Dihydrobis(methimazolyl)borate and methimazolyl complexes of titanium
Anthony F.
Hill
* and
Matthew K.
Smith
Research School of Chemistry, Institute of Advanced Studies, Australian National University, Canberra, A.C.T., Australia. E-mail: a.hill@anu.edu.au; Fax: (+61) 2 6125 3216; Tel: (+61) 2 6125 8577
First published on 27th October 2005
Abstract
The first poly(methimazolyl)borato complex of group 4, [Ti(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NCMe3){H2B(mt)2}2]
(mt = methimazolyl), results from the reaction of Na[H2B(mt)2] with [Ti(NCMe3)Cl2(py)3] and features both κ2-S,S′ and κ3-H,S,S′ coordination of H2B(mt)2 ligands coincident within the same molecule.
NCMe3){H2B(mt)2}2]
(mt = methimazolyl), results from the reaction of Na[H2B(mt)2] with [Ti(NCMe3)Cl2(py)3] and features both κ2-S,S′ and κ3-H,S,S′ coordination of H2B(mt)2 ligands coincident within the same molecule.
The poly(methimazolyl)borate ligands HnB(mt)4−n (mt = methimazolyl, Scheme 1) present themselves as ‘soft’ analogues of the more familiar poly(pyrazolyl)borates. Reglinski's HB(mt)3 ligand1 in general functions as a simple tridentate κ3-S,S′,S″ facially coordinating ligand. Examples of κ3-H,S,S′ coordination have also been encountered involving three-centre–two-electron (3c–2e) B–H–M interactions2 and these are implicated in the formation of metallaboratranes.2,3
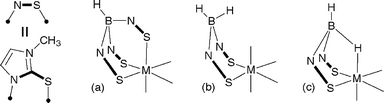 | ||
| Scheme 1 Poly(methimazolyl)borate coordination: (a) κ3-S,S′,S″; (b) κ2-S,S′; (c) κ3-H,S,S′. | ||
This type of coordination has also been observed for Parkin's bis(methimazolyl)borate ligand4 H2B(mt)2, when coordinated to molybdenum,5 rhenium6 and platinum7 centres. The affinity of these ligands for soft late transition metals in low oxidation states is to be expected from simple hard and soft acid and bases (HSAB) considerations. We have therefore turned our attention to seemingly less appropriate metals on the assumption that the ambivalence of hard metals towards sulfur donors might be overcome at least in part through the entropic advantages offered by methimazolylborate chelation. A further point is that of the wide class of facially tridentate ligands, HnB(mt)4−n have been shown to be exceptionally π-basic.8 This feature might be traced to a strong π-donor role for sulfur in which case coordination to high oxidation state metal centres might actually be favourable. Herein we report the first reactions of HnB(mt)4−n salts with high oxidation state titanium complexes.
Our initial investigations of the reactions of TiCl4 or [TiCl4(thf)2] with Na[HB(mt)3] or Na[H2B(mt)2] were spectacularly unsuccessful, providing intractable mixtures of unidentified compounds, none of which appeared to be the desired simple complexes [TiClx{HnB(mt)4−n}4−x]. Although our attempts were not exhaustive, we are inclined to suspect that both redox processes and ligand cleavage reactions interfere with the simple halide metathesis. These problems might be traced to the potent Lewis acidity of the TiIV reagents. We have recently found that similar synthetic hurdles in the chemistry of niobium(V) and tantalum(V) may be overcome by the inclusion of a π-basic imido co-ligand to reduce the electrophilicity of the metal centre thereby allowing the isolation of the complexes [M(NC6H3iPr2-2,6)Cl2{HB(mt)3}].9 We now find that a similar approach meets with success in the chemistry of TiIV: treating Mountford's versatile imido complex [Ti(NCMe3)Cl2(py)3]10 with two equivalents of Na[H2B(mt)2]5 results in the formation of the complex [Ti(NCMe3){H2B(mt)2}2]
(1: Fig. 1, Scheme 2).† The putative intermediate [Ti(NCMe3)Cl(py){H2B(mt)2}] is not observed even when a deficiency of Na[H2B(mt)2] is employed, suggesting it reacts more rapidly with Na[H2B(mt)2] than does the precursor.
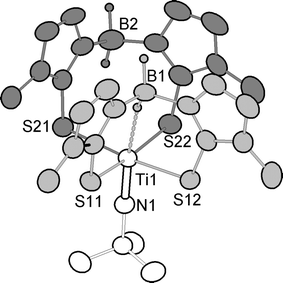 | ||
| Fig. 1 Molecular structure of 1 (40% displacement ellipsoids; methimazolyl hydrogen atoms omitted; κ2-S,S′-H2B(mt)2 in dark gray; κ3-H,S,S′-H2B(mt)2 in light grey). Selected bond lengths (Å) and angles (°): Ti1–N1 1.696(6), Ti1–S12 2.509(2), Ti1–S11 2.514(2), Ti1–S22 2.515(2), Ti1–S21 2.516(2), B1H–Ti 2.09(3), B(1)–Ti 3.124(8); N1–Ti1–S12 91.4(2), N1–Ti1–S11 93.9(2), S12–Ti1–S11 84.57(8), N1–Ti1–S22 96.5(2), N1–Ti1–S21 98.7(2), S22–Ti1–S21 92.64(8), C1–N1–Ti1 175.9(5). | ||
 | ||
| Scheme 2 Synthesis of 1 and 2. | ||
The molecular geometry of 1 is depicted in Fig. 1 and whilst the low precision of the structural model† precludes detailed discussion, the overall topology reveals an interesting bonding scenario: The two H2B(mt)2 ligands adopt different modes of coordination within the same molecule to provide a collar of sulfur π-donors, with the imido ligand on one side, trans to one 3c–2e B–H–Ti interaction. In principle the electron deficient d0-TiIV centre might be expected to accommodate two such B–H–Ti interactions, however any tendency for this is presumably outweighed by the heavy π-loading and issues of attendant steric factors and ring strain. Furthermore, the κ3-H2B(mt)2 ligand (light grey) fits snugly into a cleft provided by the alternate κ2-H2B(mt)2 ligand (dark grey).
A second trace product, 2, was isolated from the reaction, and whilst this was only obtained in sufficient quantities for crystallographic identification,† the molecular geometry is presented in Fig. 2 as it illustrates some features of note. The compound is the binuclear bis(imido) complex [Ti2(µ-NCMe3)2(µ-mt)2(κ2-mt)Cl] (2) which apparently arises from the degradation of the H2B(mt)2 pro-ligand salt. The Ti2(NCMe3)2 core is a comparatively recurrent feature of the chemistry of Mountford's complex,11 and whilst no examples have involved sulfur based ligands, the topology is somewhat reminiscent of Cotton's formamidinate derivative [Ti2(µ-NPh)2(µ-PhNCHNPh)2(κ2-PhNCHNCPh)2].12 The bridging mode of mt coordination in which the Ti–Ti vector is effectively coplanar with the mt heterocycles is unprecedented and presumably reflects the (σ + π) donor role for this heterocycle bound to d0-TiIV. The Ti1–Ti2 separation (2.7335(16) Å) is remarkably short for a TiIV–TiIV ‘non-bond’ with only the oxo-bridged complexes [Ti2(µ-O)2Cl2{C5H2(SiMe3)3}2] (2.707 Å)13 and [Ti2(µ-O)2(µ-H2CC5Me4)(C5Me5)2] (2.725 Å)14 having shorter associations.
 | ||
| Fig. 2 Molecular structure of 2 (40% displacement ellipsoids; hydrogen atoms omitted. Selected bond lengths (Å) and angles (°): Ti1–N51 1.895(4), Ti1–N41 1.897(4), Ti1–N21 2.153(4), Ti1–N11 2.181(4), Ti1–S11 2.6141(18), Ti1–C11 2.710(5), Ti1–Ti2 2.7335(16), Cl1–Ti2 2.3409(17), Ti2–N 51 1.869(4), Ti2–N41 1.877(4), Ti2–S31 2.4704(16), Ti2–S21 2.4738(16); N51–Ti1–N41 86.36(16), N51–Ti1–N21 87.35(15), N41–Ti1–N21 90.63(15), N51–Ti2–N41 87.68(16), N51–Ti2–Cl1 138.22(13), N41–Ti2–Cl1 134.07(13), N51–Ti2–S31 90.09(11), N41–Ti2–S31 92.13(11). | ||
Given the absence of the methimazolyl ligand from early transition metal chemistry, other than 2, we have investigated the reaction of Hmt with [TinBu2(η-C5H5)2].15 Rather than the anticipated TiIV complexes [TiH(mt)(η-C5H5)2] or [Ti(mt)2(η-C5H5)2], we find that inter alia the major product is the blue TiIII methimazolyl complex [Ti(κ2-mt)(η-C5H5)2] (3: Fig. 3)† in which the methimazolyl ligand adopts a bidentate coordination mode through both nitrogen and sulfur. Structural data are not available for mt complexes of early transition metals, however relative to the more familiar pyridinethiolato ligand bound to titanium,16 the methimazolyl chelate bite angle in 3 is some 10° wider, with longer Ti–S and comparable Ti–N bond lengths. Relative to 2, the chelated mt in 3 has shorter Ti–N and longer Ti–S bond lengths subtending an increased N–Ti–S bite angle.
 | ||
| Fig. 3 Molecular structure of 3 (40% displacement ellipsoids; hydrogen atoms omitted). Selected bond lengths (Å) and angles (°): Ti1–N11 2.226(2), Ti1–S11 2.499(2), S11–C11 1.809(2), N11–C11 1.272(2); C11–S11–Ti1 75.64(7), C11–N11–Ti1 97.13(13). | ||
References
- M. Garner, J. Reglinski, I. Cassidy, M. D. Spicer and A. R. Kennedy, Chem. Commun., 1996, 1975 RSC.
- M. R. St, J. Foreman, A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Organometallics, 2003, 22, 4446 CrossRef CAS.
- A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759 CrossRef CAS; M. R. St.-J. Foreman, A. F. Hill, A. J. P. White and D. J. Williams, Organometallics, 2004, 23, 913 CrossRef CAS; I. R. Crossley and A. F. Hill, Organometallics, 2004, 23, 5656 CrossRef CAS; I. R. Crossley, A. F. Hill and A. C. Willis, Organometallics, 2005, 24, 1062 CrossRef CAS; I. R. Crossley, M. R. St.-J. Foreman, A. F. Hill, A. J. P. White and D. J. Williams, Chem. Commun., 2005, 221 RSC.
- C. Kimblin, T. Hascall and G. Parkin, Inorg. Chem., 1997, 36, 5680 CrossRef CAS; C. Kimblin, B. M. Bridgewater, T. Hascall and G. Parkin, J. Chem. Soc., Dalton Trans., 2000, 891 RSC.
- M. R. St.-J. Foreman, A. F. Hill, N. Tshabang, A. J. P. White and D. J. Williams, Organometallics, 2003, 22, 5593 CrossRef CAS; R. J. Abernethy, A. F. Hill, H. Neumann and A. C. Willis, Inorg. Chim. Acta, 2005, 358, 1605 CrossRef CAS; M. R. St.-J. Foreman, A. F. Hill, N. Tshabang and M. K. Smith, Organometallics, 2005, 24, 5224 CrossRef CAS.
- R. Garcia, A. Domingos, A. Paulo, I. Santos and R. Alberto, Inorg. Chem., 2002, 41, 2422 CrossRef CAS; R. Garcia, A. Paulo, A. Domingos and I. Santos, J. Organomet. Chem., 2001, 632, 41 CrossRef CAS.
- I. R. Crossley, A. F. Hill and A. C. Willis, Organometallics, 2005, 24, 4889 CrossRef CAS.
- M. R. St.-J. Foreman, A. F. Hill, A. J. P. White and D. J. Williams, Organometallics, 2003, 22, 3831 CrossRef CAS.
- A. F. Hill and M. K. Smith, Chem. Commun., 2005, 1920 RSC; A. F. Hill, A. D. Rae and M. K. Smith, Inorg. Chem., 2005, 44, 7316 CrossRef CAS.
- A. J. Blake, P. E. Collier, S. C. Dunn, W.-S. Li, P. Mountford and O. V. Shiskin, J. Chem. Soc., Dalton Trans., 1997, 1549 RSC.
- P. J. Stewart, A. J. Blake and P. Mountford, Inorg. Chem., 1997, 36, 3616 CrossRef CAS; P. J. Stewart, A. J. Blake and P. Mountford, J. Organomet. Chem., 1998, 564, 209 CrossRef CAS.
- F. A. Cotton and W. A. Wojtczak, Polyhedron, 1994, 13, 1337 CrossRef CAS.
- J. Okuda and E. Herdtweck, Inorg. Chem., 1991, 30, 1516 CrossRef CAS.
- F. Bottomley, I. J. B. Lin and P. S. White, J. Am. Chem. Soc., 1981, 103, 703 CrossRef.
- A. F. Hill, H. D. Hönig and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1988, 3031 RSC.
- Y. Nakayama, K. Miyamoto, N. Ueyama and A. Nakamura, Chem. Lett., 1991, 391 CAS.
Footnote |
| † 1: 1H NMR data for 1: (CD2Cl2, 25 °C): δH 0.99 (s, 9 H, CCH3), 3.32, 3.33 (s × 2, 6 H × 2, NCH3), 6.67, 6.68, 6.97, 6.98 (d × 4, 2 H × 4, NCHCH, JHH
= 1.8 Hz). Crystal data for C20H33B2N9S4Ti 1, Mr
= 597.31, monoclinic, space group P21/c, a
= 16.540(3), b
= 9.2900(19), c
= 18.924(4)
Å, β
= 94.27(3)°, V
= 2899.7(10)
Å3, Z
= 4, T
= 200(2) K, yellow prism, Dc
= 1.368 Mg m−3, µ(Mo-Kα)
= 0.611 mm−1, 6611 independent reflections, F refinement, R
= 0.107, wR
= 0.248 for 4009 independent absorption corrected reflections [I > 2σ(I), 2θ
≤ 55°], 346 parameters. CCDC reference number 283377. Crystal data for C20H33N8S3ClTi2·0.5CH2Cl22, Mr
= 655.44, tetragonal, space group P43212, a
= 12.981(5), b
= 12.981(5), c
= 36.183(5)
Å, V
= 6097(3)
Å3, Z
= 8, red hexagon, Dc
= 1.428 Mg m−3, µ(Mo-Kα)
= 0.930 mm−1, T
= 200(2) K. 3097 independent reflections, F refinement, R
= 0.0315, wR
= 0.0772 for 2756 independent absorption corrected reflections [I > 2σ(I), 2θ
= 41.2°], 331 parameters, CCDC reference number 283376. Crystal data for C14H15N2STi 3: Mr
= 291.24, monoclinic, space group P21/a, a
= 13.730(3), b
= 7.9000(16), c
= 12.780(3)
Å, β
= 103.24(3)°, V
= 1349.4(5)
Å3, Z
= 4, T
= 200(2) K, blue prism, Dc
= 1.434 Mg m−3, µ(Mo-Kα)
= 0.771 mm−1, 3749 independent reflections. F refinement, R
= 0.0459, wR
= 0.114 for 2837 independent absorption corrected reflections [I > 2σ(I), 2θ
≤ 55°], 184 parameters CCDC reference number 283378. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513251g |
| This journal is © The Royal Society of Chemistry 2006 |