SPANphos: trans-spanning diphosphines as cis chelating ligands!
Cristina
Jiménez-Rodríguez
,
Francesc X.
Roca
,
Carles
Bo
,
Jordi
Benet-Buchholz
,
Eduardo C.
Escudero-Adán
,
Zoraida
Freixa
and
Piet W. N. M.
van Leeuwen
*
Institute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007, Tarragona, Spain. E-mail: pvanleeuwen@iciq.es; Fax: 34 977920221; Tel: 34 977920200
First published on 18th November 2005
Abstract
Several SPANphos ligands based on a spirobichroman backbone, introduced as a putative trans ligand, form compounds of the type [Rh(nbd)(SPANphos)]BF4 (1–6) in which both norbornadiene and SPANphos act as cis chelating ligands. The cyclooctadiene rhodium chloride derivatives form bimetallic complexes. Crystal structures for several of these compounds and free ligands are reported. Semiemperical AM1 and DFT calculations show that spirobichroman can assume several conformations, some of which are suitable for the formation of cis chelating SPANphos. All calculations on SPANphos complexes of PdCl2, PtCl2 and Rh(CO)Cl show that the trans complex is more stable by 4–10 kcal mol−1. The cis conformation in 1–6 is enforced by the cis chelating norbornadiene ligand.
Introduction
The design and synthesis of diphosphine ligands exhibiting coordination properties deviating from the standard cis chelating ligands has been an intriguing and rewarding research goal for several decades.1 Two groups of ligands were reported, viz. BISBI2 and Xantphos,3 the backbones of which favour bite angles of 110–120° according to molecular mechanics calculations. However rigid the backbone of Xantphos might seem, it still does form square planar complexes with bite angles as low as 99° in cis complexes4 and 164° in trans complexes.5 The phosphorus donor atoms are very close to one another in Xantphos and one might not expect bimetallic complexes to be formed, but nevertheless bimetallic gold complexes have been identified.6 In particular, our interest concerns the catalytic properties of such ligands; indeed, Xantphos has led to a variety of catalysts exhibiting unusual properties.Likewise, exclusively trans-coordinating ligands have been a research goal for several decades7 to which end Venanzi introduced TRANSphos.8 The ligand turned out to be very flexible and although it does form trans complexes, the full range of bite angles was found experimentally and MM2 calculations supported this. The TRAP ligands developed by Ito9 coordinate in a trans fashion, but smaller bite angles were found and in view of their activity in insertion reactions of square planar complexes one assumes that cis complexes are accessible as well.
Recently we reported on a new trans-spanning diphosphine, SPANphos, containing a spirobichroman backbone, for which several trans organometallic complexes were identified confirming a consistent preference for trans coordination.10 Simple modeling had shown that SPANphos was ideally set up for forming trans complexes. Several other examples of trans diphosphine ligands (containing slightly flexible backbones) have appeared in the literature.11 Thus, by reaction of one equivalent of SPANphos per metal with [PtCl2(cod)], [PdCl2(cod)], [PdClMe(cod)], [Pd(C6H4-3-CN)(P(o-tolyl)3)Br]2, and [Rh(CO)2Cl]2 the corresponding trans complexes [PtCl2(SPANphos)], [PdCl2(SPANphos)], [PdClMe(SPANphos)], [Pd(4-CNC6H4)Br(SPANphos)], and [Rh(CO)Cl(SPANphos)] were isolated as the only product, as confirmed by 1H and 31P NMR spectroscopy, and X-ray diffraction.12 In many instances monodentate phosphines form both cis and trans diphosphine complexes as the free energies may be very similar.13,14 Stronger phosphorus donors and polar solvent are more likely to yield cis complexes. If a carbon σ-donor is present in the complex, as in some of the examples above, this will stabilize trans complexes relative to cis complexes, and thus it is no surprise that trans complexes form. Isomerization can be very slow as shown recently by Pringle and co-workers for BISBI (substituted 2,2′-bis(phosphinomethyl)-1,1′-biphenyl) complexes of PtCl2, which gave the cis and, surprisingly, also the trans complex depending on the precursor platinum complex,15 while SPANphos gave trans complexes only, even when the cis precursors [PtCl2(cod)] and [PdCl2(cod)] were used.
The reactivity of the trans complexes gave further information about the trans preference of SPANphos. Oxidative addition of MeI to trans-[Rh(CO)Cl(SPANphos)] does not take place,16 because one of the coordination sites is blocked by the backbone and apparently isomerization to a cis diphosphine complex does not occur to relieve this. Furthermore, the absence of any activity in palladium catalysis of ethene and carbon monoxide seemed to support our idea that cis complexes were not accessible.17
With excess of metal, bimetallic complexes were isolated. The reaction of [Rh(CO)2Cl]2 with one equivalent of SPANphos gave [Rh2(CO)2Cl2(SPANphos)] in which the ligand bridges the two rhodium metals in a trans fashion over the folded Rh2Cl2 moiety. This complex undergoes oxidative addition of one molecule of MeI and provides a fast catalyst for methanol carbonylation.16
While all results strengthened our belief that SPANphos was a true trans ligand, we were awakened from this trance by several results showing the contrary. Here we report on the first examples in which SPANphos coordinates in a cis fashion and more careful molecular modeling studies show that actually the energies of cis and trans complexes differ only slightly.
Experimental
General
All reactions were carried out under an argon atmosphere using standard Schlenk techniques. Solvents were obtained from Sigma-Aldrich and dried with an SPS system of IT-inc. [Rh(nbd)2]BF4 was obtained from Alfa Aesar. The diphosphines, SPANphos, SPANPOP, SPANDBP, SPANEt, SPANiPr, SPANCy and SPANtBu were prepared by procedures analogous to those reported in the literature.10 NMR spectra unless otherwise stated were recorded at the following frequencies: 400.13 MHz (1H), 100.63 MHz (13C), 161.98 MHz (31P). 13C and 31P NMR spectra were recorded using broad band proton decoupling. Chemical shifts of 1H and 13C NMR spectra are reported in ppm downfield from TMS, used as internal standard. Chemical shifts of 31P NMR spectra are referred to H3PO4 as external standard. Signals are quoted as s (singlet), d (doublet), t (triplet) vt (virtual triplet) (average coupling constant presented J = [nJP,C + n+2JP,C]/2), m (multiplet), br (broad). Mass spectra were run by electrospray impact on a Waters LCT Premier spectrometer.Synthesis
X-Ray structure determinations
Crystals of 1, 5, 6, 7 and 8 were obtained as described in the experimental (synthesis). Crystals of SPANiPr and SPANtBu were obtained by slow evaporation at room temperature of CH2Cl2–CH3OH. SPANPOP was obtained by precipitation in THF as mother liquor. The measured crystals were prepared under inert conditions immersed in perfluoropolyether as protecting oil for manipulation.Programs used: Data collection Apex2 V. 1.0–22 (Bruker-Nonius 2004), data reduction Saint + Version 6.22 (Bruker-Nonius 2001) and absorption correction SADABS V. 2.10 (2003).
CCDC reference numbers 286677 (1), 286678 (5), 286679 (6), 286681 (7), 286682 (8), 286683 (SPANiPr), 286684 (SPANtBu), 286685 (SPANPOP).
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513870c
Computational details
The geometry of spirobichroman conformers was optimized using the semiempircal AM1 hamiltonian as implemented in ArgusLab,20 and at the B3LYP/6-31G level using Gaussian03.21 Palladium, platinum and rhodium complexes were fully optimized using the Amsterdam Density Functional (ADF2004.01) program developed by Baerends et al.22,23 We used the local VWN24 exchange-correlation potential to optimize geometries, while energies were evaluated by single point calculations including nonlocal Becke's exchange correction25 and Perdew's correlation correction26 (BP86). Relativistic corrections were introduced by scalar-relativistic Zero Order Regular Approximation (ZORA).27–29 A triple-ζ plus polarization basis set was used on all atoms. For non-hydrogen atoms a relativistic frozen-core potential was used, including 3d for palladium and rhodium, 4d for platinum, 2p for phosphorus and 1s for carbon and oxygen.Results and discussion
Synthesis and characterization
Since the first report on SPANphos as a trans spanning diphosphine, we continued our work on the synthesis of organometallic complexes to determine the scope of the ligand in both coordination chemistry and catalytic reactions. In all reactions in which no preference for a cis or trans coordination is imposed by the metal precursor, SPANphos formed exclusively trans complexes.Initially putative “cis” precursors failed to give bidentate complexes. For instance, when the SPANphos was reacted with one equivalent of [Rh(acac)(CO)2], in which the acetylacetonate ligand imposes a cis coordination, we obtained [Rh(acac)(CO)(SPANphos)] in which SPANphos acts as a monodentate ligand (Scheme 1).10
 | ||
| Scheme 1 | ||
However, when one equivalent of SPANphos (phosphine substituent = PPh2) was reacted with [Rh(nbd)2]BF4 in THF, the mononuclear compound [Rh(nbd)(SPANphos)]BF4 (1) in which SPANphos occupies two cis coordination sites was obtained in 95% yield. Likewise were obtained compounds 2–6 carrying, respectively, the substituents POP (2), DBP (3), Et (4), i-Pr (5) and Cy (6). The 1H-NMR spectra all witness the liberation of one norbornadiene molecule. All 31P NMR spectra, except the broad ones of 6 and 2 exhibit a single resonance with 2JP,Rh = 144–163 Hz as is to be expected for complexes having C2 symmetry, at least on the NMR time scale. These spectral data are in accordance with both cis and (most likely, oligomeric) trans complexes. The mass spectra all gave the mass of the molecular cation, Rh(nbd)(SPAN)+. The 13C NMR spectra of compounds 1, 3–5 show many triplets in the aromatic and aliphatic region of the backbone due to coupling of 13C with 31P nuclei, which is due to a relatively strong mutual coupling between the phosphorus nuclei and the fact that the chemical shifts are equal (virtual triplets). Often this is taken as a proof of a trans P–P arrangement,30 but simulation shows that for 2JP,P > 20 Hz and the common nJP,C and n+2JP,C coupling constants already apparent triplets will be observed. Indeed, the apparent coupling constants range from 1 through 5 Hz, the average of nJP,C (0–20) and n+2JP,C (0–10), which often have opposite signs.31,32 The appearance of virtual triplets for cis complexes of nickel has been reported before and hence care must be taken when using this as a criterion.33
The 31P NMR spectra of 6 in CDCl3 (Fig. 1) at different temperatures provide most information. At 300 K only two broad signals are observed and a trace amount of doublet at ∼17 ppm (1JP,Rh = 168.7 Hz).34 This suggests that different phosphorus nuclei of 6 or isomers of 6 are present, which rapidly equilibrate. On cooling to 250 K two broad doublets start to grow at 14.26 and 23.34 ppm. When the temperature reaches 230 K, in addition to the appearance of a minor doublet at 26.7 ppm (1JP,Rh = 146.4 Hz), it becomes evident that the two doublets are in fact doublets of doublets (1JP,Rh = 156 and 138 Hz respectively, 2JP,P = 30 Hz). The presence of two doublets of doublets proves that the two phosphorus atoms are inequivalent and the value of the coupling constant 2JP,P indicates the relative cis position to one another. Thus, at 230 K the equilibrium is slow enough to distinguish a structure of 6 in which the C2 symmetry has disappeared. At low temperature the 13C NMR spectra should show only doublets due to the coupling with one phosphorus nucleus only, as the difference in chemical shifts of 9 ppm removes the virtual coupling effect. Unfortunately, the presence of a mixture of symmetrical and unsymmetrical compounds renders a complex 13C NMR spectra in which the signals cannot be easily attributed. According to simulations already 0.1 ppm suffices to remove the triplet character, at the spectrometer frequency and coupling constants under consideration.35 At high temperature we observe not only intramolecular exchange, but also exchange with the minor absorption at 26.7 ppm. We assign a symmetrical structure to the complex giving rise to this doublet in which the C2 symmetry is retained. In compounds 1 and 3–5 the doublet observed in the 31P NMR spectra can be attributed to a static symmetric structure or to an asymmetric structure in which the exchange is faster than in 6 and 2; the latter would not be unlikely for the sterically less hindered phenyl, isopropyl or ethyl derivatives (1, 4 and 5) but less so for the bulky ligand in 3. The X-ray structures obtained elucidated how a cisC2 symmetric complex may lose its symmetry and convert into a C1 symmetric complex.
![Variable-temperature 31P spectra of [Rh(nbd)(SPANCy)]BF4 (6) in CDCl3.](/image/article/2006/DT/b513870c/b513870c-f1.gif)
Thus the cations Rh(nbd)2+ react with SPANphos derivatives under replacement of one norbornadiene ligand rendering cis complexes. Reaction of the chloro-bridged dimer [(cod)RhCl]2 usually leads to cleavage of the bridge for monodentate ligands and replacement of cod when cis diphosphines are used.36 Reaction of [(cod)RhCl]2 with SPANDBP and SPANiPr gave dinuclear compounds 7 and 8 in which the chloro bridges have been cleaved but cyclooctadiene remains coordinated to the rhodium metal. In both compounds the two phosphine donors coordinate each to one (cod)rhodium chloride moiety.
X-Ray structures
X-Ray single-crystal structures were obtained for the mononuclear cis compounds 1, 5, and 6, for the bis rhodium compounds 7 and 8, and for the free ligands SPANiPr, SPANtBu and SPANPOP, while that of SPANphos10 has been reported previously. For compound 6 two pseudopolymorphic structures with a similar arrangement of the compound were determined (6 and 6b). Due to the similarity only 6 will be reported. The obtained molecular structures are represented in Fig. 2 (the numbering of the spirobichroman, which is relevant for the discussion, is represented in 5 and is identical for all the molecules in the series). Crystal data are listed in Tables 1 and 2. Selected bond distances and angles are listed in Table 3.| Compound | 1 | 5 | 6 |
|---|---|---|---|
| Formula | C54H54BF4O2P2Rh | C44H64BCl6F4O2P2Rh | C56H80BCl6F4O2P2Rh |
| Anion/solvents in crystal | BF4/4THF (Squeeze) | BF4/2CHCl3 | BF4/2CHCl3 |
| Formula weight | 986.63 | 1089.31 | 1249.56 |
| Crystal size/mm | 0.50 × 0.50 × 0.50 | 0.40 × 0.20 × 0.20 | 0.40 × 0.30 × 0.20 |
| Crystal color | Red | Red | Red |
| T/K | 100 | 100 | 100 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/n |
| a/Å | 12.3904(5) | 13.0532(8) | 12.4171(6) |
| b/Å | 19.1948(8) | 19.6411(12) | 27.9184(14) |
| c/Å | 24.8906(9) | 19.8764(13) | 17.4627(9) |
| β/° | 93.6620(10) | 94.746(2) | 104.4700(10) |
| V/Å3 | 5907.7(4) | 5078.4(6) | 5861.7(5) |
| Z | 4 | 4 | 4 |
| D c/g cm−3 | 1.109 (with 4THF: 1.353) | 1.425 | 1.416 |
| µ/mm−1 | 0.389 | 0.764 | 0.672 |
| θ max/° | 39.54 | 39.54 | 39.64 |
| Refl. measured | 120149 | 100953 | 119031 |
| Unique reflections (Rint) | 31008 (0.0296) | 28230 (0.0335) | 30269 (0.0377) |
| Absorp. correct. | SADABS (Bruker) | SADABS (Bruker) | SADABS (Bruker) |
| Transmission min./max. | 0.7228/1.0000 | 0.8009/1.0000 | 0.6924/1.0000 |
| Parameters | 847 | 581 | 668 |
| R1/wR2 [I > 2σ(I)] | 0.0350/0.1148 | 0.0335/0.0814 | 0.0466/0.1232 |
| R1/wR2 (all data) | 0.0437/0.1191 | 0.0481/0.0899 | 0.0610/0.1342 |
| Goodness-of-fit (F2) | 1.082 | 1.036 | 1.040 |
| Peak, hole/e Å−3 | 0.835/−0.638 | 1.106/−0.809 | 2.123/−2.059 |
| Compound | 7 | 8 | SPANiPr | SPANtBu | SPANPOP |
|---|---|---|---|---|---|
| Formula | C51H78Cl2O2P2Rh2 | C66.6H69.7Cl13O2P2Rh2 | C35H54O2P2 | C39H62O2P2 | C47H42O2P2 |
| Anion/solvents in crystal | — | 4.5CHCl3 | — | — | — |
| Formula weight | 1061.79 | 1631.48 | 568.72 | 624.83 | 732.75 |
| Crystal size/mm | 0.10 × 0.10 × 0.05 | 0.20 × 0.10 × 0.10 | 0.20 × 0.20 × 0.20 | 0.20 × 0.10 × 0.05 | 0.20 × 0.20 × 0.10 |
| Crystal color | Yellow | Yellow | Colorless | Colorless | Colorless |
| T/K | 100 | 100 | 100 | 100 | 100 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
C2/c | P21/c |
| a/Å | 12.3860(15) | 14.0174(19) | 10.8544(10) | 39.0066(13) | 11.7640(5) |
| b/Å | 18.114(2) | 14.210(2) | 11.1005(11) | 11.6480(5) | 24.9675(11 |
| c/Å | 21.617(3) | 19.617(3) | 15.1731(15) | 16.9525(6) | 12.5726(6) |
| α/° | 90 | 100.226(3) | 85.280(2) | 90 | 90 |
| β/° | 97.392(4) | 109.755(3) | 89.538(2) | 101.8800(10) | 96.9550(10) |
| γ/° | 90 | 101.837(3) | 63.872(2) | 90 | 90 |
| V/Å3 | 4809.9(10) | 3467.6(8) | 1635.0(3) | 7537.4(5) | 3665.6(3) |
| Z | 4 | 2 | 2 | 8 | 4 |
| D c/g cm−3 | 1.466 | 1.563 | 1.155 | 1.101 | 1.328 |
| µ/mm−1 | 0.903 | 1.066 | 0.162 | 0.146 | 0.166 |
| θ max/° | 36.03 | 38.25 | 40.00 | 39.49 | 39.50 |
| Refl. measured | 70795 | 60660 | 32900 | 60102 | 17439 |
| Unique reflections (Rint) | 20080 (0.0645) | 33678 (0.0418) | 18323 (0.0317) | 20428 (0.0466) | 10811 (0.0251) |
| Absorp. correct. | SADABS (Bruker) | SADABS (Bruker) | SADABS (Bruker) | SADABS (Bruker) | SADABS (Bruker) |
| Transmission min./max. | 0.6919/1.0000 | 0.5817/1.0000 | 0.5978/1.0000 | 0.7870/1.0000 | 0.5381/1.0000 |
| Parameters | 546 | 874 | 366 | 406 | 484 |
| R1/wR2 [I > 2σ(I)] | 0.0683/0.1741 | 0.0806/0.1884 | 0.0536/0.1668 | 0.0417/0.1150 | 0.0402/0.1116 |
| R1/wR2 (all data) | 0.1104/0.1957 | 0.1396/0.2369 | 0.0631/0.1721 | 0.0528/0.1223 | 0.0507/0.1187 |
| Goodness-of-fit (F2) | 1.055 | 1.018 | 1.096 | 1.039 | 1.034 |
| Peak, hole/e Å−3 | 2.282/−2.311 | 3.049/−3.367 | 1.486/−1.267 | 0.691/−0.300 | 0.424/−0.282 |
| a The olefinic carbon atoms of norbornadiene have been named A, B, C and D. b The α-carbons to the phosphorous non belonging to spirobichroman have been named X and Y. | |||||||
|---|---|---|---|---|---|---|---|
| Rh1–P1 | Rh1–P2 | Rh1–C(A) a | Rh1–C(B) a | Rh1–C(C) a | Rh1–C(D) a | ||
| 1 | 2.3013(3) | 2.3463(3) | 2.1708(12) | 2.1861(12) | 2.2246(12) | 2.2444(13) | |
| 5 | 2.3854(3) | 2.3717(3) | 2.1828(11) | 2.1870(11) | 2.1937(11) | 2.2024(10) | |
| 6 | 2.3832(4) | 2.3776(4) | 2.1865(14) | 2.1925(13) | 2.1925(15) | 2.1961(13) | |
| Rh1–P1 | Rh2–P2 | ||||||
| 7 | 2.3637(10) | 2.3673(11) | |||||
| 8 | 2.2945(10) | 2.2974(9) | |||||
| P1–Rh1–P2 | C8–P1–Rh1 | CX–P1–Rh1 b | CY–P1–Rh1 b | C19–P2–Rh1 b | CX′–P2–Rh1 b | CY′–P2–Rh1 b | |
| 1 | 97.892(11) | 118.17(4) | 111.72(4) | 114.24(4) | 100.74(3) | 125.80(4) | 114.56(4) |
| 5 | 95.594(9) | 111.95(3) | 111.33(4) | 114.70(4) | 107.95(3) | 110.84(4) | 116.99(3) |
| 6 | 96.312(13) | 110.43(5) | 113.86(4) | 113.94(4) | 110.06(5) | 112.81(4) | 115.36(4) |
| C19–P2–Rh2 | CX′–P2–Rh2 b | CY′–P2–Rh2 b | |||||
| 7 | 113.70(11) | 123.12(14) | 109.65(15) | 114.69(13) | 107.24(17) | 124.20(13) | |
| 8 | 121.55(12) | 110.83(14) | 120.25(14) | 120.59(10) | 108.66(11) | 120.32(11) | |
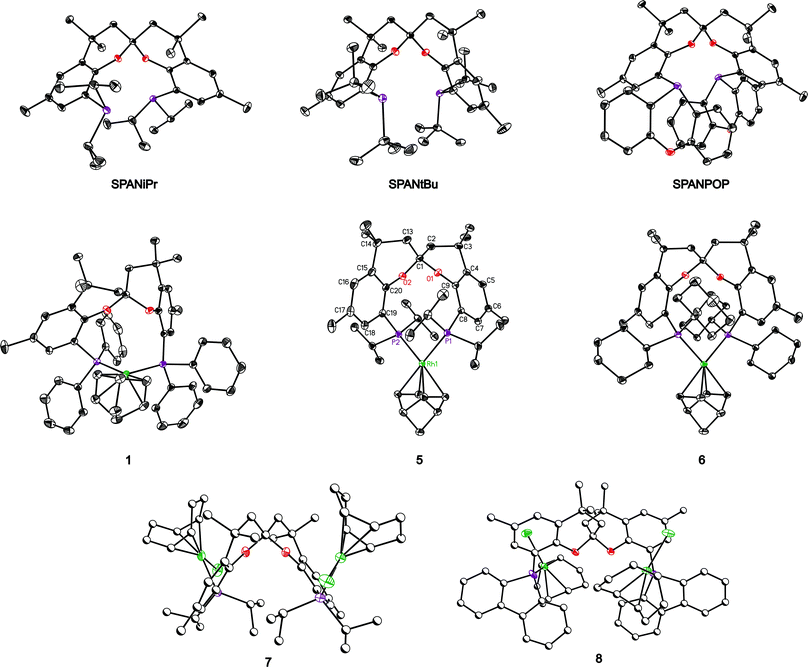 | ||
| Fig. 2 ORTEP Plots (ellipsoids drawn in 50% probability levels) of the analyzed structures. The dimeric compounds are drawn using partly spheres and hydrogen atoms are omitted to fully appreciate the geometry of the complexes. Compound 5 shows the labeling scheme of the relevant atoms used for all molecules. | ||
Compounds 5 and 6 exhibit a molecular symmetry close to C2 with the spirobichroman backbone arranged symmetrically close to the plane of coordination of the rhodium atom. The rhodium atom is coordinated in a slightly distorted square planar geometry. In 5 the planes P1–Rh1–P2 and double bond–Rh1–double bond are rotated approximately 11° and in 6 approximately 13°. In solution only a small fraction of compound 6 has the symmetric structure giving rise to a singlet in the 31P NMR spectrum; vide infra for the nonsymmetric structure. In solution symmetric structures have been found for compounds 1, 3, 4 and 5.
Compound 1 shows interesting differences in the solid state compared to compounds 5 and 6. It is arranged differently, crystallizing in a molecular C1 symmetry with the spirobichroman ligand out of plane of the planar coordination sphere of the rhodium atom, actually located at one face of the coordination plane. The Rh–P bonds in 1 have shorter distances than those in 5 and 6. Short values have also been observed in the dimeric compound 8. The shorter distances are detected in ligands containing phosphorus atoms linked to three sp2-hybridized carbon atoms, but the shorter distances may also be due to less steric hindrance in phenyl substituted phosphines compared to isopropyl and cyclohexyl substituted phosphines. These effects are probably due to the changes in the electronic properties of the phosphorous atoms. In the case of 1 the rhodium atom is also coordinated in a slightly distorted square planar geometry (The planes P1–Rh1–P2 and double bond–Rh1–double bond are rotated approximately 8°).
An interesting difference detected in compound 1 is a pyramidization of the sp2-hybridized atom connecting C19 with P2. The angle between the plane defined by the aromatic ring containing C19 and the bond P2–C19 is 17.5°. The corresponding angle with respect to P1 is 1.2°, which is within the standard values. This torsion also affects the planarity of the aromatic ring which is slightly distorted in direction of the bending out of plane of C19–P2. An explanation for the distortions of the molecule in this area is probably the CH/π interaction between the aromatic ring C15–C16–C17–C18–C19–C20 of the spirobichroman and an olefinic hydrogen atom of the norbornadiene ligand.37 The distance between the carbon atom at norbornadiene and the center of the aromatic ring is 3.26 Å (the uncorrected distance between center of the aromatic ring and the hydrogen atom is 2.47 Å). Similar distances for CH/π interaction between benzene and methane are calculated in the region of 3.6–3.8 Å.37c Additionally, a π/π interaction between the aromatic ring C4–C5–C6–C7–C8–C9 of the spirobichroman and one of the phenyl groups bonded at P2 can be detected. The approximate distance of this contact is 3.8 Å.37d Both, CH/π and π/π interaction, need to be considered in order to describe the unexpected C1 symmetry of the compound 1. Such a structure would give rise to two different absorptions in 31P NMR and most likely 2 and 6 in solution assume a conformation related to this one, rapidly equilibrating at room temperature with a C2 symmetric conformer.
The “bite angles” (P–Rh–P) of the cis chelating complexes are all around 97°, i.e. only slightly more opened than the expected angles for an ideal square planar system (see Table 3). The almost equal distances of the metal to the four olefinic carbon atoms of norbornadiene represents a symmetric arrangement without distortions.
A comparison of the Rh–P–C angles for all complexes gives similar values except for compound 1 and compounds 7 and 8. The differences in 1 may be caused by the tilting of the backbone described above. The differences in the angles of compounds 7 and 8 are probably due to steric effects of the substituents at phosphorus.
In order to understand the ability of SPANphos to form cis complexes an exact analysis of the conformations of the spiro rings in spirobichroman in the cis complexes, the free ligands and the dimeric complexes is necessary. The hexacyclic rings in the spiro compounds can adopt a boat conformation, a twist conformation and an envelope conformation. Additionally to the different conformations, the six membered rings can adopt concave or distal orientations relative to one another. In order to define the conformation of each ring it is assumed that the two sets of atoms C3–C4–C9–O1 and C14–C15–C20–O2 are each in one same plane due to the sp2-hybridization of the central atoms, and carbon atoms C1, C2 and C13 may be out of these planes. The out of plane deviations (in Å) for the single atoms C1, C2 and C1, C13 with respect to these defined planes have been calculated to determine boat, twist and envelope conformations. A boat conformation will be found if both atoms are out of plane at the same side with a deviation >0.2 Å. A twist conformation will be found if both atoms are showing small deviations out of plane at opposite sides of the plane. An envelope conformation will be found if only one atom deviates significantly from this plane. The envelope conformation can be found at the spiro atom (C1) [= envelope (spiro)] and at the CH2-atom (C2, C13) [= envelope (CH2)]. The detected conformations for the described atoms are resumed at Table 4.
| Compound | 1 | 5 | 6 | 7 | 8 | SPANiPr | SPANtBu | SPANPOP | Bichroman38 | |
|---|---|---|---|---|---|---|---|---|---|---|
| a The deviations of plane for the spiro atom or the CH2-atoms is defined for ring 1 as: deviation of C1-spiro in the plane C1–O1–C9–C4–C3 and deviation of C2 (CH2) in the plane C2–C3–C4–C9–O1. The deviations of plane for the spiro atom or the CH2-atoms is defined for ring 2 as: deviation of C1-spiro in the plane C1–O2–C20–C15–C14 and deviation of C13 (CH2) in the plane C13–C14–C15–C20–O2. The signs of the deviations are chosen arbitrarily with respect to the face of the plane. | ||||||||||
| Approximate deviation out of plane (Å) | ||||||||||
| Ring 1a | C1 (spiro) | +0.25 | +0.21 | +0.22 | +0.13 | −0.14 | +0.05 | +0.14 | +0.08 | +0.08 |
| C2 (CH2) | +0.27 | +0.26 | +0.27 | −0.03 | +0.02 | −0.08 | −0.02 | −0.05 | −0.06 | |
| Boat | Boat | Boat | Envelope (spiro) | Envelope (spiro) | Twist | Envelope (spiro) | Twist | Twist | ||
| Ring 2a | C1 (spiro) | +0.07 | +0.22 | +0.32 | +0.14 | +0.16 | +0.10 | +0.09 | +0.08 | — |
| C13 (CH2) | −0.20 | +0.26 | +0.34 | −0.03 | +0.02 | −0.04 | −0.05 | −0.06 | — | |
| Envelope (CH2) | Boat | Boat | Envelope (spiro) | Envelope (spiro) | Envelope (spiro) | Twist | Twist | Twist | ||
A comparison of the conformations in 1, 5 and 6 of the spiro rings shows in 1 a boat/envelope (CH2) conformation and in 5 and 6 a boat/boat conformation. In a spiro bichroman molecule, 4,4,4′,4′,7,7′-hexamethyl-2,2′-spirobichroman, a twist/twist conformation has been found.38 The ligands SPANiPr and SPANtBu display a twist/envelope (spiro) conformation and the ligand SPANPOP containing the large planar substituents at phosphorus has, like spirobichroman, a twist/twist conformation. The dimeric compounds 7 and 8 reveal an envelope (spiro)/envelope (spiro) conformation. All the analyzed compounds adopt a concave arrangement of the spiro rings except the dimeric compound 8 which shows a distal orientation of the rings.
The conformations found in the analyzed structures show that SPANphos ligands are highly flexible and can adopt different conformations depending on the needs of the molecules or complexes to be formed. If, due to the fixed geometry of the norbornadiene, the resulting monomeric complex allows only cis geometry, the bichroman backbone will adopt the appropriate conformation to reach the required orientation and form the cis structure.
Computational chemistry
In view of the various conformations that the two six-membered rings of SPANphos ligands can assume, a too simple approach for bite angle map calculations using MM2 can lead one easily astray. Therefore we had a look at spirobichroman compounds at higher levels of theory. Spirobichroman 9 (Scheme 2, 2,2′-spirobi[2H-1-benzopyran], 3,3′,4,4′-tetrahydro-4,4,4′,4′-tetramethyl-) was studied with the use of semiempirical (AM1) and DFT (B3LYP/6-31G) methods. | ||
| Scheme 2 | ||
Seven local minima were identified within 10 kcal mol−1 from the global minima exhibiting the ring conformations also encountered in the X-ray studies. On both levels of theory three distinct conformers show identical relative stability; the conformers can be described as envelope (spiro)/envelope (spiro), twist/twist and envelope (CH2)/envelope (CH2). The global minimum coincides with the crystal structure found for bischromane framework which has a perfect twist/twist structure, also found for the free SPANPOP ligand (Table 5, conformation D). This structure can evolve, without a significant energy change, to an envelope (spiro)/envelope (spiro) conformer which was found for free SPANphos10 as well as for compound 7 (Table 5, conformation C). The third conformer, almost degenerate to D and C, corresponds to an envelope (CH2)/envelope (CH2) conformation (Table 5, conformation E), only characterized in trans compounds. The next lowest in energy is a structure that contains one 2H,3,4-dihydrobenzopyran in a boat conformation and the other ring in a twist conformation (Table 5, conformation B). A very similar arrangement is found in the solid state structure of compound 1, which should be described as boat/envelope (CH2) (vide supra).
| A | B | C | D | E | F | G | |
|---|---|---|---|---|---|---|---|
| a Actually the crystal structure of compound 1 has boat/envelope (CH2) in our definitions, but it is close to boat/twist. b Ref. 10. c Ref. 38. | |||||||
| 2D |

|
|
|

|

|

|

|
| 3D |

|
|

|

|

|

|
|
| Conformer | Boat/boat | Boat/twist | Envelope (spiro)/envelope (spiro) | Twist/twist spirobichroman,c | Envelope (CH2)/envelope (CH2) concave | Envelope (spiro)/twist | Envelope (spiro)/envelope (spiro) distal |
| Compound(s) | 5, 6 | 1 a | 7, SPANphosb | SPANPOP | trans-SPANphosPtCl2b | SPANtBu, SPANiPr | 8 |
| d(HC8⋯HC19)/Å | 4.24 | 4.73 | 5.35 | 5.42 | 5.75 | 6.04 | 6.58 |
| Relative energy (AM1) | +1.7 | +1.0 | 0. | 0. | +0.3 | +2.4 | +5.8 |
| Relative energy (B3LYP/6-31G) | +4.2 | +2.0 | 0. | 0. | 0. | +4.3 | +9.2 |
The next lowest in energy shows both rings in a boat conformation (Table 5, conformation A), which is exactly the structure displayed by cis compounds 5 and 6 in the solid state. The asymmetric conformation F, has been observed in crystal structures of the free ligands SPANiPr and SPANtBu. Conformation G, the highest energy backbone conformation, in which the phosphorus substituents are at a large distance, was observed for bimetallic compound 8. In this fashion the interaction between the two (cod)Rh(PR2)Cl fragments is minimized, although the backbone strain is higher than in the other conformations. In summary, all the possible conformations of the spirobichroman moiety displayed in the calculated minima have been observed in crystal structures of either free ligands or SPAN-containing complexes. Thus, although four out of six atoms are retained in one plane and a rigid spiro center holds the two rings together the flexibility is high.
We have calculated the distances between the two hydrogen atoms in the spirobichroman backbone that become phosphorus atoms in SPANphos. Indeed, the distances in boat/boat conformations are much shorter than in the envelope conformations (see Table 5). Still, considerable distortions are needed to fit either a cis or a trans complex. In addition to the P–P distance one should also consider the resulting angle that the lone pairs of the donor atoms adopt in these conformations. If metal coordination requires a deviation from these values it will also imply further distortions of the backbone. Accuracy of the molecular mechanics calculations of organometallics are hampered by the fact that not many parameters have been introduced in such programs and often estimated parameters are used. In natural bite angle calculations3,39 it is assumed that it suffices to set the P–M–P bending frequency to zero and that other parameters are available to do an accurate MM2 calculation of the backbone energies. This would be true if only organic backbone atoms were involved, which are indeed accurately parameterized. Still several other force constants play a role and large errors may result. M–P–C bending force constants and dihedral force constants involving metal or phosphine should have relatively trustworthy values when the “natural” angles of the backbone don't fit the metal ligand bond.40 Furthermore, due to changes in electronics, for instance when considering cis and trans positions, the force constants will not be as general as they are in organic compounds.41
In order to estimate how much energy it might cost to enforce SPANphos to act as a cis ligand we have calculated the energies of the cis and trans isomers of two previously reported trans complexes, (SPANphos)PtCl2 and (SPANphos)Rh(CO)Cl, and a hypothetical (SPANphos)PdCl2, using a DFT method. DFT calculations were also done for the complexes containing PH2 as a model ligand instead of PPh2 in SPANphos. On the DFT level of calculation and the use of PH2 model substituents the energies of the cis and trans isomers are very similar, trans being more stable for rhodium and cis for palladium. In these PH2 models the lack of the steric bulk introduced by the phosphine substituent allows us to analyze the backbone strain and the cis/trans preference separately. Using an equivalent methodology, it was found previously that for PtCl2(PH3)2 in the gas phase the trans complex was consistently more stable, and only when solvents were included13 a preference for cis was found. In our case for PH2 model ligands, the cis/trans energy difference is quite small, and this suggests that the backbone strain in both isomers is similar. Due to the small energy difference, it may be concluded that in a polar environment also our PH2 models will prefer a cis configuration. Calculations on the full complexes revealed the additional effect of the phenyl phosphine substituents. In the gas-phase a preference for the trans configuration by 9.5 kcal.mol−1 for platinum dichloride, 14.8 kcal.mol−1 for palladium dichloride and 18 kcal mol−1 for the rhodium chloride monocarbonyl was found. The phenyl substituents affect stability and geometries as well. Note how the bite angle changed when the full systems were considered (Table 6). For the trans platinum dichoride complex the agreement between X-ray parameter (171.9°) and the computed geometry (171.5°) is excellent. Harvey calculated for PtCl2(PPh3)2 that the trans isomer should be 4.5 kcal.mol−1 more stable in the gas phase. For SPANphos we calculate a difference of 9.5 kcal.mol−1, part of which may result from the interaction of the neighbouring phenyl groups in the cis complex. From these data we conclude that at most 10 kcal.mol−1 backbone strain may be needed to enforce cis coordination of SPANphos. Normally such a difference would preclude the formation of such a complex, but the presence of another strongly bound ligand such as norbornadiene to cationic Rh(I) suffices to obtain cis complexes. A second provision to be made is that both calculations and experiments supply ample evidence that in polar solvents cis complexes are by far more stable, making cis complexes more readily accessible.
| (SPANphos)MCl2 | (SPAN phos)RhCl(CO) | ||||
|---|---|---|---|---|---|
| Relative energy/kcal mol−1 | trans | cis | trans | cis | |
| M = Pd | SPAN-PH2 | 2.4 | 0.0 | 0.0 | 2.2 |
| M = Pd | Full system | 0.0 | 14.8 | 0.0 | 17.9 |
| M = Pt | Full system | 0.0 | 9.5 | ||
| β angle/° | trans | cis | trans | cis | |
|---|---|---|---|---|---|
| M = Pd | SPAN-PH2 | 159.2 | 94.9 | 156.6 | 92.6 |
| M = Pd | Full system | 171.6 | 102.9 | 170.0 | 98.4 |
| M = Pd | Full system | 171.5 | 100.7 | ||
As mentioned in the introduction, for complexes that prefer a trans configuration for monodentate phosphines, such as Rh(CO)Cl and Pd(CH3)Cl, it is obvious that we find trans complexes when a bidentate ligand is used that can assume the trans conformation. The non-reactivity of trans-(SPANphos)Pd(CH3)+ in the common insertion chemistry of palladium cis diphosphine complexes suggested that cis configurations were not in reach energetically.17 Catalytic activity drops rapidly even when only a few kcal mol−1 are added as extra barrier. In view of the above results and the observations by Eberhard et al.15 it may be worthwhile to investigate whether the absence of activity is due to a high barrier of cis–trans isomerization, or a high energy of the cis complex.
Conclusions
The conformations found in the analyzed structures show that SPANphos ligands are highly flexible and can adopt different orientations depending on the needs of the formed molecules or complexes. The flexibility of the spirobichroman backbone is well-known, but initially we had thought that by the introduction of the bulky PR2 only trans or nearly trans complexes or bimetallic with transitions metals would be obtained. If the complex to be formed due to either a fixed geometry, (nbd)Rh+ providing an extreme case, or an intrinsic preference for cis complexes of some transition metal complexes, the spirobichroman ligand will adopt a conformation enabling the required orientation to form the cis structure. Everything being equal, SPANphos retains a preference for trans complexes, but the energetic preference is not as high as we initially thought.From a viewpoint of reactivity in catalysis the flexibility of the ligands may be even more interesting, as too rigid complexes may not be willing to undergo any reaction, as for instance the absence of oxidative addition of MeI to (SPANphos)Rh(CO)Cl or insertion reaction of palladium(II) SPANphos complexes. At present we are investigating ligands that may be more strongly trans directing than SPANphos and the reaction with (nbd)2Rh+ reported above provides a quick test to see whether we are on the right track!
References
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895 CrossRef CAS.
- T. J. Devon, G. W. Phillips, T. A. Puckette, J. L. Stavinoha and J. J. Vanderbilt, US Pat., 4, 694,109, 1987 (to Eastman Kodak) ( Chem. Abstr. , 1988 , 108 , 7890 ) Search PubMed.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081 CrossRef CAS.
- M. L. Parr, C. Perez-Acosta and J. W. Faller, New J. Chem., 2005 Search PubMed.
- A. J. Sandee, L. A. van der Veen, J. N. H. Reek, P. C. J. Kamer, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 3231 CrossRef CAS.
- A. Pintado-Alba, H. De la Riva, M. Nieuwhuyzen, D. Bautista, P. R. Raithby, H. A. Sparkes, S. J. Teat, J. M. Lopez-de-Luzuriaga and M. C. Lagunas, Dalton Trans., 2004, 3459 RSC.
- C. A. Bessel, P. Aggarwal, A. C. Marchilok and K. J. Takeuchi, Chem. Rev., 2001, 101, 1031 CrossRef CAS , and references therein.
- N. J. DeStefano, D. K. Johnson and L. M. Venanzi, Helv. Chim. Acta, 1976, 59, 2683 CAS.
- M. Sawamura, H. Hamashima and Y. Ito, Tetrahedron: Asymmetry, 1991, 2, 593 CrossRef CAS.
- Z. Freixa, M. S. Beentjes, G. D. Batema, C. B. Dieleman, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, J. Fraanje, K. Goubitz and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2003, 42, 1284 CrossRef CAS.
- R. C. Smith and J. D. Protasiewicz, Organometallics, 2004, 23, 4215 CrossRef CAS; R. C. Smith, C. R. Bodner, M. J. Earl, N. C. Sears, N. E. Hill, L. M. Bishop, N. Sizemore, D. T. Hehemann, J. J. Bohn and J. D. Protasievich, J. Organomet. Chem., 2005, 690, 477 CrossRef CAS; C. M. Thomas, R. Mafua, B. Therrien, E. Rusanov, H. Stoeckli-Evans and G. Süss-Fink, Chem. Eur. J., 2002, 8, 3343 CrossRef CAS; C. M. Thomas and G. Süss-Fink, Coord. Chem. Rev., 2003, 243, 125 CrossRef CAS; S. Burger, B. Therrien, Bruno and G. Süss-Fink, Helv. Chim. Acta, 2005, 88, 478 CrossRef CAS; J. I. van der Vlugt, R. Sablong, R. A. M. Mills, H. Kooijman, A. L. Spek, A. Meetsma and D. Vogt, Dalton Trans., 2003, 4690 RSC; N. H. T. Huy, P. Chaigne, I. Déchamps, L. Ricard and F. Mathey, Heteroat. Chem., 2005, 16, 44 CrossRef CAS.
- Some of these structures are unpublished results; namely [PdCl2(SPANphos)], [PdClMe(SPANphos)], [Pd(4-CNC6H4)Br(SPANphos)].
- J. N. Harvey, K. M. Heslop, A. G. Orpen and P. G. Pringle, Chem. Commun., 2003, 278 RSC.
- A. W. Verstuyft and J. H. Nelson, Inorg. Chem., 1975, 14, 1501 CrossRef CAS.
- M. R. Eberhard, K. M. Heslop, A. G. Orpen and P. G. Pringle, Organometallics, 2005, 24, 335 CrossRef CAS.
- Z. Freixa, P. C. J. Kamer, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2005, 44, 4385 CrossRef CAS.
- P. W. N. M. Van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, J. Am. Chem. Soc., 2003, 125, 5523 CrossRef CAS.
- G. M. Sheldrick, SHELXTL Crystallographic System Ver. 5.10, Bruker AXS, Inc., Madison, WI, 1998 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, C34; A. L. Spek, Platon - a Multipurpose Crystallographic Tool, Utrecht University; Utrecht, The Netherlands, 2003 Search PubMed.
- ArgusLab 4.0. Mark A. Thompson. Planaria Software LLC, Seattle, WA. http://www.arguslab.com Search PubMed.
- Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CrossRef.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At. Mol. Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- (a) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 34, 7406–7406 CrossRef; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- M. Wada and K. Sameshima, J. Chem. Soc., Dalton Trans., 1981, 240 RSC.
- A. W. Verstuyft, J. H. Nelson and L. W. Cary, Inorg. Chem., 1976, 15, 732 CrossRef CAS.
- P. S. Pregosin and R. Kunz, Helv. Chim. Acta, 1975, 58, 423 CrossRef CAS.
- R. T. Boeré, C. D. Montgomery, N. C. Payne and C. J. Willis, Inorg. Chem., 1985, 24, 3680–7 CrossRef CAS.
- The structure of the trace amount observed at δ ≈ 17 ppm remains undefined.
- W. H. Hersh, J. Chem. Educ., 1997, 74, 1485 Search PubMed.
- D. P. Fairlie and B. Bosnich, Organometallics, 1988, 7, 936 CrossRef CAS.
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS; (b) J. D. Dunitz and A. Gavezzotti, Angew. Chem., Int. Ed., 2005, 44, 1766 CrossRef CAS; (c) S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 3746 CrossRef CAS; (d) S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2002, 124, 104 CrossRef CAS; (e) M. O. Sinnokrot, E. F. Valeev and C. D. Sherill, J. Am. Chem. Soc., 2002, 124, 10887 CrossRef CAS; (f) M. O. Sinnokrot and C. D. Sherill, J. Phys. Chem. A, 2004, 108, 10200 CrossRef CAS; (g) X. Ye, Z.-H. Li, W. Wang, K. Fan, W. Xu and Z. Hua, Chem. Phys. Lett., 2004, 397, 56 CrossRef CAS; (h) E. J. Meijer and M. Sprink, J. Chem. Phys., 1996, 105, 8684 CrossRef CAS.
- K. Ejsmont, J. Kyziol, E. Nowakowska and J. Zaleski, Acta Crystallogr., Sect. C, 2000, 56, 93 CrossRef.
- C. P. Casey and G. T. Whiteker, Isr. J. Chem., 1990, 30, 299 CAS.
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519 RSC.
- D. Balcells, G. Drudis-Sole, M. Besora, N. Doelker, G. Ujaque, F. Maseras and A. Lledos, Faraday Discuss., 2003, 124, 429 RSC.
| This journal is © The Royal Society of Chemistry 2006 |