.5 mL toluene). The reaction vessel was closed and purged several times with hydrogen before final pressurizing to 50 bar. The reaction mixture was continuously stirred with a magnetic stirrer (ca. 850 rpm) and heated at 100 °C for 24, 48 or 72 h. After a cooling period of approx. 30 min., the reaction vessel was carefully depressurized and opened. The homogeneous reaction mixture was transferred to a 50 mL flask and concentrated under vacuum. The conversions for the catalysis runs were calculated on the basis of NMR analyses. Samples for NMR were taken both before and after concentration of the reaction mixture. The cluster species and 2-methyl butyric acid were separated from the concentrated sample using HPLC, and analyzed by IR and NMR spectroscopies. The enantiomeric excess of the product was determined by derivatizing 2-methylbutyric acid with (S)-methyl mandelate and analyzing the diastereomeric product mixture by NMR, as described by Tyrrell et al.10X-Ray data collection and structure solutions
The diffraction data for 1–4 were collected using a Nonius Kappa CCD diffractometer. The Denzo and Scalepack programs11 were used for cell refinements and data reductions. The diffraction data for 5–7 were collected using a Bruker-AXS SMART CCD diffractometer. The initial cell parameters and an orientation matrix were obtained from least-squares refinement on reflections measured in three different sets of 20 frames each, in the range −15 < θ < 15°. The intensity data comprising a full sphere were collected using the ω-scan technique with frame width set at 0.3°. The structures of 1–7 were solved using the SIR9712 or SHELXS 97 programs,13 and refined with full-matrix least-squares on Fo2 using SHELXTL.14 Anisotropic displacement parameters were assigned to all non-hydrogen atoms in the structure. All hydrogens except the hydrides were placed in idealized positions. The positions of the hydrides for 1–4 were calculated using the XHYDEX program.15 The hydrides for 5 and 6 were experimentally located and their locations were confirmed by the XHYDEX program. The absolute structures for complexes 1–7 were determined on the basis of the absolute structure parameter (Flack index)16 during the refinement process. Clusters 2, 5, 6 and 7 crystallize as solvated [H4Ru4(CO)10(NMDPP)2]·CH2Cl2, [H4Ru4(CO)10(DIOP)]·0.6CHCl3, [{H4Ru4(CO)10(DIOP)}2]·1.6C6H14 and 1,1-[H4Ru4(CO)10(DIPAMP)]·0.4H2O. Cluster 3 crystallizes in the space group P212121, with two independent molecules (A and B) in the asymmetric unit. The crystallographic data for the reported clusters are collected in Table 1.| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| Compound | C33H33O11PRu4 | C54H62O10P2Ru4·CH2Cl2 | C28H32O10P2Ru4 | C28H32O11P2Ru4 | C41H36O12P2Ru4 ·0.6CHCl3 | C82H72O24P4Ru8 ·1.6C6H14 | C38H32O12P2Ru4 ·0.4H2O |
| a R 1 = ∑‖Fo| − |Fc|/∑|Fo|. b wR 2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2 where w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3. | |||||||
| M | 1040.84 | 1422.18 | 994.76 | 1010.76 | 1306.29 | 2460.01 | 1162.86 |
| T/K | 120(2) | 120(2) | 293(2) | 120(2) | 293(2) | 293(2) | 293(2) |
| λ/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal symmetry | Orthorhombic | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | P212121 (no. 19) | P21 (no. 4) | P212121 (no. 19) | P21 (no. 4) | P212121 (no. 19) | P21 (no. 4) | P212121 (no. 19) |
| a/Å | 9.3074(1) | 9.309(2) | 11.651(2) | 9.727(2) | 12.7512 (2) | 15.9624(5) | 11.8868(4) |
| b/Å | 17.7513(3) | 19.380(4) | 15.180(3) | 16.058(3) | 18.7601 (4) | 19.8385(6) | 16.0305(6) |
| c/Å | 22.3268(3) | 15.867(3) | 38.529(8) | 12.132(2) | 21.1114 (4) | 17.5353(5) | 22.8822(8) |
| β/° | 90 | 90.51(3) | 90 | 113.35(3) | 90 | 106.296(1) | 90 |
| V/Å3 | 3689(1) | 2862(1) | 6814(2) | 1739.9(6) | 5050.1(2) | 5329.8(3) | 4360.2(3) |
| Z | 4 | 2 | 8 | 2 | 4 | 2 | 4 |
| D c/Mg m−3 | 1.874 | 1.650 | 1.939 | 1.929 | 1.718 | 1.533 | 1.771 |
| µ(Mo-Kα)/mm−1 | 1.71 | 1.24 | 1.884 | 1.85 | 1.450 | 1.224 | 1.492 |
| F(000) | 2040 | 1428 | 3888 | 988 | 2568 | 2436 | 2280 |
| Crystal size/mm | 0.40 × 0.20 × 0.10 | 0.30 × 0.10 × 0.10 | 0.30 × 0.10 × 0.10 | 0.25 × 0.25 × 0.20 | 0.30 × 0.15 × 0.10 | 0.40 × 0.20 × 0.10 | 0.30 × 0.23 × 0.20 |
| θ limits/° | 3–27 | 4–27 | 3–26 | 2–26 | 1–30 | 3–28 | 2–30 |
| Reflections collected, hkl | 41845 | 12823 | 13756 | 7035 | 69624 | 61602 | 57874 |
| Unique observed reflections [Fo > 4σ(Fo)] | 8385 | 12823 | 13756 | 7035 | 14751 | 25688 | 13019 |
| Goodness of fit on F2 | 1.072 | 1.086 | 1.033 | 1.090 | 0.794 | 0.802 | 1.086 |
| R 1 (F)a | 0.0226 | 0.0169 | 0.0428 | 0.0181 | 0.0422 | 0.0452 | 0.0411 |
| wR 2 (F2)b | 0.0427 | 0.0399 | 0.0615 | 0.0433 | 0.0792 | 0.0893 | 0.0976 |
| Weighting scheme | a = 0.0139, b = 0.5566b | a = 0.0000, b = 0.6777b | a = 0.0000, b = 4.2044b | a = 0.0000, b = 0.0961b | a = 0.0336, b = 0.0000b | a = 0.0600, b = 8.1281b | a = 0.0366, b = 7.5109b |
| Largest diff. peak, hole/e Å−3 | 0.360, −0.617 | 0.362, −0.421 | 0.697, −0.601 | 0.489, −0.671 | 0.691, −0.971 | 0.600, −0.828 | 0.952, −1.079 |
| Absolute structure parameter | −0.03(2) | −0.02(1) | −0.01(3) | 0.0024(2) | −0.05(3) | −0.01(2) | −0.02(4) |
CCDC reference numbers 287842–287848.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b515273a
Results and discussion
Cluster synthesis
In order to obtain good yields of the phosphine-substituted tetraruthenium clusters, the coordination of the phosphine ligands via both thermal and oxidative decarbonylation of the parent cluster was investigated. The optimal synthetic method was found to vary with the nature of the phosphine and the reaction conditions. In general, thermal ligand substitution reactions were inefficient when solvents with relatively low boiling points were used. For the monodentate phosphine NMDPP, thermal substitution in refluxing toluene gave the best yields and led to the formation of an approximate 1 : 1 mixture of the mono- and disubstituted products [H4Ru4(CO)11(NMDPP)] (1) and [H4Ru4(CO)10(NMDPP)2] (2) (Fig. 2; combined yield ca. 70%). When the same reaction was carried out in a dichloromethane/acetonitrile solution using Me3NO as a decarbonylation reagent, the yields were markedly lower and the product distribution changed to yield a 2 : 1 mixture of mono- and disubstituted species (combined yield ca. 40%). | ||
| Fig. 2 Schematic structures of the phosphine coordination modes in the new ruthenium clusters that have been prepared in this study. | ||
In contrast, the diphosphine DUPHOS could be most efficiently coordinated to [H4Ru4(CO)12] by oxidative decarbonylation of the parent cluster. Only the disubstituted cluster 1,1-[H4Ru4(CO)10(DUPHOS)] (3) was isolated, although usually several minor products/fractions were observed upon purification of the product with TLC. Higher ligand to cluster molar ratios gave the same result. Thermal ligand substitution reactions in refluxing toluene led to extensive decomposition of the starting materials, also when reactions were carried out under H2 atmosphere. Further attempts to coordinate DUPHOS to the parent cluster in refluxing THF caused partial oxidation of the diphosphine ligand, most likely because of the presence of peroxides or residual water in the solvent. The cluster 1,1-[H4Ru4(CO)10(O-DUPHOS)] (4), which contains a six-membered chelate ring, could be separated by fractional crystallization from the reaction mixture, mainly containing cluster 3, and was characterized by single-crystal X-ray diffraction analysis (vide infra).
Whereas only the 1,1-isomer of [H4Ru4(CO)10(DUPHOS)] could be isolated, the analogous reaction with DIOP led to the formation of the corresponding 1,2-isomer, 1,2-[H4Ru4(CO)10(DUPHOS)] (5), in which the DIOP ligand bridges one Ru–Ru edge. It appears that the formation of the eight-membered “dimetallacycle” that results from the coordination of DIOP in 5 is favoured over the corresponding seven-membered metallacycle that would be formed if the phosphine were to coordinate in the chelate mode found in the 1,1-isomer. In addition to 5, the dimeric cluster [{H4Ru4(CO)10(µ-DIOP)}2] (6) could be isolated as a minor product in the above-mentioned reaction. In the latter cluster, the coordination of the phosphorus donor atoms to the H4Ru4 framework differs from what is found in 5 and other similar tetraruthenium diphosphine clusters in the respect that the two phosphorus atoms are in axial and equatorial positions relative to each other (vide infra). The isolation of 6 provides further indirect evidence for the DIOP ligand preferring to coordinate in a bridging rather than chelating mode.
Reasonably good yields were obtained via both thermal substitution and oxidative decarbonylation when the diphosphine DIPAMP was coordinated to [H4Ru4(CO)12]. The most striking feature was the difference in the product distribution. Thermal decarbonylation gave mainly the cluster 1,1-[H4Ru4(CO)10(DIPAMP)] (7), containing a five-membered chelate ring, and only a fraction of 1,2-[H4Ru4(CO)10(DIPAMP)] (8), which contains a six-membered “dimetallacycle”. On the other hand, oxidative decarbonylation gave mainly cluster 8 with a small quantity of 7. The combined yields of 7 and 8 were found to be roughly the same for both these methods. It was found that 8 slowly converts into 7 in solution, probably because of the relative ring strain present in 8. Thus, attempts to crystallize 8 invariably resulted in a mixture of 7 and 8. A slow increase of the concentration of 7 in the crystallization mixture was observed using spot TLC (CH2Cl2–n-hexane 2 : 3). This slow conversion of 8 into 7 could be monitored for several days/weeks (depending on the storage temperature for the specific sample). Keeping the compound(s) in solution for such a relatively long period of time resulted in (competing) cluster decomposition, as evidenced by a brown stationary spot in the spot TLC that increased in intensity with time. The interconversion of 8 into 7 is very similar to that which has been established previously for the directly analogous isomers of [H4Ru4(CO)10(dppe)],17 which are essentially isostructural to 7 and 8.
General description of the crystal structures of clusters 1–7
All clusters investigated by single-crystal X-ray diffraction are based on the tetrahedral geometry of the metal frameworks and all clusters obey the EAN rule (60 valence electrons). The coordination geometry of the Ru4 tetrahedra are completed by four bridging hydrides, eleven or ten terminal carbonyl ligands and one or two monodentate chiral phosphine ligands to give [H4Ru4(CO)11(NMDPP)] (1) and [H4Ru4(CO)10(NMDPP)2] (2), respectively. In the case of the diphosphine-substituted clusters, the chelating (1,1-[H4Ru4(CO)10(DUPHOS)] (3), 1,1-[H4Ru4(CO)10(O-DUPHOS)] (4) and 1,1-[H4Ru4(CO)10(DIPAMP)] (7)) and bridging ([H4Ru4(CO)10(DIOP)] (5)) or doubly bridging ([{H4Ru4(CO)10(µ-DIOP)}2] (6)) coordination modes are present (cf.Fig. 2) with ten terminal carbonyl ligands. In 1–7, the CO ligands are staggered with respect to the Ru–Ru vectors in contrast to the eclipsed conformation of the carbonyls in [H4Re4(CO)12] where the hydrides are face-bridging.18 Although not all the hydrides have been directly located from the Fourier-maps, the usual pattern of two “short” and four “long” Ru–Ru separations indicate that the latter bonds are spanned by bridging hydrides. Further evidence of the positions of the hydrides is given by the orientations of the carbonyls bound to ruthenium atoms that are also connected to bridging hydrides; these carbonyl ligands are bent away from the hydride positions. In the Ru4(µ-H)4 cores, the distribution of the hydrides along the Ru–Ru edges conforms to the idealized D2d symmetry (2, 6) of the precursor [H4Ru4(CO)12] or to Cs symmetry (1, 3, 4, 5, 7) in the solid state (Fig. 3). It should be pointed out that the energy difference between D2d and Cs symmetry of the Ru4(µ-H)4 core is usually very small and thus, the hydrides are usually fluxional in solution at ambient temperature (vide infra);19 however, exceptions to this rule are found, notably the cluster 1,1-[H4Ru4(CO)11(BDPP)] which gives high enantioselectivity in asymmetric hydrogenation of tiglic acid.8 | ||
| Fig. 3 Schematic depiction of the D2d and Cs conformations of the Ru4(µ-H)4 cluster core. | ||
Molecular structures of [H4Ru4(CO)11(NMDPP)] (1) and [H4Ru4(CO)10(NMDPP)2] (2)
The molecular structures of clusters 1 and 2 are shown in Fig. 4 and 5 and relevant bond lengths and angles are reported in Table 2. In the solid state, the monosubstituted phosphine derivative 1 differs from the previously reported [H4Ru4(CO)11(PR3)] species (R = OMe,20,21 Ph,22,23 SC4H3,24 C6F5,21 OEt21) and dimeric [{H4Ru4(CO)11}2(µ-Ph2P–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CC–PPh2)]24 with respect to the symmetry of the Ru4(µ-H)4 core which becomes Cs instead of D2d. This lower symmetry arrangement of the edge bridging hydrides has been found only in [H4Ru4(CO)11(PMe2Ph)]21 and in both cases the ruthenium bearing the phosphine is that bound to three bridging hydrides and transoid to one of the short Ru–Ru vectors. The trans Ru(1)–Ru(3)–P(1) angle deviates more from linearity [159.84(2)°] than the analogous angles in the three aforementioned species and is strictly comparable to the value found in [H4Ru4(CO)11(PMe2Ph)]21 [159.03(5)°]. The disubstituted cluster [H4Ru4(CO)10(NMDPP)2] (2) adopts a D2d geometry of the Ru4(µ-H)4 core with the two phosphines transoid to the same short (unbridged) Ru–Ru bond [Ru(1)–Ru(2) 2.790(1) Å] as has been found in [H4Ru4(CO)10(PPh3)2]25 and in [H4Ru4(CO)10(PFu3)2]26 (Fu = furyl). Interestingly, the latter cluster exhibits also an isomer which differs with regard to the phosphine positions that are transoid to two different short Ru–Ru interactions, as has been observed in [H4Ru4(CO)10(POEt3)2].19
C–CC–PPh2)]24 with respect to the symmetry of the Ru4(µ-H)4 core which becomes Cs instead of D2d. This lower symmetry arrangement of the edge bridging hydrides has been found only in [H4Ru4(CO)11(PMe2Ph)]21 and in both cases the ruthenium bearing the phosphine is that bound to three bridging hydrides and transoid to one of the short Ru–Ru vectors. The trans Ru(1)–Ru(3)–P(1) angle deviates more from linearity [159.84(2)°] than the analogous angles in the three aforementioned species and is strictly comparable to the value found in [H4Ru4(CO)11(PMe2Ph)]21 [159.03(5)°]. The disubstituted cluster [H4Ru4(CO)10(NMDPP)2] (2) adopts a D2d geometry of the Ru4(µ-H)4 core with the two phosphines transoid to the same short (unbridged) Ru–Ru bond [Ru(1)–Ru(2) 2.790(1) Å] as has been found in [H4Ru4(CO)10(PPh3)2]25 and in [H4Ru4(CO)10(PFu3)2]26 (Fu = furyl). Interestingly, the latter cluster exhibits also an isomer which differs with regard to the phosphine positions that are transoid to two different short Ru–Ru interactions, as has been observed in [H4Ru4(CO)10(POEt3)2].19
| 1 | 2 | 3 a | 4 | 5 | 7 | |
|---|---|---|---|---|---|---|
a Two independent molecules are present in the crystal of complex 3 (A and B).
b Ru–Ru bonds bridged by hydrides.
c P(2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(1) for 4.
d C(31)C(76) for 4. O(1) for 4.
d C(31)C(76) for 4.
|
||||||
| Ru(1)–Ru(2) | 2.9693(3)b | 2.790(1) | 2.994(1)b(A), 2.959(1)b (B) | 2.895(1)b | 3.0295(7)b | 2.9796(6)b |
| Ru(1)–Ru(3) | 2.9720(3)b | 2.962(1)b | 2.932(1)b (A), 3.058(1)b (B) | 3.029(1)b | 2.9894(7)b | 3.0439(6)b |
| Ru(1)–Ru(4) | 3.0069(3)b | 3.005(1)b | 3.038(1)b (A), 2.956(1)b (B) | 3.015(1)b | 3.0245(7)b | 2.9570(6)b |
| Ru(2)–Ru(3) | 2.7765(3) | 2.977(1)b | 2.801(1) (A), 2.786(1) (B) | 2.783(1) | 2.7823(7) | 2.9396(6)b |
| Ru(2)–Ru(4) | 2.9162(3)b | 2.952(1)b | 2.911(1)b(A), 2.805(1)b (B) | 2.779(1) | 2.9448(7)b | 2.7935(6) |
| Ru(3)–Ru(4) | 2.7830(3) | 2.789(1) | 2.789(1) (A), 2.943(1) (B) | 2.939(1)b | 2.7803(7) | 2.7823(6) |
| Ru(1)–P(1) | 2.372(1) | 2.383(1) | 2.306(2) (A), 2.311(2) (B) | 2.297(1) | 2.357(2) | 2.325(1) |
| Ru(1)–P(2)c | 2.314(2) (A), 2.305(2) (B) | 2.140(2) | 2.303(2) | |||
| Ru(2)–P(2) | 2.377(1) | 2.360(2) | ||||
| P(1)–Ru(1)–P(2)c | 83.70(6) (A), 85.28(6) (B) | 85.61(6) | 85.20(5) | |||
| Ru(1)–P(1)–C(31)c | 108.0(2) (A), 109.5(2) (B) | 113.2(1) | ||||
| Ru(1)–P(2)–C(36) | 108.9(2) (A), 109.0(2) (B) | |||||
| Ru(1)–O(1)–P(2) | 121.9(1) | |||||
![An ORTEP drawing of the molecular structure of [H4Ru4(CO)11(NMDPP)] (1) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f4.gif) | ||
| Fig. 4 An ORTEP drawing of the molecular structure of [H4Ru4(CO)11(NMDPP)] (1) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
![An ORTEP drawing of the molecular structure of [H4Ru4(CO)10(NMDPP)2] (2) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f5.gif) | ||
| Fig. 5 An ORTEP drawing of the molecular structure of [H4Ru4(CO)10(NMDPP)2] (2) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
Molecular structures of 1,1-[H4Ru4(O)10(DUPHOS)] (3) and [H4Ru4(CO)10(O-DUPHOS)] (4)
Cluster 3, which is shown in Fig. 6 (relevant bond lengths and angles are reported in Table 3), has a DUPHOS ligand chelating the ruthenium atom that is coordinated by three bridging hydrides, and adopts the Cs symmetry of the Ru4(µ-H)4 core that is usually found in the case of chelating and bridging diphosphines. It crystallizes with two independent molecules in the asymmetric unit that mostly differ in the conformation of the five-membered RuPCCP metallacycle. This ring is planar in one molecule (maximum deviation from planarity for molecule B is 0.02 Å) and slightly skewed in the other but presumably these differences are due to packing effects. The unusual asymmetric chelating P–Ru–O–P coordination of DUPHOS in compound 4 (Fig. 7) gives bond lengths of 2.297(1), 2.140(2) and 1.511(2) Å for Ru(1)–P(1), Ru(1)–O(1) and O(1)–P(2), respectively, while the observed Ru(1)–O(1)–P(1) bond angle was 121.9(1)° (bond lengths and angles are summarized in Table 3). As expected, the bite angle of 85.6(1)° for O(1)–Ru(1)–P(1) in the six-membered chelate ring was somewhat larger than the corresponding values in the five-membered chelate rings in cluster 3 (P(1)–Ru(1)–P(2) 83.70(6)°, 85.28(6)° for molecule A and B, respectively). A similar six-membered ring consisting of a dimetallacycle formed by DUPHOS bridging an Os–Os bond has been observed in [Os3(CO)10(µ-DUPHOS)].27| Ru(1)–Ru(2) | 2.9839(6) | Ru(5)–Ru(8) | 2.7918(6) |
| Ru(1)–Ru(3) | 2.7941(5) | Ru(6)–Ru(7) | 2.7941(5) |
| Ru(1)–Ru(4) | 2.9808(6) | Ru(6)–Ru(8) | 2.9931(6) |
| Ru(2)–Ru(3) | 2.9719(5) | Ru(7)–Ru(8) | 2.9476(6) |
| Ru(2)–Ru(4) | 2.7857(6) | Ru(5)–Ru(7) | 2.9790(6) |
| Ru(3)–Ru(4) | 2.9616(6) | Ru(5)–Ru(6) | 2.9914(6) |
| P(1)–Ru(1) | 2.357(2) | P(2)–Ru(5) | 2.361(2) |
![An ORTEP drawing of the molecular structure of 1,1-[H4Ru4(CO)10(DUPHOS)] (3) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f6.gif) | ||
| Fig. 6 An ORTEP drawing of the molecular structure of 1,1-[H4Ru4(CO)10(DUPHOS)] (3) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
![An ORTEP drawing of the molecular structure of [H4Ru4(CO)10(O-DUPHOS)] (4) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f7.gif) | ||
| Fig. 7 An ORTEP drawing of the molecular structure of [H4Ru4(CO)10(O-DUPHOS)] (4) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
Molecular structures of 1,2-[H4Ru4(CO)10(DIOP)] (5) and [{H4Ru4(CO)10(µ-DIOP)}2] (6)
In cluster 5 (shown in Fig. 8) the DIOP diphosphine is bridging one Ru–Ru vector giving rise to an eight-membered ring and the Ru4(µ-H)4 core conforms to a Cs symmetry as is found in other [H4Ru4(CO)10(diphosphine)] clusters. The two Ru–P distances are almost identical [2.357 and 2.360(2) Å] and the P⋯P separation [4.870(2) Å] compares well to the value found in another tetranuclear cluster with a bridging DIOP ligand, [Ir4(CO)10(DIOP)]28 [4.75 Å], and in the trinuclear [Os3H2(CO8(DIOP)]29 [4.529(7) Å] but is much shorter than that in [Ru3(CO)10(DIOP)] [5.09 Å]. The solid state structure of 6 (Fig. 9) consists of two tetrahedral H4Ru4(CO)10 units linked together by two diphosphines bridging the two cluster units, thus forming a 16-membered ring. Each tetrahedral cluster retains the D2d symmetry of the H4Ru4 core typical of the parent [H4Ru4(CO)12] and most of the [H4Ru4(CO)11(PR3)] and [H4Ru4(CO)10(PR3)2] clusters (vide supra). In addition, the coordination of the P atoms of the DIOP ligand to the Ru atoms is more similar to that of the phosphorus atoms in [H4Ru4(CO)10(POEt3)2]26 than to the analogous interaction in the related compound 5 where the diphosphine is bridging one Ru–Ru bond. The two P atoms in each cluster unit are transoid to two different short Ru–Ru vectors, as previously observed in [H4Ru4(CO)10(POEt3)2],19 in order to allow further coordination using the second P atoms of the DIOP ligands which would be prevented by a conformation similar to that in 2. Another effect of this unusual coordination of the diphosphine resides in the long P⋯P separations in each DIOP unit [P(1)⋯P(2) 6.51 and P(3)⋯P(4) 6.30 Å, respectively] compared to a distance of 4.87 Å in 5 but very close to the separations found in the polymeric [Ag(NO3)(DIOP)]n30 and dinuclear [{OsH2(PiPr3)2}2(µ-DIOP)]31 complexes [6.79 and 6.63 Å, respectively].![An ORTEP drawing of the molecular structure of 1,2-[H4Ru4(CO)10(DIOP)] (5) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f8.gif) | ||
| Fig. 8 An ORTEP drawing of the molecular structure of 1,2-[H4Ru4(CO)10(DIOP)] (5) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
![An ORTEP drawing of the molecular structure of [{H4Ru4(CO)10(µ-DIOP)}2] (6) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f9.gif) | ||
| Fig. 9 An ORTEP drawing of the molecular structure of [{H4Ru4(CO)10(µ-DIOP)}2] (6) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
Molecular structure of 1,1-[H4Ru4(CO)10(DIPAMP)] (7)
In the cluster 1,1-[H4Ru4(CO)10(DIPAMP)] (7) (shown in Fig. 10) the Ru4(µ-H)4 core conforms to Cs symmetry and the chelating coordination mode of the diphosphine generates a five-membered ring with a P(1)–Ru(1)–P(2) bite angle of 85.20(5)° which is almost identical to that found in cluster 3 (vide infra) containing the five-membered DUPHOS ligand. The conformation of the five-membered RuPCCP ring is δ and the absolute configuration of the two P atoms becomes S,S by coordination. The chiral DIPAMP ligand displays an edge-face edge-face chiral conformation of the phenyl and o-anisyl groups in addition to the chirality imposed by the P atoms. The oxygen atom of the o-anisyl group points towards the Ru(1) centre but no bonding interactions are present [Ru(1)⋯O(12) 3.77, Ru(1)⋯O(11) 3.96 Å].![An ORTEP drawing of the molecular structure of 1,1-[H4Ru4(CO)10(DIPAMP)] (7) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted.](/image/article/2006/DT/b515273a/b515273a-f10.gif) | ||
| Fig. 10 An ORTEP drawing of the molecular structure of 1,1-[H4Ru4(CO)10(DIPAMP)] (7) showing the atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. For the sake of clarity, all hydrogen atoms except the hydrides have been omitted. | ||
A number of previous studies have shown that the hydrides in [H4Ru4(CO)12] and its derivatives are fluxional; variable temperature 1H and 31P NMR measurements have revealed hydride scrambling pathways both for bridging and chelating diphosphine derivatives.3b,17,32 In clusters 1 and 2, the observed hydride resonances are fairly sharp in contrast to the significantly broadened signals in 1,1-[H4Ru4(CO)10(DUPHOS)] (3), which again indicates tautomerization between the various edge-bridging hydride positions on the Ru4 tetrahedron. In addition to these resonances, a fairly sharp triplet at −19.6 ppm is found for cluster 3, which may be assigned as the hydride located cis to both phosphines, i.e. spanning Ru(1) and Ru(4) in Fig. 6. A variable temperature NMR study32 on [H4Ru4(CO)10(dppe)] has shown the non-fluxional character of this hydride. In the solid state, identical hydride locations are found for cluster 3; in both molecules A and B the basal hydrides coordinate similar Ru–Ru edges, i.e. Ru(2)–Ru(4) in Fig. 6. Although variable temperature investigations have not been carried out, the 31P NMR measurements are fully consistent with the observed solid-state structures for 1–3, suggesting that the phosphine ligands do not give rise to fluxionality or existence of other isomeric structures in these clusters. In the case of the DIOP-containing clusters 5 and 6, the 31P NMR spectra indicated the existence of conformers for the former and stereochemical nonrigidity (on the NMR time scale) for the latter. Furthermore, the above-mentioned isomerization of 8 to 7 could be detected via31P NMR spectroscopy.
Asymmetric hydrogenation of tiglic acid
Most successful asymmetric catalytic hydrogenations involving both mono- and polynuclear ruthenium compounds have been carried out with diphosphine-derivatized species;1,33 in this study, monodentate phosphine derivatives were also tested as potential catalysts. The catalysis experiments were carried out at a relatively modest H2 pressure of 50 bar, instead of the high H2 pressure (130 bar) commonly used in cluster-based catalysis tests. The lower pressure was expected to enhance the chiral induction, as demonstrated in our previous study with BDPP derivatives.8 Severe decomposition of the cluster derivatives was observed when temperatures above 120 °C were used. At these temperatures, heterogeneous black solid particles were formed and these appeared to be responsible for the catalytic action which led to the formation of considerable amounts of cyclohexenes and cyclohexane in addition to the desired 2-methyl butyric acid, as evidenced by GC-MS analyses. When lower temperatures were used, all reaction mixtures remained homogeneous, and the above-mentioned hydrogenation products of toluene were not detected. In order to obtain reasonable conversions, a temperature of 100 °C and longer reaction times of 72 h were required (conversion rates were found to decrease appreciably below ca. 80 °C).The results for the catalysis tests for complexes 1–3, 5, 7, 8 (cluster 4 was not tested because of the exceedingly low yield) are presented in Table 4. All clusters yielded moderate to low conversions, even when extended reaction times (72 h) and substrate to catalyst molar ratios of 250 to 500 were used. After catalysis runs, clusters 1 and 3, and 7/8 could be recovered unaltered with ca. 50% yields. As was found in our study of hydrogenation catalyzed by [H4Ru4(CO)10(BDPP)],8 and in our efforts to crystallize cluster 8 (vide supra), the didentate DIPAMP ligand in 8 changes its coordination mode from bridging two different Ru metal atoms into the more energetically favored chelating coordination mode, thus forming 7 during the catalytic tests. This was found to have no apparent influence on the results of the catalytic experiments, w.r.t. efficiency, regio- and enantioselectivity. The same results were obtained regardless of whether cluster 7 or 8 was used as the initial catalyst.
| Catalyst/mg | R a/react. time (h) | Conversion (%) | Selectivityb (%) | ee (%) | Configuration |
|---|---|---|---|---|---|
| a R = n(substrate)/n(catalyst), p(H2) = 50 bar, T = 100 °C, solvent = EtOH–toluene 1 : 1 [5 mL (R = 250); 6 mL (R = 500)]. b 2-Methyl-2-butenoate is also formed. c Isomerization between 7 and 8 is observed (see text). | |||||
| [H4Ru4(CO)12]/5 | 250/48 | 21 | 99 | — | — |
| 1/5 | 250/48 | 13 | 98 | — | — |
| 1/5 | 250/72 | 32 | 97 | 11 | R |
| 2/5 | 250/48 | 29 | 100 | — | — |
| 2/5 | 250/72 | 42–68 | 97–99 | 6 | R |
| 3/5 | 300/48 | 23 | 100 | — | — |
| 3/5 | 300/72 | 13–24 | 97–99 | 5–11 | R |
| 5/10 | 500/24 | 70–95 | 92–93 | 10–23 | S |
| 7, 8c/10 | 500/24 | 49–98 | 89 | 0 | — |
For both [H4Ru4(CO)10(NMDPP)2] (2) and 1,2-[H4Ru4(CO)10(DIOP)] (5), partial decomposition of the catalyst was observed. In several tests, the final reaction mixtures obtained from catalysis by 2 contained both the mono- and disubstituted species 1 and 2, respectively. Nevertheless, the observed conversion rates remained low in most experiments, the decomposition causing only slightly larger deviations to the product distributions, i.e. formation of 2-methyl-2-butenoate. Larger deviations of the product distribution could be observed for clusters 7/8, even though very little cluster decomposition was detected.
While reasonable improvements in conversion rates relative to the parent cluster [H4Ru4(CO)12] could be observed for clusters 1, 2, 5 and 7/8, relatively low enantiomeric excesses were observed (5–23%) and in the case of 7/8 no enantioselectivity could be detected. These results indicate weak interactions between the substrate and these cluster derivatives. Furthermore, the catalytic inefficiency of the possible decomposition products under the used reaction conditions provides indirect evidence that the cluster species may be the actual catalysts.8 Both the catalytic activity and the chiral induction appear to be mainly dependent on the chemical nature of the phosphine ligand used, although the exact reaction mechanism for the cluster-based catalytic hydrogenation is still uncertain.8
In order to obtain reasonable chiral induction, it is likely that bulkier phosphine ligands must be used to introduce the chirality to cluster derivatives of [H4Ru4(CO)12]. This has been demonstrated by the results obtained with cluster derivatives of BINAP7a and BDPP,8 where the chiral centers are in the vicinity of phosphine-coordinated phenyl rings. It is likely that these groups also suitably modify the electronic properties of the cluster core to establish efficient catalytic cycles.
Conclusions
In summary, we have synthesized and characterized eight new chiral tetranuclear ruthenium clusters containing the ligands NMDPP, DUPHOS, DIOP and DIPAMP, and we have tested their behaviour in asymmetric catalytic hydrogenation of tiglic acid. On the basis of these results and those obtained in previous investigations, it appears that the cluster-coordinated phosphines play a key role for the performance of these catalysts. The best results are obtained when the parent cluster [H4Ru4(CO)12] is derivatized with sterically demanding didentate phosphines such as BDPP or BINAP, which additionally favour the chelating coordination mode. We are currently trying to use in situ high-pressure spectroscopic methods to investigate catalytic hydrogenation reactions that involve these types of clusters.Acknowledgements
This research has been sponsored by the Swedish Research Council (VR), the European Union TMR network Metal Clusters in Catalysis and Organic Synthesis, the Nordic Council of Ministers, University of Bologna (M. M., S. S.) and MIUR (M. M. and S. S.). We thank Prof. Carlaxel Andersson for the generous loan of an autoclave.References
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998 CrossRef CAS; W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS.
- (a) M. J. Burk, M. F. Gross and J. P. Martinez, J. Am. Chem. Soc., 1995, 117, 9375–9376 CrossRef CAS; (b) M. J. Burk, Y. M. Wang and J. R. Lee, J. Am. Chem. Soc., 1996, 118, 5142–5143 CrossRef CAS.
- (a) T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3176 CrossRef CAS; (b) T. Ohta, H. Takaya and R. Noyori, Inorg. Chem., 1988, 27, 566 CrossRef CAS; (c) H. Takaya, T. Ohta, K. Mashima and R. Noyori, Pure Appl. Chem., 62, 1135 Search PubMed.
- (a) D. Braga, U. Matteoli, P. Sabatino and A. Scrivanti, J. Chem. Soc., Dalton Trans., 1995, 419 RSC; (b) J. Puga, A. Arce, D. Braga, N. Centritto, F. Grepioni, R. Castillo and J. Ascanio, Inorg. Chem., 1987, 26, 867 CrossRef CAS; (c) U. Matteoli, P. Frediani, M. Bianchi, C. Botteghi and S. Gladiali, J. Mol. Catal., 1981, 12, 281 CrossRef; (d) U. Matteoli, G. Menchi, P. Frediani, M. Bianchi and F. Piacenti, J. Organomet. Chem., 1985, 285, 281 CrossRef CAS.
- U. Matteoli, M. Bianchi, P. Frediani, G. Menchi, C. Botteghi and M. Marchetti, J. Organomet. Chem., 1984, 263, 243 CrossRef CAS.
- F. Piacenti, P. Frediani, U. Matteoli, G. Menchi and M. Bianchi, Chim. Ind. (Milan), 1986, 68, 53 Search PubMed.
- (a) U. Matteoli, V. Beghetto and A. Scrivanti, J. Mol. Catal. A., 1996, 109, 45 CrossRef; (b) A. Salvini, P. Frediani, M. Bianchi, F. Piacenti, L. Pistolesi and L. Rosi, J. Organomet. Chem., 1999, 582, 218 CrossRef CAS.
- P. Homanen, R. Persson, M. Haukka, T. A. Pakkanen and E. Nordlander, Organometallics, 2000, 19, 5568 CrossRef CAS.
- S. A. R. Knox, W. J. Koepke, M. A. Andrews and H. D. Kaez, J. Am. Chem. Soc., 1975, 97, 3942 CrossRef CAS.
- E. Tyrrell, M. W. H. Tsang, G. A. Skinner and J. Fawcett, Tetrahedron, 1996, 52, 9841 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CrossRef CAS.
- A. Altomare, C. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, M. C. Burla, G. Polidori, M. Camalli and R. Spagna, SIR97, A Package for Crystal Structure Solution by Direct Methods and Refinement, University of Bari, Italy, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS97, Program for Crystal Structure Determination, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- A. G. Orpen, XHYDEX, J. Chem. Soc., Dalton Trans., 1980, 2509 RSC.
- H. D. Flack, Acta Crystallogr., Sect. A., 1983, 39, 876 CrossRef.
- M. R. Churchill, R. A. Lashewycz, S. I. Richter and J. R. Shapley, Inorg. Chem., 1980, 19, 1277 CrossRef CAS.
- R. D. Wilson, S. M. Wu, R. A. Love and R. Bau, Inorg. Chem., 1978, 17, 1271 CrossRef CAS , and references therein.
- S. Aime, M. Botta, R. Gobetto, L. Milone, D. Osella, R. Gellert and E. Rosenberg, Organometallics, 1995, 14, 3693 CrossRef CAS.
- R. D. Wilson and R. Bau, J. Am. Chem. Soc., 1976, 98, 2434 CrossRef CAS.
- M. G. Ballinas-López, E. V. García-Báez and M. G. Rosales-Hoz, Polyhedron, 2003, 22, 3403 CrossRef CAS.
- R. D. Wilson and R. Bau, J. Am. Chem. Soc., 1976, 98, 4687 CrossRef CAS.
- A. U. Härkönen, M. Ahlgrén, T. A. Pakkanen and J. Pursiainen, J. Organomet. Chem., 1997, 530, 191 CrossRef.
- T. M. Räsänen, S. Jääskelainen and T. A. Pakkanen, J. Organomet. Chem., 1998, 553, 453 CrossRef CAS.
- C. J. Adams, M. I. Bruce, E. Horn, B. W. Skelton, E. R. T. Tiekink and A. H. White, J. Chem. Soc., Dalton Trans., 1993, 3299 RSC.
- W.-Y. Wong, F.-L. Ting and Z. Lin, Organometallics, 2003, 22, 5100 CrossRef CAS.
- S. Selva, PhD Thesis, University of Bologna, 2001.
- D. Tranqui, A. Durif, M. N. Eddine, J. Lieto, J. J. Rafalko and B. C. Gates, Acta. Crystallogr., Sect. B, 1982, 38, 1916 CrossRef.
- M. J. Stchedroff, V. Moberg, E. Rodriguez, A. E. Aliev, J. Böttcher, J. W. Steed, E. Nordlander, M. Monari and A. J. Deeming, Inorg. Chim. Acta DOI:10.1016/j.ica.2005.06.048 , in press.
- B. Wu, W.-J. Zhang and S.-Y. Yu, J. Chem. Soc., Dalton Trans., 1997, 1795 RSC.
- C. Schlünker, M. E. Esteruelas, F. J. Lahoz, L. A. Oro and H. Werner, Eur. J. Inorg. Chem., 2004, 2477.
- J. R. Shapley, S. I. Richter, M. R. Churchill and R. A. Lashewycz, J. Am. Chem. Soc., 1977, 99, 7384 CrossRef CAS.
- R. D. Adams and F. A. Cotton, in Catalysis by Di-and Polynuclear Metal Cluster Complexes, Wiley-VCH, Weinheim, 1998 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |