Engineering emissive europium and terbium complexes for molecular imaging and sensing†
Shashi
Pandya
,
Junhua
Yu
and
David
Parker
*
Department of Chemistry, Durham University, South Road, Durham, UK DH1 3LE. E-mail: david.parker@durham.ac.uk; Tel: +44 191 3342033
First published on 28th April 2006
Abstract
Emissive f-block coordination complexes constitute an important class of optical probes, with applications ranging from sensing of bioactive species, high throughput assays and screening protocols in vitro, to time-resolved imaging studies in cellulo or in vivo. The key chemistry issues to be addressed in complex design and characterisation are defined, with an emphasis on the use of emissive europium and terbium complexes and their conjugates in molecular imaging. Both luminescent ‘tags’ useful in energy transfer studies and ‘responsive’ systems for sensing are discussed.
 Shashi Pandya Shashi Pandya | Shashi Pandya obtained her B.Sc. (Chemistry) from Saurashtra University, India in 1991, her M.Sc. (Organic Chemistry) in 1993 and Ph.D. (Chemistry) in 1997 from Sardar Patel University, India. She then took up a lectureship (1997–2005) in Gujarat, India. She was on sabbatical leave (2000–2002) with Prof. Yannick Vallee (LEDSS-1, Grenoble, France). Recently she joined Prof. David Parker's group as a post-doctoral fellow. Her research area involves the synthesis and studies of molecules/complexes of diagnostic interest. |
 Junhua Yu Junhua Yu | Junhua Yu was born in China. He received his B.S. degree at Peking University in 1998, and his Ph.D. degree in 2003 from the Technical Institute of Physics and Chemistry, Chinese Academy of Sciences with Professor Baowen Zhang as his advisor. He is currently working at Georgia Institute of Technology in Atlanta following a period in Durham as a postdoctoral fellow. His research interests include photoinduced energy transfer and its application to chemical probe design. |
 David Parker David Parker | David Parker was educated in Durham, Oxford and Strasbourg, gaining a D.Phil. in Oxford with John Brown. Following a NATO Fellowship with Jean-Marie Lehn, he took up a Lectureship at Durham in 1982 and was promoted to a Chair in Chemistry ten years later. He completes his second term as Departmental Chairman this year. His research interests span many aspects of functional complexation chemistry, with particular attention to stereochemical phenomena and metal complex synthesis and design. |
Introduction
Luminescent molecular probes that are used as sensor materials or in imaging applications are mostly based on well established condensed heterocyclic dyes, such as fluorescein or rhodamine.1,2 They are fluorescent and typically possess a short lived singlet excited state with a lifetime of up to a few nanoseconds. Absorption and emission bands are broad, with bandwidths of the order of tens of nm, and they usually possess rather small shifts between these bands, i.e. a small Stokes shift. The wide range of useful and commercially important fluorescent probes has been expanded and enhanced over the past two decades by the introduction of Alexa Fluor® and BOPIPY® dyes,2,3 the latter based on the charge neutral 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene core, 1. These dyes are more resistant to photobleaching (a light induced change in a chromophore, resulting in loss of absorption of light at a given wavelength), exhibit a pH-independent fluorescence and possess high extinction coefficients (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) with emission quantum yields that are often >0.7. Absorption and emission wavelengths can be determined by appropriate substitution of the core fluorophore.3 These dyes, or their bio-conjugates, have some limitations. They are prone to interference from light scattering in biological media (Rayleigh, Tindall or Raman) and from the natural fluorescence of certain biomolecules, as well as being limited by occasional technical problems (e.g. autofluorescence) associated with their small Stokes shift. Such limitations have driven the search for improvements in luminescent probes over the past few years. Important advances have been made in the controlled synthesis and enhancing photo-stability of the strongly absorbing (ε > 106 M−1 cm−1) semi-conductor quantum dots’ based on e.g. InN, CdS and CdSe. These materials also possess emission bands whose bandwidth is a function of particle size, allowing an element of control that is absent in the traditional organic dyes.4–6
000 M−1 cm−1) with emission quantum yields that are often >0.7. Absorption and emission wavelengths can be determined by appropriate substitution of the core fluorophore.3 These dyes, or their bio-conjugates, have some limitations. They are prone to interference from light scattering in biological media (Rayleigh, Tindall or Raman) and from the natural fluorescence of certain biomolecules, as well as being limited by occasional technical problems (e.g. autofluorescence) associated with their small Stokes shift. Such limitations have driven the search for improvements in luminescent probes over the past few years. Important advances have been made in the controlled synthesis and enhancing photo-stability of the strongly absorbing (ε > 106 M−1 cm−1) semi-conductor quantum dots’ based on e.g. InN, CdS and CdSe. These materials also possess emission bands whose bandwidth is a function of particle size, allowing an element of control that is absent in the traditional organic dyes.4–6
Metal coordination complexes comprise another important class of luminescent probes and considerable efforts have been made to exploit their unique excited state properties.7 In this article, attention is focussed on lanthanide complexes, and consideration of the key issues to be addressed before coordination complexes can be engineered to act as practicable sensors or as imaging probes for microscopy.8–14
The excited state lifetimes of lanthanide ions range from the order of microseconds for the rear-IR emitting lanthanides (Yb(III), Er(III) and Nd(III))8 to milliseconds for Eu(III) and Tb(III) complexes. These long lifetimes immediately offer an advantage as they allow time-resolved spectroscopy and microscopy to be used. The introduction of a time delay (say 1 or 100 µs), prior to detection of the emitted light eliminates interference from light scattering and auto-fluorescence and greatly enhances the signal/noise ratio (detection limits are conservatively in the 10−12–10−15 M range) and hence the reliability of detection and monitoring.15,16 Such features encouraged the development in the 1980's of various time-resolved assays, using lanthanide components, first commercialised by Wallac-Oy in Finland following the innovative work of Hemmila and Soini in heterogeneous systems.16 Homogeneous assays followed, developed mainly by CIS-bio International in France.15
Lanthanide emission bands are very sharp compared to organic fluorophores, typically with a full width at half maximum of less than 10 nm. To a first approximation, the metal-based emission profile is similar for each lanthanide complex. However, for Eu(III) and Yb(III) complexes in particular, the form and relative intensity of the main emission bands are a sensitive function of the local coordination environment, allowing the signalling of changes in metal complex speciation12c,17–20 by ratiometric analysis.
Every lanthanide complex requires the incorporation into the ligand structure of a sensitising chromophore, capable of transferring its excited state energy to the encapsulated lanthanide(III) ion (Scheme 1). This is essential as direct excitation of the Ln3+ ion is very inefficient, leading to low values of ε (≤1 M−1 cm−1) as the relevant f–f transitions are parity (Laporte) forbidden. Since the early work on single component emissive systems of Lehn, Mathis and Alpha,21 an extensive range of aromatic and heterocyclic chromophores has been examined. These moieties need to be engineered into the ligand and serve as an antenna to harvest incident light. Detailed analyses of their structural and mechanistic features have been reviewed in depth8,12,13,15–17,20 elsewhere.
 | ||
| Scheme 1 | ||
The essential requirements for this sensitising moiety can be summarised as follows: a high extinction coefficient at an excitation wavelength appropriate for the application—e.g. in the range 337–420 nm for single photon excitation of biological samples with Eu3+/Tb3+ systems; a fast inter-system crossing step to ensure the aromatic triplet is populated efficiently—minimising ligand fluorescence; a fast energy transfer step leading to population of the lanthanide excited state; a seven- to nine-coordinating ligand to encapsulate the Ln(III) ion, minimising the quenching of the lanthanide excited state by vibrational energy transfer to matched oscillators, e.g. O–H, N–H or C–H stretching vibrations.
The lanthanide excited state may persist for several milliseconds, during which it is prone to quenching by electron or energy transfer processes. The 5D0 and 5D4 excited states of Eu3+ and Tb3+, for example, lie at 206 (17200 cm−1) and 244 kJ mol−1 (20400 cm−1) above the ground state. This free energy can be used to drive various electron transfer processes, for example in a collisional encounter with electron rich species such at I−, urate or ascorbate (Eox < 0.5 V).22 The latter two anions are ubiquitous low molecular weight anti-oxidants in many biosystems, typically existing in 0.1 to 2 mM levels in different cell types.23 Inhibition of this quenching process requires that electron transfer to the complex be disfavoured, either by anionic repulsion, e.g. introducing peripheral negatively charged groups to suppress collisional encounters, by adding a very high local concentration of a non-oxidisable anion to the medium (e.g. > 0.1 M F−),15 or by reducing the electron affinity of the overall complex.
Recent examples of highly emissive lanthanide complexes absorbing above 340 nm, 2,24 [Ln·3]14 and 4,22 serve to illustrate these design issues of desirable sensitiser characteristics and appropriate ligand structure. The absorption and total emission spectrum of 2 (Fig. 1), highlights the relatively long excitation wavelength (λexc 384 nm), shows that inter-system crossing is competitive with fluorescence from the aza-thiaxanthone singlet excited state (residual ligand fluorescence at 450 nm) and reveals the highly structured red europium emission spectrum, which can be observed selectively upon application of a 100 µs time delay in acquisition.
 | ||
| Fig. 1 Absorption and total emission spectra for the aqua–europium complex, 2, (λexc 384 nm, ϕem 9% H2O) highlighting the short-lived ligand-based fluorescence and the long-lived europium luminescence, the latter being observed uniquely when a 100 µs time delay is used (right). | ||
Critical design features for luminescent probes
Emissive lanthanide complexes that are to be used either as ‘responsive’ systems or as luminescent ‘tags’ in bio-conjugates, for example as fluorescence resonant energy transfer (FRET) components, must be designed with the following features in place, in order to gain generic utility. A high overall emission quantum yield (≥10% in aqueous media) is needed following excitation into a strongly absorbing chromophore at wavelengths greater than 337 nm. Of particular interest are excitation wavelengths that correspond to readily available laser lines (337, 355 or 405 nm for Eu/Tb; 532 nm for Yb/Nd), or are close in energy to the growing range of powerful light emitting diodes, e.g. 365 or 390 nm. Some of these LEDs now have power-ratings in the 100 milliwatt range. This requires a small singlet triplet energy gap for Eu- and especially Tb-sensitised emission. Suitable chromophores include quinolones, tetraazatriphenylenes,25 substituted bipyridyls/terpyridyls and their N-oxides,9,15,16,26 xanthones and their derivatives24 (acridones are often limited by inefficient triplet population in polar media).27,28 The ligand should be synthesised efficiently, from readily available precursors and at reasonable cost, and the complex should be thermodynamically and kinetically stable with respect to metal ion loss, photofading and oxidative degradation in air or in aqueous media. When the lanthanide complex is to be used as an emissive ‘tag’, it must possess a suitable functional group to expedite conjugation, e.g. a pendant acid or primary amine group. Recent examples include 5,96 (Fig. 2) and 7. The simple permutation of the lanthanide ion allows bi- and multimodal analyses to be undertaken, e.g. using emissive Eu/Tb/Sm and Dy analogues differing in emission lifetime and spectral output for multi-parametric analysis.8,30,31 In addition, Eu/Tb and Gd complexes of a given ligand may be expected to show similar biodistribution and/or cellular uptake profiles so that combined MRI and optical imaging protocols become possible with a ‘common’ complex or conjugate.32–34 A recent example is provided by 8, in which the conjugated o-chlorophenylquinoline moiety targets (with apparent nanomolar affinity) a peripheral benzodiazepine receptor that is over-expressed in glioblastoma, breast cancer and Alzheimer's disease.29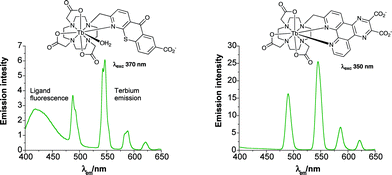 | ||
| Fig. 2 Emission spectra for selected terbium complexes (298 K, 0.1 M NaCl), showing residual ligand fluorescence from the aza-thiaxanthone chromophore (left). | ||
Using lanthanide complexes for imaging in cellulo
Given the favourable impact that time-resolved homogenous assays has had in analytical biochemistry and high throughput screening,15,16 a topical challenge is to devise complexes that luminesce efficiently within living cells. Extensions to in vivo work can then be confidently undertaken, whilst appreciating that light in the 710–820 nm window penetrates skin and tissue most effectively. This may limit developments in vivo to the use of near-IR emitting luminophores (e.g. cyanine dyes and Nd3+/Yb3+ complexes), unless effective two-photon excitation protocols are devised for Eu3+/Tb3+ luminescent probes. Two different classes of probe may be distinguished at the outset: stable luminescent complexes that serve as ‘tags’ in bioconjugates for use in FRET or ‘tracking’ assays, and responsive systems wherein the spectral emission profile, lifetime or circular polarisation varies as a function of the local concentration of a target intracellular analyte. In each case, the complex or conjugate must resist excited state quenching by local donors/acceptors. In this respect, suppressing quenching of the lanthanide excited state by photoinduced electron transfer from ascorbate, urate or glutathione is again likely to be important in defining practicable systems.22An obvious pre-requisite for in cellulo imaging is that the complex must be non-toxic and cell permeable, preferably with a distinctive compartmentalisation profile, i.e. a preference to localise in a given organelle.2 For conjugates, this localisation is likely to be determined by the nature of the protein/peptide/synthetic vector, with cell-penetrating peptides, e.g. penetratin and its analogues offering much scope in this respect.35 However, many of these systems recognise a cell surface receptor and use receptor mediated endocytosis as the mechanism of cell entry, so that the conjugate then resides in an endosome (or a related vesicle structure). Endosomal escape is then essential, if an organelle is being targeted or if the cytosol needs to be labelled, otherwise the endosome will age to become a lysosomal compartment (low pH, enhanced likelihood of degradation), isolated from the rest of the cell. For example, the use of 8 (Eu complex, λexc 320 nm) to label glioblastoma cells (Fig. 3) is believed to follow an endocytosis mechanism,29 consistent with the finding that uptake was minimal at 4 °C. It was suggested that this conjugate might localise in the outer mitochondrial membrane, where the target receptors are expressed, although this was not unequivocally demonstrated. Earlier work from the same group had highlighted the opportunities in optical imaging of surface lesions using a related Tb–pyridyl complex (λexc 280 nm), following topical administration using a hamster oral cancer model.36
 | ||
| Fig. 3 Epifluorescence microscopy images of C6 glioblastoma cells; (left) following incubation with 8 (Ln = Eu, λexc 320 nm, 60 minute incubation 25 µm complex); (right) is a blank control. | ||

Tracking changes in Zn2+ concentrations in real time in living cells is an important current area of study in bio-inorganic chemistry.37,38 Bifunctional europium complexing agents have been defined incorporating a zinc binding moiety so that the observed luminescence emission intensity increases as a function of [Zn2+].39,40 Using non-ratiometric methods, proof-of-principle zinc-dependent imaging studies have been reported with 9 (λexc 320–340 nm) following direct injection of the complex into HeLa cells. Other workers interested in plant nutrition have examined direct uptake of Eu3+ aqua species in oat roots and interrogated the process using laser excitation time-resolved confocal microscopy.41
Predicting cellular compartmentalisation profiles for low MW luminescent species is not straightforward. A good example is provided by the differing behaviour of the constitutionally isomeric complexes 10 and 11.42,43 In mouse skin fibroblasts (NIH-3T3 cells), confocal microscopy has been used to examine the intracellular distribution of these complexes. They were developed to report upon local concentrations of HCO3− by measuring the changes in the intensity ratio of two (or more) Eu emission bands. Bicarbonate is an important intracellular species as it binds reversibly to the enzyme adenylylcyclase, thereby regulating release of the key secondary messenger cyclic-AMP. The two isomeric complexes 10 and 11 (λexc 405 nm) exhibit very different behaviour in cellulo! The ‘C-linked’ isomer (Fig. 4) distributes quite well in the cytosol and shows a tendency to stain the endoplasmic reticulum revealed in co-staining experiments with a conjugate of Brefeldin A. The ‘N-linked’ complex 11, in contrast, is taken up by an endosonal pathway, and in co-localisation studies with Lysotracker-green, exhibits good correspondence with this organic dye that is known to localise in the acidic lysosomal compartment. Useful bicarbonate analyses are therefore precluded in this case (pKa ∼6.2 for HCO3−/H2O–CO2).
 | ||
| Fig. 4 Comparison of confocal fluorescence microscopic images of constitutionally isomeric Eu complexes 10 and 11 in NIH-3T3 cells highlighting their differing localisation profiles. | ||
The citrate anion is a key component of many biological systems including fatty-acid synthesis, nitrogen metabolism and photorespiration. Simple terbium salts, such as the citrate, have been noted to bind in vitro to single-stranded nucleic acids, as revealed by the enhancement of terbium emission intensity following binding to RNA.44 In addition, terbium citrate has been used to examine the visualisation of RNA by electron microscopy. Using ultra-thin sections staining of nucleoli, interchromatin and perichromatin granules and coiled bodies in the cell nucleus have been revealed, whereas ribosomes are the only contrasted structures in the cytoplasm.45 A pH-insensitive ratiometric chemosensor for citrate has been reported,46 in which the europium emission spectral form changes as a function of local citrate concentration in competitive media (Kd = 0.28 mM), allowing ratiometric analysis (λexc 384 nm). This complex is also taken up by live cells via a non-endocytotic pathway, and stains the intranuclear cell nucleoli with some selectivity (Fig. 5). Control experiments suggested that the complex is not binding to RNA itself (addition of RNA quenches Eu emission in vitro). The local speciation of the europium complex, 2, has been probed by examining the spectroscopic signature of the complex in cells, and the integrity of the sensitiser in the complex confirmed by excitation spectroscopy. The species observed in cellulo by confocal microscopy is certainly not a simple RNA, aqua or citrate adduct, but may be a protein-bound entity.47
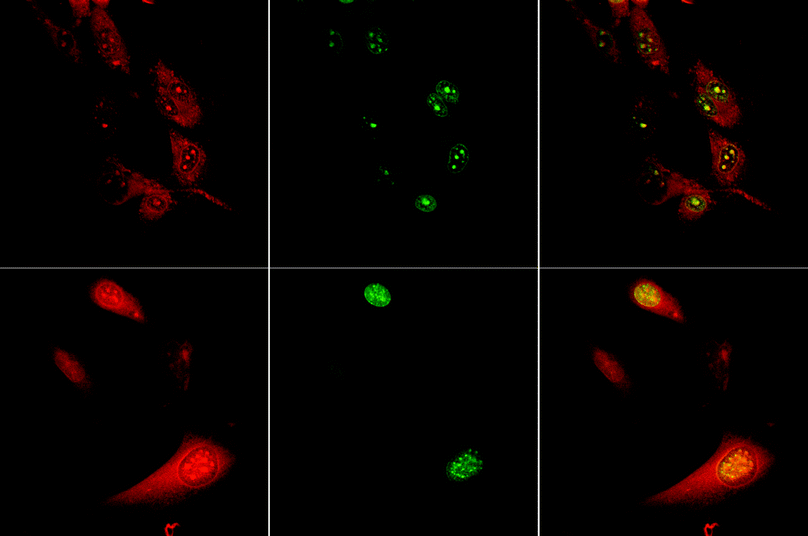 | ||
| Fig. 5 Confocal fluorescence microscopy study showing the cellular localisation behaviour of the europium complex 2 (λexc 405 nm) in living (upper row) and fixed (lower) cells, revealing the staining of the nucleolus inside the cell nucleus, consistent with the observed co-localisation of the green fluorescent dye ‘SYTO-RNA-select’. | ||
The amphipathic cationic lanthanide complexes related to 12 and incorporating an efficient tetraazatriphenylene chromophore, (λexc 350 nm) also exhibit good cell uptake and a strong intracellular emission profile.22 Originally, these enantiopure complexes were examined in vitro for their ability to bind the most common helical form of DNA (B-DNA) stereoselectively48 (Ka > 106 M−1 with different affinities for Δ
vs. Λ isomers), but cell localisation work in HEK (embryonic kidney) 3T3, COS and CHO cells revealed good cell penetration. At higher concentrations of complex in the incubating medium, the complex localises in the cell nucleus (perhaps DNA-bound), and egress of the complex from the nucleus back to the extranuclear organelles has been observed by confocal microscopy. Cell uptake is almost non-existent for the corresponding charge neutral and anionic analogues of 12 (i.e. with carboxymethyl or ‘glutarate’ side chains). Given that the cationic complex also localises well at 4 °C and binds fairly strongly to albumin in the cell growth medium (effective Kd
∼ 0.1 mM), it may be entering the cell via a pore or channel possibly as a reversibly bound protein adduct.

Many examples have been reported of the use of conjugates of europium complexes in high throughput screening, cell surface labelling or FRET studies.8b,15,16 Several examples15 relate to the use of the europium cryptates (λexc 337 nm), for example in FRET systems labelled with Alexa-Fluor 647 (as an acceptor) to analyse membrane protein–protein interactions at the surface of living cells.15d Examples include the use of europium-labelled annexin V to study the cell-death cascade in adherent cell cultures49 and a ‘europium tagged’ antibody in the screening of inhibitors that block phosphorylation of the tumour-suppressor retinoblastoma protein.50 In contrast to some earlier work using the effective DTPA–carbostyril8b,51,52 conjugates described by Selvin(λexc 337 nm), some reports use very inefficient sensitisers such as 4-aminosalicylate53a or a simple anilide53b (λexc 325/320 nm), exciting at the long-wavelength edge of each absorption band. Finally, the lipophilic europium complex europium tris(thenoyltrifluoro-acetonate), has been shown to stain liposomal membranes.53c The emission intensity was temperature dependent in the range 15–40 °C, suggesting its use in cellular thermal imaging, for example as a consequence of the local enthalpy change following activation of the muscarinic receptor by introduction of acetyl choline.54 Issues of calibration (observed intensity is a function of local complex concentration) and europium speciation have not been overtly clarified.
Lanthanide complexes as functional components of sensors operating in aqueous media
In devising good sensors based on the long lived emission of lanthanide complexes additional design criteria and practical considerations need to be borne in mind and several recent reviews have highlighted some of these issues.12,13,55–57 Most published reports are concerned with issues of chemo-selectivity in binding, often under particular solvent conditions which bear little relation to the analysis of a target species in an operational sensor. Two different cases can be considered. First, sensors that are spatially localised are required for flow-analysis and immobilisation in laterally structured thin layers, e.g. on silicon with pre-determined integrated electronics for signal read-out, or in a sol–gel glass for extended operational lifetimes. An intrinsic feature here is that a wide range of signal transduction mechanisms can be employed, provided that the sensing moiety (Ln complex) is stable to storage, adheres to the support and does not leach, resists photo-degradation and gives a strong signal with high signal/noise ratio. Intensity, lifetime, phase modulation or emission polarisations are all practicable parameters to monitor if these criteria are met. For systems that involve ‘freely mobile’ probes in solution—as needed for certain in cellulo/in vivo and environmental applications—then the analytical signal must be referred intrinsically to another signal. This avoids a dependence on concentration for emission intensity measurements and can be another emission or excitation wavelength, thereby giving rise to a ratiometric sensor.Immobilised lanthanide sensors
An optical sensor for hydrogen peroxide has been devised by immobilising a rather ill-defined europium tetracycline complex (λexc 405 nm) in a polyurethane hydrogel. The europium emission intensity increases linearly in the range 2–200 µM and is accompanied by a lifetime increase, but is subject to interference by citrate and phosphate.58 Coupled assays for glucose and glucose-oxidase have been developed, as well as a fluorescence imaging method for monitoring glucose oxidase activity in vitro.59 Flow-injection analyses for the broad-spectrum antimicrobial agents norfloxacin and ciprofloxacin (each is a quinolone) have been developed.60,61 Here, terbium ions are simply immobilised on a cationic Sephadex resin and luminesce when reversibly bound to these quinolones in the flowing analyte, with a working range of 10–150 ng L −1.Sol–gel matrices are excellent supports for sensor immobilisation due to their inherent mechanical and chemical stability, optical transparency and case of formation. Typical fabrication conditions require ageing of the sol–gel precursors (e.g. Si(OR)4, R = Me or Et) under acidic conditions (effective pH in range 2–3), so that kinetic stability of the complex with respect to acid-catalysed dissociation is essential. Robust sensors for pH62,20 based on Eu and Tb complexes with a phenanthridinium chromophore have been devised (e.g.13, Fig. 6) for use in static or flow analyses and a stable pH response (range of pH 5 to 8) was observed for periods of at least 6 months. In this case, protonation of the phenanthridine N leads to suppression of the photoinduced electron transfer (PET) process that quenches the sensitiser singlet excited state,20 resulting in enhanced Eu emission. Related examples of ratiometric changes in excitation spectra have been defined with similar immobilised terbium complexes,62 where the 370/311 nm intensity ratio varied by a factor of 8 over the pH range 4 to 8. The sol–gel matrix is highly suitable for pH and oxygen sensors, due to the ease of proton or O2 transport through the medium. Examples of oxygen sensors include the terbium complex 14, for which excellent sensor stability and performance was reported,63 based on the analysis of changes in the lifetime of terbium emission intensity of 545 nm (λexc 370–375 nm). In this example, reversible back-energy transfer from the terbium ion to the phenanthridinium triplet leads to competitive dynamic quenching of the long-lived triplet by oxygen, signalled by variations in terbium intensity and lifetime (N.B. the Eu analogue is insensitive to pO2 variations, suggesting a simple ratiometric analysis of emission intensity in co-complexes).30
 | ||
| Fig. 6 Variation of the europium emission intensity of complex 13 (λexc 365 nm, λem 617 nm) with pH, following immobilisation of the complex in a sol–gel matrix and fabrication of an optically transparent thin film sensor for flow-injection analysis.20,62 | ||
Thin film applications have also been devised with more lipophilic lanthanide complexes. An ultrathin polymer optode has been reported involving Langmuir-Blodgett (LB) films of poly-dodecylacrylamide and the Eu–phen complex 15,64 which operates as a temperature sensor through variations of the intensity of the hypersensitive Eu emission band at 613 nm. Related work has explored LB films of octabutyl bis(phthalocyaninate) rare earth complexes (Pr, Gd, Ln) as components of fibre optic sensors.65 The near-IR absorption spectra of the Ln complexes changed form in the presence of nitrogen oxide gases and certain volatile aldehydes and esters. No emission variations were reported.
Lanthanide sensory systems operating in solution
The photophysical pathway defining sensitised lanthanide luminescence (Fig. 1 above) offers three ‘windows of opportunity’ for sensing, as either the two short-lived excited states of the sensitiser or the lanthanide excited state may be perturbed. A more detailed analysis has been described66,12,20 and further clear reviews of the chemistry of such systems reported.56,13,16 Related work highlighting the scope of chiroptical analyses has also been defined,67,68 encouraged by the strong circularly polarised luminescence of chiral lanthanide complexes.12,69 In addition, systems exhibiting well defined sensitivity to two or more analytes, either intrinsically30,70 or by the introduction71–73 of 2 receptor sites in the complexing agent have been studied. For example, complexes 1672 and 1773 exhibit emission intensity variations in the presence of H+/Cu2+ and H+/K+ respectively, allowing their consideration as components of optical logic gates.71Perhaps the most challenging work at present addresses anion sensing in aqueous media, as the large free energies of anion hydration, pH-dependent speciation and geometrical similarity of many common anions renders the development of chemoselective systems difficult. Various strategies are being explored and many involve reversible binding to the lanthanide centre.74,75 They range from a europium-containing iron–sulfur cluster species showing apparent ‘selectivity’ for phosphate species,76 to allegedly selective sensors for 2,3-bisphosphoglycerate based on a ditopic receptor77 (λexc 280 nm, MeOH–MeCN). A suitable heptadentate ligand incorporating an appropriate sensitiser will usually form either a CN = 9 diaqua or a CN = 8 mono-aqua complex. In each case, the bound waters may be displaced by certain anions. Anion affinity is determined by the charge,43 polarisability18 and chelating ability of the different species.19,75
The charge density and steric demand at the lanthanide centre are also key issues and are complex dependent, giving rise to differing anion affinity profiles for each lanthanide complex. Thus, complex 2 binds citrate strongly and selectively46 and has a low affinity for HCO3− whereas 10 and 11 favour HCO3− in ratiometric analysis (λexc 405 nm). Complexes related to 10 and 11, but lacking the ring N-alkyl substituent show ca. 40 : 1 chemoselectivity for phosphorylated-tyrosine residues over phospho-serine/threonine sites in phosphorylated-peptides.78,79
A key point to emphasise is that the desired chemoselectivity and affinity can be engineered into the lanthanide complex, according to the problem posed by the nature of the interferents presented in the chosen analyte. Thus, caution must be exercised in assessing the significance of a complex reputedly ‘sensing’ a given anion, unless the potential anionic interferents in the sample to be addressed are properly examined.80–82
Finally, a europium complex, 18, has been designed to report on the presence of singlet oxygen.83 The reaction of singlet oxygen, 1ΔO2, with substituted anthracenes generates the related endo-peroxide. In complex 18, europium emission is quenched by intramolecular triplet–triplet energy transfer between the terpyridyl moiety and the anthracene. Triplet anthracene is unable to sensitise Eu emission as its energy lies well below that of the 5D0 Eu emissive state (206 kJ mol−1). This quenching pathway is not available in the endo-peroxide adduct so that each encounter of 18 with a molecule of 1ΔO2 switches on the Eu luminescence. Detection limits of 2–8 nM were reported (λexc 337 nm), rendering the system suitable for snapshot imaging purposes, provided that the retro-cyloaddition reaction is not competitive on the experimental timescale. The absence of reversibility of the cyclo-addition, of course, limits the application, as an in situ calibration of measured Eu emission intensity is essential.

Conclusions
Developments in molecular imaging optical, MRI and PET probes are set to enhance our ability to monitor key biological processes in cellulo and assess injury and disease states in vivo. There remains much to be done to devise emissive probes based on metal-coordination complexes, taking advantage of time-resolved sensing and imaging methodology. Key issues will be to understand better excited-state quenching of the lanthanide excited state, to gain a working structure/activity appreciation of the permeability of emissive, responsive complexes in cellulo, to encourage the development of available instrumentation for time resolved multiphoton microscopy and to work in harmony with bioscientists, addressing their challenges by using systems in which the understanding of the chemistry of the probes is secure.Acknowledgements
The work in Durham is supported by the Royal Society, EPSRC, the EC networks of excellence EMIL and DIMI, and the STBE/Durham County ONE-NE partnership.References
- Y. Urano, M. Kamiya, K. Kanda, T. Ueno, K. Hirose and T. Nagano, J. Am. Chem. Soc., 2005, 127, 4888 CrossRef CAS; T. Ueno, Y. Urano, K. Setsukinai, H. Takakusa, H. Kojima, K. Kikuchi, K. Ohkubo, S. Fukuzumi and T. Nagano, J. Am. Chem. Soc., 2004, 126, 14079 CrossRef CAS.
- http://www.probes.com and http://www.invitrogen.com; R. P. Haugland, A Guide to Fluorescent Probes and Labelling Technologies, Molecular Probes, Eugene, Oregon, 10th edn, 2005 Search PubMed.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- W. C. W. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40 CrossRef CAS.
- (a) P. Mitchell, Nat. Biotechnol., 2001, 19, 1013 CrossRef CAS; (b) U. Hasegawa, S. M. Nomura, S. C. Kaul, T. Hirano and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2005, 331, 917 CrossRef CAS.
- Many important d-block complexes of Ru, Pd with bipyridines/porphyrins have found use as oxygen sensors and are being examined for use in vivo, e.g.: M. Paxian, S. A. Keller, B. Cross, T. T. Huyuh and M. G. Clemens, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 286, 637 Search PubMed; J. N. Demas and B. A. DeGraff, Coord. Chem. Rev., 2001, 211, 317.
- (a) S. Faulkner, S. J. A. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1 CrossRef CAS; (b) P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275 CrossRef CAS.
- (a) R. Ziessel and L. J. Charbonniere, J. Alloys Compd., 2004, 374, 283 CrossRef CAS; (b) N. Weibel, L. J. Charbonniere, M. Guardigli, A. Roda and R. Ziessel, J. Am. Chem. Soc., 2004, 126, 4888 CrossRef CAS.
- M. Q. Tan, Z. Q. Ye, G. L. Wang and J. L. Yuan, Chem. Mater., 2004, 16, 2494 CrossRef CAS.
- Z. Q. Ye, M. Q. Tan, G. L. Wang and J. L. Yuan, Talanta, 2005, 65, 206 CAS.
- (a) D. Parker, Coord. Chem. Rev., 2000, 205, 109 CrossRef CAS; (b) D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977 CrossRef CAS; (c) D. Parker, Chem. Soc. Rev., 2004, 33, 156 RSC.
- T. Gunnlaugsson and J. P. Leonard, Chem. Commun., 2005, 3114 RSC.
- S. Petaud, S. M. Cohen, J.-C. G. Bunzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324 CrossRef CAS.
- (a) G. Mathis, Clin. Chem. (Washington, D. C.), 1993, 39, 1953 CAS; (b) G. Mathis, Clin. Chem. (Washington, D. C.), 1995, 41, 1391 CAS; (c) H. Bazin, M. Préaudat, E. Trinquet and G. Mathis, Spectrochim. Acta, 2001, A57, 2197 CrossRef; (d) D. Maurel, J. Kriazeff, G. Mathis, E. Trinquet, J. P. Pin and H. Ansanay, Anal. Biochem., 2004, 329, 253 CrossRef CAS; (e) M. Gabourdes, V. Bourgine, G. Mathis, H. Bazin and B. Alpha-Bazin, Anal. Biochem., 2004, 333, 105 CrossRef CAS.
- I. Hemmila and V.-M. Mukkala, Crit. Rev. Clin. Lab. Sci., 2001, 38, 441 Search PubMed; V. W. W. Yam and K. K. Lo, Coord. Chem. Rev., 1999, 184, 157 CrossRef ; for earlier work see: E. Soini and I. Hemmila, Clin. Chem. (Washington, D. C.), 1979, 25, 353 Search PubMed; H. Sitari, I. Hemmila, K. Pettersson and T. Lovgren, Nature (London), 1983, 301, 258 CAS.
- K. Matsumoto and J. Yuan, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker, New York and Basle, 2003, vol. 40, p. 191 Search PubMed.
- R. S. Dickins, A. S. Batsanov, J. A. K. Howard, D. Parker, H. Puschmann and S. Salamano, Dalton Trans., 2004, 70 RSC; R. S. Dickins, J. I. Bruce, D. Parker and D. J. Tozer, Dalton Trans., 2003, 1264 RSC.
- R. S. Dickins, S. Aime, A. S. Batsanov, A. Beeby, M. Botta, J. I. Bruce, J. A. K. Howard, C. S. Love, D. Parker, R. D. Peacock and H. Puschmann, J. Am. Chem. Soc., 2002, 124, 12697 CrossRef CAS.
- D. Parker and J. A. G. Williams, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker, New York and Basle, 2003, vol. 40, p. 233 Search PubMed.
- B. Alpha, J.-M. Lehn and G. Mathis, Angew. Chem., Int. Ed. Engl., 1987, 26, 266 CrossRef.
- R. A. Poole, G. Bobba, M. J. Cann, J.-C. Frias, D. Parker and R. D. Peacock, Org. Biomol. Chem., 2005, 3, 1013 RSC.
- J. Chen, L. Gorton and B. Ahessan, Anal. Chim. Acta, 2002, 474, 137 CrossRef CAS; J. M. Zen and C. T. Hsu, Talanta, 1998, 46, 1363 CrossRef CAS; S. Chevion, E. M. Berry, N. Kitrosky and R. Kohen, Free Radical Biol. Med., 1997, 22, 411 CrossRef CAS.
- D. Parker and J. Yu, Chem. Commun., 2005, 3141 RSC.
- B. H. Bakker, M. Goes, N. Hoebe, H. J. van Ramesdonk, J. W. Verhoeven, M. H. V. Werts and J. W. Hofstraat, Coord. Chem. Rev., 2000, 208, 3 CrossRef CAS.
- G. Song, G. Wang and J. Yuan, Chem. Commun., 2005, 3553 RSC.
- A. Dadabhoy, S. Faulkner and P. G. Sammes, J. Chem. Soc., Perkin Trans. 2, 2000, 2359 RSC; A. Dadabhoy, S. Faulkner and P. G. Sammes, J. Chem. Soc., Perkin Trans. 2, 2002, 348 RSC.
- Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Chem. Commun., 2002, 1930 RSC.
- H. C. Manning, T. Goebel, R. C. Thompson, R. R. Price, H. Lee and D. J. Bornhop, Bioconjugate Chem., 2004, 15, 1488 CrossRef CAS.
- D. Parker, in Supramolecular Science: Where it is and Where it is Going, ed. R. Ungaro and E. Delcanale, Kluwer, Dordrecht, 1999, vol. C527, pp. 53–67 Search PubMed.
- A. E. Soini, A. Kuusisto, N. J. Meltola, E. Soini and L. Sevens, Microsc. Res. Tech., 2003, 62, 396 CrossRef CAS; J. Karvinen, A. Elomaa, M. L. Makinen, H. Hakala, V. M. Mukkala, J. Peuralahti, P. Hurskainen and I. Hemmila, Anal. Biochem., 2004, 325, 317 CrossRef CAS.
- M. M. Huber, A. B. Staubli, K. Kustedjo, M. H. B. Gray, J. Shih, S. E. Fraser, R. E. Jacobs and T. J. Meade, Bioconjugate Chem., 1998, 9, 242 CrossRef CAS.
- M. Modo, D. Cash, K. Mellodew, S. C. R. Williams, S. E. Fraser, T. J. Meade, J. Brie and H. Hodges, NeuroImage, 2002, 17, 803 CrossRef.
- L. Josephson, M. F. Kircher, U. Mahmood, Y. Tang and R. Weissleder, Bioconjugate Chem., 2002, 13, 554 CrossRef CAS.
- P. E. G. Thoren, D. Persson, E. K. Esbjorner, M. Goksor, P. Lincoln and B. Norden, Biochemistry, 2004, 43, 3471 CrossRef CAS.
- D. J. Bornhop, J. M. M. Griffin, T. S. Goebel, M. R. Sudduth, B. Bell and M. Motamedi, Appl. Spectrosc., 2003, 57, 1216 CrossRef CAS.
- K. Komatsu, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2005, 127, 10197 CrossRef CAS.
- K. Hanaoka, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2004, 126, 12470 CrossRef CAS.
- K. Hanaoka, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, Angew. Chem., Int. Ed., 2003, 42, 2996 CrossRef CAS.
- O. Reany, T. Gunnlaugsson and D. Parker, J. Chem. Soc., Perkin Trans. 2, 2000, 1819 RSC; O. Reany, T. Gunnlaugsson and D. Parker, Chem. Commun., 2000, 473 RSC.
- R. J. Fellows, Z. M. Wang and C. C. Ainsworth, Environ. Sci. Technol., 2003, 37, 5247 CrossRef CAS.
- Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Chem. Commun., 2002, 1930 RSC.
- Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Org. Biomol. Chem., 2004, 1624 RSC.
- P. K. L. Fu and C. Turro, J. Am. Chem. Soc., 1999, 121, 1 CrossRef.
- M. Biggiogera and S. Fakan, J. Histochem. Cytochem., 1998, 46, 389 Search PubMed.
- D. Parker and J. Yu, Chem. Commun., 2005, 3141 RSC.
- J. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc, 2006, 128, 2294 CrossRef CAS.
- G. Bobba, J.-C. Frias and D. Parker, Chem. Commun., 2002, 890 RSC; J.-C. Frias, G. Bobba, M. J. Cann, D. Parker and C. J. Hutchison, Org. Biomol. Chem., 2003, 1, 905 RSC; G. Bobba, S. D. Kean, D. Parker, A. Beeby and G. Baker, J. Chem. Soc., Perkin Trans. 2, 2001, 1738 RSC.
- P. Engbers-Buijtenhuis, M. Kamphuis, G. V. Veer, C. Haanen, A. A. Poot, J. Feijen and I. Vermes, Apoptosis, 2005, 10, 429 Search PubMed.
- S. E. Barrie, E. Eno-Amoquaye, A. Hardcastle, G. Platt, J. Richards, D. Bedford, P. Workman, W. Aherne, S. Mittnacht and M. D. Garrett, Anal. Biochem., 2003, 320, 66 CrossRef.
- J. Chen and P. R. Selvin, J. Am. Chem. Soc., 2000, 122, 657 CrossRef CAS; P. R. Selvin and J. E. Hearst;, Proc. Natl. Acad. Sci. USA, 1994, 91, 10024 CAS; M. Xiao and P. R. Selvin, J. Am. Chem. Soc., 2001, 123, 7067 CrossRef CAS.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1581 RSC.
- (a) S. Phimphivang and S. S. Saavedra, Bioconjugate Chem., 1998, 9, 350 CrossRef CAS; (b) J. Inglese, P. Samama, S. Patel, J. Burbalim, I. L. Stroke and K. C. Appel, Biochemistry, 1998, 37, 2372 CrossRef CAS; (c) H. Goicochea, B. C. Roy, M. Santos, A. D. Campiglia and S. Mallik, Anal. Biochem., 2005, 336, 64 CrossRef CAS.
- O. Zohar, M. Ikeda, H. Shinagawa, H. Inoue, H. Nakamura, D. Elbaum, D. L. Alkan and T. Yoshioka, Biophys. J., 1998, 74, 82 CrossRef CAS.
- O. Wolfbeis, J. Mater. Chem., 2005, 15, 2657 RSC.
- L. Prodi, New J. Chem., 2005, 29, 20 RSC.
- A. P. de Silva, B. McCaughan, B. O. F. McKinney and M. Querol, Dalton Trans., 2003, 1902 RSC.
- O. S. Wolfbeis, A. Durkop, M. Wu and Z. Lin, Angew. Chem., Int. Ed., 2002, 41, 4495 CrossRef CAS ; for another example of hydrogel immobilisation, see: T. Gunnlaugsson, C. P. McCoy and F. Sorneo, Tetrahedron Lett., 2004, 45, 8403 Search PubMed.
- M. Wu, Z. H. Lin, M. Schaferling, A. Durkop and O. S. Wolfbeis, Anal. Biochem., 2005, 340, 66 CrossRef CAS.
- E. J. L. Martinez, J. F. G. Reyes, P. O. Barrales and A. M. Diaz, Anal. Chim. Acta, 2005, 532, 159 CrossRef.
- J. Sokolnicki, J. Legendiewicz, G. Muller and J. P. Riehl, Opt. Mater. (Amsterdam), 2005, 27, 1529 Search PubMed.
- S. Blair, M. P. Lowe, C. E. Mathieu, D. Parker, P. K. Senanayake and R. Kataky, Inorg. Chem., 2001, 40, 5860 CrossRef CAS ; for an example of a ratiometric change in a europium pyrroyl complex in solution (λexc 300 nm): M. Woods and A. D. Sherry, Inorg. Chem., 2003, 42, 4401 Search PubMed.
- S. Blair, R. Kataky and D. Parker, New J. Chem., 2002, 26, 530 RSC.
- M. Mitsuishi, S. Kikuchi, T. Miyashita and Y. Amao, J. Mater. Chem., 2003, 13, 2875 RSC.
- M. L. Rodriguez-Mendez, Y. Gorbunova and J. A. de Saja, Langmuir, 2002, 18, 9560 CrossRef CAS.
- D. Parker, K. P. Senanayake and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2129 RSC.
- H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389 CrossRef CAS.
- H. Tamiaki, S. Ueno, E. Takeuchi, N. Tameshige, S. Shinoda and H. Tsukube, Tetrahedron, 2003, 59, 10477 CrossRef CAS.
- J. I. Bruce, S. Lopinski, D. Parker and R. D. Peacock, Chirality, 2002, 14, 502 CrossRef.
- D. Parker and J. A. G. Williams, Chem. Commun., 1998, 245 RSC.
- T. Gunnlaugsson, D. A. MacDonaill and D. Parker, J. Am. Chem. Soc., 2001, 123, 12866 CrossRef CAS ; see also: A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, Nature (London), 1993, 364, 42 Search PubMed.
- T. Gunnlaugsson, J. P. Leonard, K. Senechal and A. J. Harte, Chem. Commun., 2004, 782 RSC.
- C. Li, E.-L. Law and W.-T. Wang, Org. Lett., 2004, 6, 4841 CrossRef CAS.
- R. S. Dickins, T. Gunnlaugsson, D. Parker and R. D. Peacock, Chem. Commun., 1998, 1643 RSC.
- J. I. Bruce, R. S. Dickins, L. J. Govenlock, T. Gunnlaugsson, S. Lopinski, M. P. Lowe, D. Parker, R. D. Peacock, J. J. B. Perry, S. Aime and M. Botta, J. Am. Chem. Soc., 2000, 122, 9674 CrossRef CAS.
- S. H. Li, C. W. Yu, W. T. Yuan and J. G. Xu, Anal. Sci., 2004, 20, 1375 CAS.
- M. D. Best and E. V. Anslyn, Chem. Eur. J., 2003, 9, 51 CrossRef CAS.
- P. Atkinson, Y. Bretonniere and D. Parker, Chem. Commun., 2004, 430 RSC.
- P. Atkinson, Y. Bretonniere, D. Parker and G. Muller, Helv. Chim. Acta, 2005, 88, 391 CrossRef CAS.
- T. Gunnlaugsson, A. J. Horte, J. P. Leonard and M. Niewenhuyzen, Chem. Commun., 2002, 2134 RSC.
- S. Mameri, L. J. Charbonniere and R. Ziessel, Inorg. Chem., 2004, 43, 1819 CrossRef CAS.
- S.-H. Li, W.-T. Yuan, C.-Q. Zhu and J. C. Xu, Anal. Biochem., 2004, 331, 235 CrossRef CAS.
- G. Song, G. Wang and J. Yuan, Chem. Commun., 2005, 3553 RSC.
Footnote |
| † Based on the presentation given at Dalton Discussion No. 9, 19–21st April 2006, Hulme Hall, Manchester, UK. |
| This journal is © The Royal Society of Chemistry 2006 |