Control of molecular interactions by the hollow of coordination cages†
Victor
Maurizot
,
Michito
Yoshizawa
,
Masaki
Kawano
and
Makoto
Fujita
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, and CREST, Japan Science and Technology Agency (JST), 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: mfujita@appchem.t.u-tokyo.ac.jp; Fax: +81 3 5841 7257; Tel: +81 3 5841 7259
First published on 11th May 2006
Abstract
Coordination self-assembly entities have been demonstrated to be useful and powerful alternatives for the construction of predefined and well-organized architectures. Self-assembled coordination cages, which possess an inner hydrophobic cavity, can be utilized to move substrates closer by encapsulation so enabling their interaction with each other and exhibiting of non-classical physical properties.
 Victor Maurizot Victor Maurizot | Victor Maurizot is a JSPS Postdoctoral fellow at The University of Tokyo, Japan. He received his PhD degree from the University of Bordeaux (France) in 2003. |
 Michito Yoshizawa Michito Yoshizawa | Michito Yoshizawa is Research Associate of the Department of Applied Chemistry, School of Engineering, The University of Tokyo, Japan. He received his PhD degree from Nagoya University in 2002. After working as a JSPS Postdoctoral fellow at The University of Tokyo, he moved to his current position in 2003. His current research focuses on chemical phenomena within self-assembled coordination cages |
 Masaki Kawano Masaki Kawano | Masaki Kawano is Associate Professor of the Department of Applied Chemistry, School of Engineering, The University of Tokyo, Japan. He received his PhD degree from Waseda University in 1992. After working at Waseda University (1991–1993, as Assistant Professor), University of Wisconsin-Madison in the US (1993–1996, as Postdoctoral follow), Tokyo Institute of Technology (1996–2003, as Postdoctoral follow of CREST), and the University of Tokyo (2003-2004, as lecturer), he moved to his current position in 2004. His major research interests include coordination chemistry, reactive intermediates, and in situ crystallography. |
 Makoto Fujita Makoto Fujita | Makoto Fujita is Professor of Department of Applied Chemistry, School of Engineering, The University of Tokyo, Japan. He received his PhD degree from the Tokyo Institute of Technology in 1987. After working in Chiba University (1988–1997, as Assistant Professor, Lecturer, and then Associate Professor), Institute for Molecular Science (IMS) at Okazaki (1997–1999, as Associate Professor), and Nagoya University (1999–2002, as Full Professor), he moved to his current position in 2002. He is a recipient of Progress Award in Synthetic Organic Chemistry Japan, Division Award of Chemical Society of Japan, IBM Science Award, Nagoya Silver Medal, and International Izatt–Christensen Award. His major research interests include self-assembly of well-defined coordination architectures and their functionalization. |
Introduction
From the beginning of organic chemistry and the discovery of the first simple reactions, much attention have been paid to the preparation of bigger and more complex synthetic molecules. During the last decades, chemists have shown a particular interest in synthetic self-assembly systems as an alternative way to build high molecular weight complex 3D-molecules. This powerful convergent procedure involves non-covalent linking of organic units via metal coordination, hydrogen bonds or aromatic interactions. Finite architectures can be readily obtained by mixing precursors as a result of minimization of the energy via consecutive bond formation and dissociation.Our strategy is to use metal coordination as the “tool” for such assembly. These bonds are directional and relatively strong but can be broken by thermal activation, which gives the opportunity for the system to reach thermodynamically or kinetically-controlled stable products. In the same way, the use of template molecules can also drive the assembly process and, after the complex formation, these molecules can be removed without deterioration of the architecture. Many reviews about designing and preparation of complex metal assembly structures have been published.1
Such systems can be used in various media such as polar solvents, in a basic condition or biological systems, with retention of frameworks. According to their robustness, self-assembled systems are good candidates for material precursors or biological mimics of enzymes. For example, the well-studied assembled complex, M6L43, composed of four triazine ligands 1 and six end-capped metal(II) ions 2 (Fig. 1), is an intriguing reaction cavity where organic reactions can be performed inside the hydrophobic cavity of the complex. The multi-molecular reaction process involves that compounds get closer in the cavity which allows the reaction to occur.2,3 This encapsulation of the starting compounds arises not only from the recognition of the substrates with the cavity but also from mutual interactions.4
 | ||
| Fig. 1 Construction of M6L4 self-assembled cages 3a and 3b. | ||
Encapsulation and confinement of molecules inside the hydrophobic pocket of the coordination cage can also lead to some interesting intermolecular interactions between the guest molecules, or between guest molecules and its host complex, leading to some new or non-trivial physical properties.5 This paper will describe guest–guest interactions observed in our laboratory that could be enhanced by encapsulation in a series of coordination assemblies.
Prism-shaped complexes or discrete staking of aromatic molecules
Planar aromatic molecules tend to stack and to form infinite columnar assemblies. These supramolecular entities have been commonly studied and used for the construction of simple discotic liquid crystals which have unique chemicophysical properties,6 as well as the elaboration of organic electroconductive materials.7 The length of such entities cannot be controlled, unless aromatic units are covalently linked. The DNA strand is the most common example where such control is present and it has inspired many analogous foldamers.8 Unlike infinite assemblies or polyaromatic foldamers, discrete assemblies of stacked independent aromatic molecules have been much less studied.9 Nevertheless, it can be expected that such size-controlled assemblies would present new properties different from those of isolated or infinite stacked π-systems.In previous work, we described the preparation of a prism-like multi-component assembly which is able to accommodate one aromatic molecule in its large flat cavity.10 This assembly 9 arises from the coordination, through the end-capped-PtII ions 4a, of two large tridentate panel-like ligands 1 and three pyrazine pillars 6. This complex could encapsulate one flat aromatic molecule which is stabilized by π–π stacking interactions with the host. One extension of this work was to increase the space between the two flat panels constituting the “roof” and the “floor” of the cage allowing more than one aromatic molecule to insert in the space inside the coordination box. This enlargement of the cavity could be easily done by lengthening of the pillar-like ligands. The use of a linear bis- or a tris-aromatic ligand 7 or 8 led to the formation of boxes containing two and three flat aromatic compounds in the hollow of the complexes 10 and 11, respectively (Fig. 2).11
 | ||
| Fig. 2 Construction and schematic representation of prism-like inclusion complexes 9⊃5, 10⊃(5)2 and 11⊃(5·1·5). | ||
The presence and the nature of the guest also significantly influence on the properties and the stability of the complexes. Indeed, guests act as templates for the formation of the boxes and avoid the side homotropic assembly of the different ligands. A coronene (5) is encapsulated in complex 9, two in complex 10 and surprisingly two coronene and one free planar ligand 1 in structure 11. The MII-coordinated planar ligand is highly electron deficient (A), due to the metal coordination at the three pyridine sites, and the presence of coronene (5), which is electron-rich (D), stabilized the global structure through donor–acceptor charge transfer interactions forming an A–D–A aromatic stacking. The same interactions are present in the structure 11⊃(5·1·5) where the intercalated triazine forms a CT complex with the two stacked coronenes and making, in this way, an overall A–D–A–D–A aromatic stacking system.
The odd numbered π-stacking system of 10 composed of two coronenes has an intriguing optical property observed by UV/Vis absorption spectroscopy (Fig. 3). The close range of the two electron-rich molecules leads to a stronger and red-shifted CT absorption band in the A–D–D–A system than in the A–D–A stack due to the higher HOMO level of the stacked (5)2 dimer compared with the monomer 5. The construction of these discrete, number-controlled, multiple stacking systems, could provide an interesting way to assemble or move closer aromatic molecules that can exhibit unique photochemical and electrochemical properties which would be different from the isolated or the infinite stacked state.
 | ||
| Fig. 3 UV-Vis spectra of the multi-stacked aromatic assemblies 9⊃5, 10⊃(5)2 and 11⊃(5·1·5) (RT, H2O). | ||
Metal–metal interaction
The same approach of moving planar compounds closer via the hydrophobic cavities of the sandwich-shaped complexes, 10 and 11, can be done for the stacking of planar transition-metal complexes via d–d interaction between their metal centers.12 These interactions could promise unique optical, electroconductive, and magnetic properties.13 Unless bridged by organic ligands, common planar sixteen-electron metal complexes interact very weakly at the metal centers and rarely form metal–metal bonds via discrete/infinite stacking.14After removing the aromatic guests of complex 10⊃(5)2, the guest-free assembly was mixed with an excess of the well known complexes MII(acac)2 where M are Pt, Pd or Cu, to form inclusion complexes 10⊃(12a)2, 10⊃(12b)2 or 10⊃(12c)2 in quantitative yields (Fig. 4). These isolated acetylacetonato complexes have never been shown to form an intermolecular M–M interaction. The X-ray analysis of complex 10⊃(12a)2 shows that the distance between the two metal atoms of the enclathrated complexes is 3.32 Å which is characteristic of a PtII–PtII d–d interaction (<3.5 Å) (Fig. 5).13,14
 | ||
| Fig. 4 Schematic representation of the formation a discrete stacking of planar metal complexes within the coordination cage 10. | ||
 | ||
| Fig. 5 X-Ray crystal structure of 10⊃(12a)2 and selected structure of 12a within the cavity of complex 10. | ||
This interaction in the solid state was confirmed by UV-visible absorption spectroscopy. The spectrum of powdered 10⊃(12a)2, as well as 10⊃(12b)2, showed an absorption band at 500 and 450 nm, respectively, which is featured by d–d metal interactions (Fig. 6(a)). The color change of the orange solid to an almost colorless solution upon dissolution of the crystal in water suggests a weaker interaction of the M–M bond in solution. However, the 195Pt NMR of 10⊃(12a)2 showed a broad signal at −280 ppm indicating an up-fielding (Δδ = 130 ppm) compared to the free 12a. This shift indicates that the M–M interaction still remains in solution.
 | ||
| Fig. 6 (a) UV-Vis spectra of 10, 12a and inclusion assembly 10⊃(12a)2 in the solid state, (b) EPR spectra (H2O, 103 K, MnO as external standard) of 10⊃(12c)2, and (c) its forbidden transition. | ||
Even clearer evidence of the M–M interaction in solution can be obtained from the d9 copper complex which exhibits a broad EPR signal (Fig. 6(b), (c)) when two molecules are packed in complex 10. The signals are attributed to the Cu–Cu interaction of 10⊃(12c)2 for S = 1. The large splitting of ca. 60 mT is due to spin–spin interaction and indicates an average interspin distance of 3.6 Å. The longer distance is comparable to that obtained by X-ray analysis of the Pt(acac)2 inclusion complex having a different type of d–d interaction. The distance variation points out the flexibility and the adaptability of these prism-shaped assemblies that can be slightly twisted to accommodate to the thickness of planar compounds or assemblies.
Mixed-valence states of organic dimers
Moving organic compounds closer, as has already been widely described in our laboratory, can promote reactions and keep compounds close together after reaction. This proximity can lead to interesting regiospecific reactions and isolation of intermediates in unstable states.3Whereas mixed-valence states are often observed for metal–metal bonded or metal–conjugated compounds,15 they are much less common for fully organic systems and have attracted much attention.16 The packing of two TTF molecules (TTF = tetrathiafulvalene) in the planar cavity of cage 10 and their oxidative reaction could lead to the observation of the mixed valence dimer [(TTF)2]+˙ in solution at room temperature.17 Such a mixed-valence state has not been observed at ambient temperature, unless two functionalized TTF molecules were covalently linked.18
Inclusion complex 10⊃(TTF)2, which was prepared by mixing empty cage 10 with an excess of TTF in water, showed a strong absorption band around 740 nm (Fig. 7) which could be assigned to the charge-transfer interaction between the electron-rich TTF and the electron-deficient triazine ligand of the cage.11
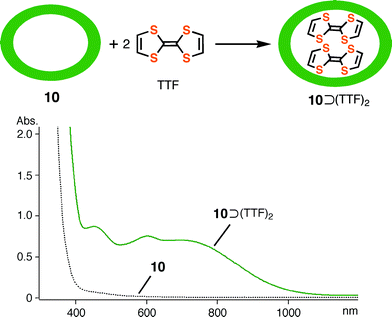
The cyclic voltammogram of 10⊃(TTF)2 shows, to a first approximation, two oxidation waves at 200 and 500 mV which could be assigned to the formation of the cation radical TTF+˙ and the dication TTF2+ (Fig. 8(a)). However, the 200 mV wave can be decomposed, judging from the square-wave voltammogram, into two distinct waves at 152 and 304 mV (Fig. 8(b)). This indicates that half of the TTF enclathrated into the prism complex is subject to oxidation, implying the formation of the mixed valence dimer of TTF ([(TTF)2]+˙). This species was further oxidized into cation radical dimer (TTF+˙)2 at 304 mV and to the dication TTF2+ at 552 mV. While the first oxidation potential of the enclathrated TTF was positively shifted compared to the free TTF+˙/TTF oxidation (178 mV, ΔE = 126 mV), probably due to the cationic repulsion between the host and the guest, the dication formation potential was comparable to the free TTF2+/TTF+˙ oxidation (555 mV, ΔE = 3 mV). The fact suggests that the dication TTF2+ was liberated from the cage, because of cationic repulsion with the guest assembly.19 The sequential oxidations and the most probable transformation can be summarized in Fig. 8(d). However, the two-electron oxidation/reduction of the (TTF)2 within complex 10 occurred reversibly in the range 0–400 mV, showing the stability of [(TTF)2]+˙ and (TTF+˙)2 as well as the absence of declathration during the redox measurement (Fig. 8(c)).
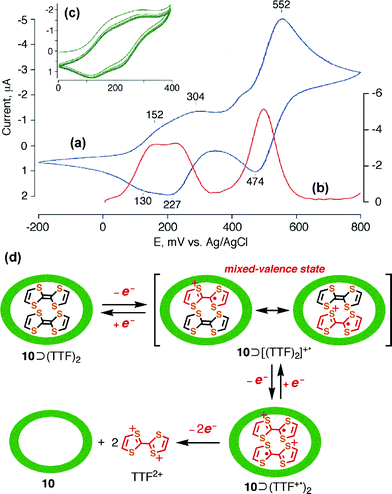 | ||
| Fig. 8 Cyclic voltammogram of 10⊃(TTF)2 (glassy carbon disk electrode vs. Ag/AgCl, NaNO3 (aq), RT) in the range of (a) −200 to 800 mV and (c) 0 to 400 mV (the five first cycles). (b) Square wave voltammogram. (d) Schematic equation of the oxidation–declathration of the encapsulated TTF dimer in coordination complex 10. | ||
The direct electronic absorption spectroscopic observation of the in situ oxidation of inclusion complex 10⊃(TTF)2 strongly supports the proposed mechanism. A new absorption band in the near-IR region arose upon a constant voltage of 180 mV, which is below the [(TTF+˙)2/(TTF)2+˙] oxidation potential (Fig. 9). The new band corresponds to mixed valence dimer formation. Surprisingly, this species was remarkably stable, and the absorption band decayed very slowly (t1/2 = 1 day) at room temperature even under air. This stability can be attributed to the efficient stacking of the two TTF molecules in the prism cage. Further oxidation at 400 mV generated the TTF+˙ dimer that exhibits characteristic absorption bands in the UV-visible region at 406, 534 and 758 nm.20 These bands gradually disappeared which suggests that the cationic guests were liberated from the cationic cage.
 | ||
| Fig. 9 In situ electronic absorption spectra of 10⊃(TTF)2 at various applied potentials of (a) 180 and (b) 400 mV (ITO electrode vs. Ag/AgCl, NaNO3 (aq), RT). | ||
This study highlights the ability for prism-shaped coordination cages to create strong dimeric species within their hydrophobic cavity and to isolate them from the solution. This can allow us to create and observe various weak interactions between “flat” molecules.
Octahedral complex
The observation of weak interaction enhancement inside the hydrophobic pocket of a coordination cage is not exclusive to prism-shaped complexes but can be expanded to the different coordination cages synthesized in our laboratory.1The octahedral M6L4 cage 3 which has been widely used in our research can readily encapsulate various sizes and shapes of organic molecules and can accommodate more than one molecule in the cavity. This ability allowed us to use it as a molecular flask, a logic gate or to isolate unstable intermediates.3,4 It is also obvious that this cage can be used, similarly to prism-shaped complexes, to generate substrate–substrate interactions and “molecular communication”.
Spin–spin interaction
Stable organic radicals have attracted great attention for the last two decades because of possible use in the construction of molecular-scale magnetic devices. The main interest of these organic compounds is that they can be readily synthesized and designed to control the physical properties of such materials.21 Whereas spin–spin interactions of organic radicals via covalent or coordination bonds have been well studied, through-space interactions have been seldom controlled. Radical centers have to be located at very short distances, because this interaction is proportional to 1/r3 (r = spin–spin distance). The enclathration of two organic radical molecules in the cavity of 3 can reduce the distance and keep the radical center close enough to produce spin–spin interactions.22An inclusion complex 3a⊃(13)2 (Fig. 10) was obtained by suspending an excess of powdered 13 in an aqueous solution of cage 3a and filtration of the non-solubilized compound after 1 h at rt. The solution could be slowly evaporated to obtain single crystals of 3a⊃(13)2.
 | ||
| Fig. 10 Schematic representation of the encapsulation of radical 13 in the hydrophobic hollow of complex 3a. | ||
X-Ray analysis elucidated the presence of two radical molecules in the cage and the close contact between two radicals (Fig. 11). The shortest and the longest intermolecular distances between the radical centers are 3.2 and 8.5 Å, respectively.
 | ||
| Fig. 11 X-Ray crystal structure of complex 3a⊃(13)2. | ||
This interaction in the solid state was confirmed by EPR (Fig. 12), where signals at 320 and 160 mT were observed corresponding to the Δms = 1 and 2 transition, respectively. Interestingly, these interactions were not observed for pure 13 in the solid state, because 13 forms antiparallel sheets in a crystal where the radical centers are apart from each other. The fine structure constant (D′) of 14 mT confirmed the average distance of ∼5.8 Å between the radical centers.
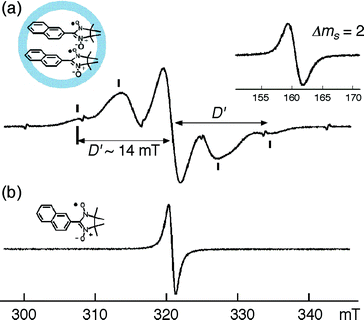 | ||
| Fig. 12 Powder EPR spectra of (a) inclusion complex 3a⊃(13)2 and (b) free radical 13. | ||
The solution study of the complex 3a⊃(13)2 shows that the spin–spin interaction was preserved even in solution. An EPR spectrum of the complex in solution at 293 K showed, in addition to a sharp signal of 13 in the doublet state, a broad signal of the triplet species similar to those observed in the solid state (but slightly shifted and broadened) (Fig. 13). Temperature changes had an impact on the fine structure constant, decreasing the constant when the solution was warmed and increasing it to the solid-state constant when the sample was cooled down to 103 K, according to Curie's law. The central doublet dramatically became broader below 273 K due to the freezing of the molecular motion. This study shows that close packing through hydrophobic forces of two organic radicals can be achieved in solution and leads to spin–spin interactions.
 | ||
| Fig. 13 EPR spectra of (a) radical 13 (saturated solution) and (b)–(e) inclusion complex 3b⊃(13)2 (5 mM in water) at different temperatures (asterisks show Mn2+ external reference). | ||
The use of organic radicals that have switchable affinity for the cage, for example, by changing its hydrophobicity, can lead to the construction of molecular devices that are external stimuli responsive.23 Radical 14, which can be encapsulated in coordination M6L4 cage 3b, has an amine group that may be protonated, depending on the pH of the solution. The charged compound 14H+, due to cationic repulsion with the complex and to its less hydrophobic nature, has a lower affinity for the cage (Fig. 14). Protonation of the enclathrated 14 released the guest in solution and quenched the magnetic interaction that was enhanced inside the cavity of the complex 3b.24
 | ||
| Fig. 14 Schematic representation of the enclathration–declathration pH responsive process of radical 14 within the cavity of complex 3b. | ||
Similarly to the radical 13, the EPR spectra of the initial solution of the complex 3b⊃(14)2, at pH ≈ 6.5, that was prepared according to the same procedure of 3b⊃(13)2, showed broad signals at 320 and 160 mT illustrating the spin–spin interaction observed in the crystal (Fig. 15(a)). After acidification of the solution to pH = 1.3, the signal of the triplet state disappeared (Fig. 15(b)), due to the declathration of the radicals. Reversible reencapsulation occurred by neutralization around pH = 7 and triplet signals reappeared (Fig. 15(c)).
 | ||
| Fig. 15 ESR spectra of inclusion complex 3b⊃(14)2 (0.5 mM, H2O, 103 K): (a) the initial solution (pH ≈ 6.5), (b) the acidified solution (pH ≈ 1.3 with HNO3), and (c) the neutralized solution (pH ≈ 6.5 with K2CO3). | ||
This study can be extended to the development of molecular magnetic devices via external stimuli such as photo-excitation, redox, specific molecular recognition, and so on.
Conclusion
The development of new “construction tools” for the elaboration of self-organized, and self-assembled molecular entities has been, and still remains, of great interest for the control of molecular architecture formation. This control of organization can be used for designing molecular materials with predictable and well-defined structure. Control of function at the molecular level is one of the most challenging tasks that chemists can tackle.Molecular self-assembly can be used to accumulate or organize functional groups but can also control molecular interactions between substrates by enclathration and in this way exhibit new physical properties. Both approaches can also be combined to provide communication between the host and the guest and develop new or better controlled properties.
References
- G. F. Swiegers and T. J. Malfetse, Chem. Rev., 2000, 100, 3483–3537 CrossRef CAS; B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS; M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 500–518 Search PubMed; S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–963 CrossRef CAS; M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371–380.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 351–360.
- M. Yoshizawa and M. Fujita, Pure Appl. Chem., 2005, 77, 1107–1112 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, J. Am. Chem. Soc., 2004, 126, 6846–2847 CrossRef CAS.
- C. H. M. Amijs, G. P. M. van Klink and G. van Koten, Dalton Trans., 2006, 308–327 RSC.
- For a recent review of discotic liquid crystals, see: R. J. Bushby and O. R. Lozman, Curr. Opin. Colloid Interface Sci., 2002, 7, 343–354 Search PubMed.
- C. D. Simpson, J. Wu, M. D. Watson and K. Müllen, J. Mater. Chem., 2004, 14, 495–504 Search PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS.
- G. S. Papaefstathiou and L. R. MacGillivray, Org. Lett., 2001, 3, 3835–3838 CrossRef CAS; D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295, 2403–2407 CrossRef CAS; P. S. Mukherjee, K. S. Min, A. M. Arif and P. J. Stang, Inorg. Chem., 2004, 43, 6345–6350 CrossRef CAS.
- K. Kumazawa, K. Biradha, T. Kusukawa, T. Okano and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3909–3913 CrossRef CAS.
- M. Yoshizawa, J. Nakagawa, K. Kumazawa, M. Nagao, M. Kawano, T. Ozeki and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 1810–1813 CrossRef CAS.
- M. Yoshizawa, K. Ono, K. Kumazawa, T. Kato and M. Fujita, J. Am. Chem. Soc., 2005, 127, 10800–10801 CrossRef CAS.
- K. Krogmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 35–42 CrossRef CAS; T. W. Thomas and A. E. Underhill, Chem. Soc. Rev., 1972, 1, 99–120 RSC; G. Gilemann and H. Yersin, Struct. Bonding (Berlin), 1985, 62, 87–153 CAS; R. Palmans, D. B. MacQueen, C. G. Pierpont and A. J. Frank, J. Am. Chem. Soc., 1996, 118, 12647–12653 CrossRef CAS.
- S.-W. Lai, T.-C. Cheung, M. C.-W. Chan, K.-K. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 2000, 39, 255–262 CrossRef CAS; A. J. Goshe, I. M. Steele and B. Bosnich, J. Am. Chem. Soc., 2003, 125, 444–451 CrossRef CAS; W. Lu, M. C.-W. Chan, N. Zhu, C.-M. Che, C. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CAS; G. C. Allen and N. S. Hush, Prog. Inorg. Chem., 1967, 8, 357–389 CAS; K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS.
- T. Jørgensen, T. K. Hansen and J. Becher, Chem. Soc. Rev., 1994, 23, 41–51 RSC; R. Rathore, A. S. Kumar, S. V. Lindeman and J. K. Kochi, J. Org. Chem., 1998, 63, 5847–5856 CrossRef CAS; J. K. Kochi, R. Rathore and P. Le Maguères, J. Org. Chem., 2000, 65, 6826–6836 CrossRef CAS; M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153–164 RSC; M. Iyoda, M. Hasegawa, Y. Kuwatani, H. Nishikawa, K. Fukami, S. Nagase and G. Yamamoto, Chem. Lett., 2001, 1146–1147 CrossRef CAS; M. Iyoda, M. Hasegawa and Y. Miyake, Chem. Rev., 2004, 104, 5085–5113 CrossRef CAS.
- M. Yoshizawa, M. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456–13457 CrossRef CAS.
- J. B. Torrance, B. A. Scott, B. Welber, F. B. Kaufman and P. E. Seiden, Phys. Rev. B., 1979, 19, 730–741 CrossRef CAS.
- W.-Y. Sun, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 2002, 124, 11570–11571 CrossRef CAS.
- D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 1584–1586 RSC; P. M. Schmidt, R. S. Brown and J. H. T. Luong, Chem. Eng. Sci., 1995, 50, 1867–1876 CrossRef CAS; A. Credi, M. Montalti, V. Balzani, S. J. Langford, F. M. Raymo and F. Stoddart, New J. Chem., 1998, 1061–1065 RSC; A. Y. Ziganshina, Y. H. Ko, W. S. Jeon and K. Kim, Chem. Commun., 2004, 806–807 RSC.
- K. Awaga and Y. J. Maruyama, Chem. Phys., 1989, 91, 2743–2747 CrossRef CAS; R. Chiarell, A. Novak, A. Rassat and J. L. Tholence, Nature, 1993, 363, 147–149 CrossRef CAS; S. Nakatsuji and H. Anzai, J. Mater. Chem., 1997, 7, 2161–2174 RSC; N. Yoshioka, M. Irisawa, Y. Mochizuki, T. Kato, H. Inoue and S. Ohba, Chem. Lett., 1997, 3, 251–252 CrossRef; K. Matsuda and M. Irie, J. Am. Chem. Soc., 2000, 122, 8309–8310 CrossRef CAS; Y. Hosokoshi, K. Katoh, Y. Nakazawa, H. Nakano and K. Inoue, J. Am. Chem. Soc., 2001, 123, 7921–7922 CrossRef CAS; K. Doi, T. Ishida and T. Nogami, Chem. Lett., 2003, 32, 544–545 CrossRef CAS.
- K. Nakabayashi, M. Kawano, M. Yoshizawa, S. Ohkoshi and M. Fujita, J. Am. Chem. Soc., 2004, 126, 16694–16695 CrossRef CAS.
- For examples on external stimuli modification of magnetic device in the solid state: W. Fujita and K. Awaga, Science, 1999, 286, 261–262 Search PubMed; M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443–1445 CrossRef CAS.
- K. Nakabayashi, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 5322–5325 CrossRef CAS.
Footnote |
| † Based on the presentation given at Dalton Discussion No. 9, 19–21st April 2006, Hulme Hall, Manchester, UK. |
| This journal is © The Royal Society of Chemistry 2006 |