Copolymerization of cyclohexene oxide and carbon dioxide using (salen)Co(III) complexes: synthesis and characterization of syndiotactic poly(cyclohexene carbonate)†
Claire T.
Cohen
,
Christophe M.
Thomas
,
Kathryn L.
Peretti
,
Emil B.
Lobkovsky
and
Geoffrey W.
Coates
*
Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, New York 14853-1301, USA. E-mail: gc39@cornell.edu
First published on 24th November 2005
Abstract
Synthetic routes to a series of new (salen)CoX (salen = N,N′-bis(salicylidene)-1,2-diaminoalkane; X = halide or carboxylate) species are described and the X-ray crystal structures of two (salen-1)CoX (salen-1 = N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-diaminocyclohexane; X = Cl, I) complexes are presented. (R,R)-(salen-1)CoX (X = Cl, Br, I, OAc, pentafluorobenzoate (OBzF5)) catalysts are active for the copolymerization of cyclohexene oxide (CHO) and CO2, yielding syndiotactic poly(cyclohexene carbonate) (PCHC), a previously unreported PCHC microstructure. Variation of the salen ligand and reaction conditions, as well as the inclusion of [PPN]Cl ([PPN]Cl = bis(triphenylphosphine)iminium chloride) cocatalysts, has dramatic effects on the polymerization rate and the resultant PCHC tacticity. Catalysts rac-(salen-6)CoX (salen-6 = N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-diaminopropane; X = Br, OBzF5) have high activities for CHO/CO2 copolymerization, yielding syndiotactic PCHCs with up to 81% r-centered tetrads. Using Bernoullian statistical methods, PCHC tetrad and triad sequences were assigned in the 13C NMR spectra of these polymers in the carbonyl and methylene regions, respectively.
Introduction
The correlation between a polymer's main-chain stereochemistry and its physical properties has stimulated many research efforts in the field of stereospecific polymerization. Ordered polymer architectures, where sequential stereocenters are of the same (isotactic) or alternating (syndiotactic) relative configuration are generally more crystalline than their atactic counterparts. Although heterogeneous catalysts can be used to control polymer microstructure in some cases, homogeneous systems are more commonly employed for this purpose. Notably, these discrete polymerization catalysts can be designed at the molecular level, providing for a tunable active site.The alternating copolymerization of epoxides and CO2 to generate biodegradable polycarbonates has been a subject of much interest as CO2 is a readily available, inexpensive, non-toxic and nonflammable reagent (Scheme 1).1 The first epoxide/CO2 copolymerization was achieved by Inoue using ZnEt2/H2O catalysts in 1969.2 Following this discovery, several other heterogeneous systems were developed for this transformation; however, catalyst activities remained low with little control of polymer microstructure. To improve catalyst performance, recent research has focused on single-site epoxide/CO2 copolymerization catalysts.1,3–18 To date, these homogeneous systems have achieved high activities for epoxide/CO2 copolymerization and in some cases stereo- and/or regiospecific monomer enchainment.5,6,11–18
 | ||
| Scheme 1 Copolymerization of epoxides and CO2 to yield polycarbonates. | ||
The asymmetric alternating copolymerization of cyclohexene oxide (CHO) and CO2 has been demonstrated first by Nozaki using ZnEt2/chiral amino alcohol catalysts and later by our group using hybrid imine-oxazoline zinc-based catalysts (Fig. 1).11–14,18 In each system, isotactic poly(cyclohexene carbonate) (PCHC) with an enantiomeric excess (ee) ∼70% is obtained. These isotactic PCHCs have been structurally characterized by 13C NMR spectroscopy using both model compounds and statistical methods.14,19 These systems show that chiral ligands allow the generation of isotactic PCHC, consistent with a site-control copolymerization mechanism.11–14
 | ||
| Fig. 1 Catalysts for the isospecific copolymerization of cyclohexene oxide and CO2; R1 = Me, CF3, or iPr; R2 = (R)-Ph, (S)-iPr, or (S)-tBu; R3 = Et or iPr. | ||
Recently we reported that (salen-1)CoX complexes (salen-1 = N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-diaminocyclohexane; X = Cl, Br, I, OAc, pentafluorobenzoate (OBzF5); Fig. 2) are highly active catalysts for the copolymerization of propylene oxide (PO) and CO2 to yield poly(propylene carbonate) (PPC) with no detectable propylene carbonate byproduct.5,6,17 Using these catalyst systems, we observed a variety of PPC microstructures, in which the regio- and stereochemical composition of the resultant PPC was dependent on the relative stereochemistry of the catalyst and monomer. Furthermore, through the addition of bulky, organic ionic cocatalysts to these systems unprecedented activities were observed, while maintaining excellent regioselectivity.6,17b Based on our success with (salen-1)CoX catalysts for PO/CO2 copolymerization, we likewise applied these systems to CHO/CO2 copolymerization, with a focus on maximization of catalyst activity and control of product PCHC microstructure.
 | ||
| Fig. 2 (salen-1)CoX catalysts for the copolymerization of epoxides and CO2 (X = Cl, Br, I, acetate, pentafluorobenzoate). | ||
Herein, we report the synthesis of a series of (salen)CoX (salen = N,N′-bis(salicylidene)-1,2-diaminoalkane; X = halide or carboxylate) complexes that are active for CHO/CO2 copolymerization, and we present the X-ray crystal structures of (R,R)-(salen-1)CoCl and rac-(salen-1)CoI. In addition, we address the effects of the salen ligand and catalyst axial ligand/initiator (X) used, the reaction conditions, and the inclusion of [PPN]Cl ([PPN]Cl = bis(triphenylphosphine)iminium chloride) cocatalysts on activity and stereospecificity for the copolymerization. With these catalysts, we obtain PCHC with various stereochemical architectures, including syndiotactic PCHC, a previously unreported PCHC microstructure. Using Bernoullian statistical methods, we assign PCHC tetrad and triad sequences for the 13C NMR spectra of these polymers in the carbonyl and methylene regions, respectively.
Results and discussion
Catalyst synthesis
Oxidation of commercially available (R,R)-(salen-1)Co readily occurs in the presence of organic acids to yield the corresponding (R,R)-(salen-1)CoOAc,20 (R,R)-(salen-1)CoOTs (OTs = p-tolylsulfonate),21 and (R,R)-(salen-1)CoOBzF5.6 (R,R)-(salen-1)CoOTs can be further modified through metathesis reactions with the desired NaX salt, affording (R,R)-(salen-1)CoX (X = Cl, Br, I).6,21Rac-(salen-1)CoX catalysts were prepared by a 50 : 50 mixture of (R,R)-(salen-1)Co and (S,S)-(salen-1)Co. A variety of other (salen)CoX complexes were synthesized, starting with an aldehyde/diamine Schiff-base condensation to generate the desired salen-ligand, followed by metallation with cobalt(II) acetate tetrahydrate and subsequent oxidation reactions. The complete series of (salen)CoX complexes used in this study is given in Fig. 3. Although the majority of the (salen)CoX complexes were obtained as powders, in limited cases X-ray quality crystals were obtained from slow evaporation of benzene ((R,R)-(salen-1)CoCl) or by slow evaporation of a methylene chloride/hexane (1:1) solution (rac-(salen-1)CoI). The X-ray crystal structures of (R,R)-(salen-1)CoCl and the (R,R)-(salen-1)CoI component of rac-(salen-1)CoI with selected bond angles and bond lengths are shown in Fig. 4 and 5, respectively. In both cases, the (salen-1)CoX complex is pseudo square-pyramidal with the Co located slightly above the plane of the salen ligand in the direction of the axial ligand. | ||
| Fig. 3 (salen)CoX catalysts for cyclohexene oxide/CO2 copolymerization (X = halide or carboxylate; cumyl = CMe2Ph). | ||
 | ||
| Fig. 4 ORTEP drawing of (R,R)-(salen-1)CoCl (non-hydrogen atoms) with thermal ellipsoids drawn at the 40% probability level. Selected bond lengths (Å) and angles (°): Co(1)–Cl(1) 2.330(2), Co(1)–O(1) 1.845(5), Co(1)–O(2) 1.844(5), Co(1)–N(1) 1.882(6), Co(1)–N(2) 1.887(6); O(1)–Co(1)–Cl(1) 102.7(2), O(2)–Co(1)–Cl(1) 98.3(2), N(1)–Co(1)–Cl(1) 93.9(2), N(2)–Co(1)–Cl(1) 95.6(2). | ||
 | ||
| Fig. 5 ORTEP drawing of the (R,R)-(salen-1)CoI component of rac-(salen-1)CoI (non-hydrogen atoms) with thermal ellipsoids drawn at the 40% probability level. Selected bond lengths (Å) and bond angles (°): Co(1)–I(1), 2.714(1), Co(1)–O(1) 1.828(3), Co(1)–O(2) 1.858(3), Co(1)–N(1) 1.877(4), Co(1)–N(2) 1.866(4); O(1)–Co(1)–I(1) 100.6(1), O(2)–Co(1)–I(1) 95.8(1), N(1)–Co(1)–I(1) 92.8(1), N(2)–Co(1)–I(1) 98.2(1). | ||
 | ||
| Fig. 6 Carbonyl region of the 13C NMR spectrum (CDCl3, 125 MHz) of poly(cyclcohexene carbonate) generated using catalyst (R,R)-(salen-1)CoI. | ||
The CHO/CO2 copolymerization data for (R,R)-(salen-1)CoX (X = Cl, Br, I, OAc, OBzF5) catalysts are listed in Table 1. While most of the catalyst activities were quite similar (entries 1–3, 5), when acetate was used as an initiating group, catalyst activity decreases substantially (entry 4). Overall, (R,R)-(salen-1)CoX catalysts had turnover frequencies (TOF)s ≤16 h−1 for CHO/CO2 copolymerization. As much higher activities were observed with these catalyst systems for PO/CO2 copolymerization under comparable reaction conditions,6 we suspect that reaction rates are hindered by the more sterically bulky, less reactive CHO monomer. Generally, variation of the axial ligand/initiator had little influence on the syndiotacticity of the resultant PCHC, however, when iodide was used, the resultant PCHC was exceptionally syndiotactic. Furthermore, the role of the iodide axial ligand in the CHO/CO2 copolymerization is under current investigation.
| Entry | Catalyst | Yieldb (%) | TOFc/h−1 | M n d/kg mol−1 | M w/Mnd | r-Centered tetradse (%) |
|---|---|---|---|---|---|---|
| a Copolymerizations run in neat cyclohexene oxide (CHO) at 22 °C for 20 h at 54.4 atm of CO2 with [CHO] : [Co] = 500 : 1. In all cases, cyclohexene carbonate byproduct is not observed and product poly(cyclohexene carbonate)s (PCHC)s have ≥96% carbonate linkages as determined by 1H NMR spectroscopy (CDCl3, 300 MHz). b Based on isolated PCHC yield. c Turnover frequency = mol CHO (mol Co)−1 h−1. d Determined by gel permeation chromatography calibrated with polystyrene standards in tetrahydrofuran at 40 °C. e Determined by 13C NMR spectroscopy (CDCl3, 125 MHz). | ||||||
| 1 | (R,R)-(salen-1)CoCl | 62 | 15 | 19.7 | 1.27 | 68 |
| 2 | (R,R)-(salen-1)CoBr | 66 | 16 | 20.3 | 1.23 | 66 |
| 3 | (R,R)-(salen-1)CoI | 51 | 13 | 17.3 | 1.30 | 75 |
| 4 | (R,R)-(salen-1)CoOAc | 27 | 7 | 9.8 | 1.28 | 64 |
| 5 | (R,R)-(salen-1)CoOBzF5 | 53 | 13 | 16.4 | 1.30 | 66 |
In all cases, the PCHCs we obtained exhibit Mn values lower than the corresponding theoretical values despite the absence of chain transfer agents. Notably, this was not observed with these catalyst systems for PO/CO2 copolymerization under comparable reaction conditions.6 As such, we are currently looking into the possibilities of chain transfer to monomer or trace water and/or the formation of cyclic polymer. The syndiotactic PCHCs we obtain are amorphous, with no observable melting point transition. In an effort to obtain more crystalline PCHC we are presently synthesizing syndiotactic PCHCs with higher Mn values.
It has been shown previously that minor alterations of the salen ligands in their chromium-, cobalt- or aluminium-based complexes have dramatic effects on epoxide/CO2 copolymerization activity.6,8–10,16,17 To explore the relationship of the structural features of the salen ligand to catalyst performance in our systems, we synthesized a diverse series of (salen)CoX complexes (Fig. 3) for use in CHO/CO2 copolymerization. Table 2 lists the CHO/CO2 copolymerization data for a series of (salen)CoBr catalysts, in which systematic changes in the diimine backbone have been employed in order to study their impact on polymerization behavior. In all cases a [CHO] : [Co] loading of 500 : 1 was applied while using high CO2 pressures (54.4 atm). Catalyst (R,R)-(salen-1)CoBr produces PCHC with 66% r-centered tetrads and a TOF of 16 h−1 (entry 1). Alternatively, its racemic analogue (rac-(salen-1)CoBr) is more syndiospecific, generating PCHC with 78% r-centered tetrads (entry 2). Increasing the steric bulk on the catalyst backbone ((R,R)-(salen-2)CoBr) reduces the TOF to 2 h−1 and yields PCHC with only 61% r-centered tetrads (entry 3). Complex (salen-3)CoBr, with a phenylene diimine backbone, has very low activity for the CHO/CO2 copolymerization with only trace PCHC observed after 20 h (entry 4). Although the similar complex (salen-3)CoOAc is highly active for isospecific PO homopolymerization,23 (salen-3)CoBr does not catalyze this transformation. The CHO/CO2 copolymerization rate is also reduced compared to that observed with (R,R)-(salen-1)CoBr when a 1,1-dimethyl substituted backbone is used ((salen-4)CoBr) (entry 5). However, decreasing the steric bulk with (salen-5)CoBr or rac-(salen-6)CoBr results in a substantial increase in catalyst activity while maintaining the high syndiospecificity initially observed (entries 6 and 7). In fact, complex rac-(salen-6)CoBr produces PCHC with 81% r-centered tetrads, which is the greatest syndiotacticity that we have observed in PCHC formation to date. Interestingly, use of the most active catalysts ((salen-5)CoBr or rac-(salen-6)CoBr) results in very high conversions even though the polymerization is run in neat CHO (entries 6 and 7). We suspect that at 54.4 atm, CO2 plays a role as a solvent and/or lowers the viscosity of the reaction mixture such that the polymerization can proceed to high PCHC yields. Finally, based on a shorter reaction time, catalyst rac-(salen-6)CoBr exhibited high activity (TOF = 98 h−1) (entry 8). In general, we found that for (salen)CoBr catalysts, the highest activity and syndiospecificity were both achieved by using a racemic ligand backbone with minimal steric hindrance.
| Entry | Catalyst | Time/h | Yieldb(%) | TOFc/h−1 | M n d/kg mol−1 | M w/Mnd | r-Centered tetradse (%) |
|---|---|---|---|---|---|---|---|
| a Copolymerizations run in neat cyclohexene oxide (CHO) at 22 °C with [CHO] : [Co] = 500 : 1 at 54.4 atm of CO2. In all cases, cyclohexene carbonate byproduct is not observed and product poly(cyclohexene carbonate)s (PCHC)s have ≥96% carbonate linkages as determined by 1H NMR spectroscopy (CDCl3, 300 MHz). b Based on isolated PCHC yield. c Turnover frequency = mol CHO (mol Co)−1 h−1. d Determined by gel permeation chromatography calibrated with polystyrene standards in tetrahydrofuran at 40 °C. e Determined by 13C NMR spectroscopy (CDCl3, 125 MHz). | |||||||
| 1 | (R,R)-(salen-1)CoBr | 20 | 66 | 16 | 20.3 | 1.23 | 66 |
| 2 | rac-(salen-1)CoBr | 20 | 64 | 16 | 23.7 | 1.22 | 78 |
| 3 | (R,R)-(salen-2)CoBr | 20 | 8 | 2 | 7.7 | 1.46 | 61 |
| 4 | (salen-3)CoBr | 20 | <1 | NA | NA | NA | NA |
| 5 | (salen-4)CoBr | 20 | 35 | 9 | 14.2 | 1.35 | 75 |
| 6 | (salen-5)CoBr | 20 | 90 | 22 | 26.3 | 1.24 | 75 |
| 7 | rac-(salen-6)CoBr | 20 | 87 | 22 | 30.3 | 1.28 | 80 |
| 8 | rac-(salen-6)CoBr | 3 | 59 | 98 | 21.0 | 1.32 | 81 |
Using the highly active rac-propyldiimine ligand backbone, we investigated the influence of the phenolate ortho and para substituents on (salen)CoX catalyzed CHO/CO2 copolymerization (Table 3). It is notable that the catalyst axial ligand X = OBzF5 was used in this study (instead of X = Br as above) because the complexes rac-(salen-8)CoBr and rac-(salen-9)CoBr proved too soluble to purify by washing with pentane. The CHO/CO2 copolymerization using catalyst rac-(salen-6)CoOBzF5 is highly active with a TOF of 93 h−1 yielding PCHC with 76% r-centered tetrads (entry 1). These values are similar to those observed with the analogous X = Br complex (Table 2, entry 8). Increasing the steric bulk of the phenolate ortho and para substituents using rac-(salen-7)CoOBzF5 is detrimental, decreasing catalyst activity to a TOF of 5 h−1 and reducing syndiotacticity to 65% (entry 2). Alternatively, using catalyst rac-(salen-8)CoOBzF5 where the steric bulk of the ortho and para positions is virtually nonexistent results in a complete loss in stereospecificity yielding atactic PCHC (entry 3). In the intermediate cases which incorporating tert-butyl groups in the ortho positions while varying the para positions of the phenolate to either H or Br (rac-(salen-9)CoOBzF5 or rac-(salen-10)CoOBzF5, respectively) catalyst activity and syndiospecificity are decreased (entries 4 and 5). Largely, the salen ligand with tert-butyl groups in both the ortho and para positions of the phenolate moiety provides the ideal steric bulk for use in the CHO/CO2 copolymerizations, giving high activity and syndiospecificity.
| Entry | Catalyst | Time/h | Yieldb (%) | TOFc/h−1 | M n d/kg mol−1 | M w/Mnd | r-Centered tetradse(%) |
|---|---|---|---|---|---|---|---|
| a Copolymerizations run in neat cyclohexene oxide (CHO) at 22 °C with [CHO] : [Co] = 500 : 1 at 54.4 atm of CO2. In all cases, cyclohexene carbonate byproduct is not observed and product poly(cyclohexene carbonate)s (PCHC)s have ≥ 96% carbonate linkages as determined by 1H NMR spectroscopy (CDCl3, 300 MHz). b Based on isolated PCHC yield. c Turnover frequency = mol CHO (mol Co)−1 h−1. d Determined by gel permeation chromatography calibrated with polystyrene standards in tetrahydrofuran at 40 °C. e Determined by 13C NMR spectroscopy (CDCl3, 125 MHz). f 93% carbonate linkages. | |||||||
| 1 | rac-(salen-6)CoOBzF5 | 3 | 56 | 93 | 22.3 | 1.35 | 76 |
| 2 | rac-(salen-7)CoOBzF5 | 20 | 22 | 5 | 11.7 | 1.40 | 65 |
| 3 | rac-(salen-8)CoOBzF5 | 3 | 19 | 32 | 10.9 | 1.22 | 50 |
| 4 | rac-(salen-9)CoOBzF5 | 3 | 39 | 65 | 14.5 | 1.42 | 66 |
| 5f | rac-(salen-10)CoOBzF5 | 3 | 30 | 50 | 14.6 | 1.38 | 75 |
As enantiomerically pure, racemic and achiral (salen)CoX catalysts are all syndiospecific for CHO/CO2 copolymerization, we suspect that monomer enchainment proceeds by a chain-end control mechanism where the stereogenic center from the last enchained monomer unit influences the stereochemical outcome of subsequent monomer addition. Notably, this propagation scheme becomes competitive with an enantiomorphic-site control pathway in the case of some (salen)CoX catalysts, resulting in a loss of syndiospecificity. Based on the results presented above, we believe that an enantiomorphic-site control mechanism is competitive for (salen)CoX catalysts with a sterically bulky diimine backbone or cumyl ortho and para phenolate substituents. Also, in the case of catalyst rac-(salen-8)CoOBzF5, where the steric bulk of the salen ligand is minimal, all stereochemical direction is lost, showing that moderate ligand steric bulk is necessary for chain-end control. Overall, the optimized ligand structure for maximum catalyst activity and syndiospecificity is achieved with minimal steric bulk on the salen diimine backbone and tert-butyl moieties present in the ortho and para positions of the phenolate.
In an effort to investigate the relationship between PCHC tacticity and the applied CO2 pressure, we proceeded to explore a series of (salen)CoX catalysts for the CHO/CO2 copolymerization at low CO2 pressures (6.8 atm) (Table 4). In all cases, catalyst activity decreased notably and reaction times were increased to 48 h or longer in order to achieve appreciable conversion. In addition, the CHO/CO2 copolymerizations at low CO2 pressures generally resulted in a loss of syndiospecificity and decreased carbonate incorporation in the resultant PCHC. Catalyst (R,R)-(salen-1)CoBr showed no syndiospecificity yielding atactic PCHC with only 90% carbonate incorporation (entry 1). Using our most sterically bulky complex (R,R)-(salen-11)CoBr, however, surprisingly produced isoenriched PCHC with 65% m-centered tetrads and 99% carbonate incorporation (entry 2). Interestingly, the sterically bulky catalyst (R,R)-(salen-2)CoBr incorporated very little CO2, producing predominantly poly(cyclohexene oxide) (entry 3). One of our best catalysts at high pressure (rac-(salen-6)CoOBzF5) showed a decrease in syndiospecificity with CO2 at 6.8 atm generating PCHC with only 60% r-centered tetrads and 88% carbonate linkages (entry 4), whereas changing its axial ligand to Br completely eliminated its syndiospecificity (entry 5). To further correlate CO2 pressure to catalyst activity and syndiospecificity, we continued to investigate the CHO/CO2 copolymerization using rac-(salen-6)CoOBzF5 at a series of different CO2 pressures. Results from these experiments are presented graphically in Fig. 7. It is notable that 54.4 atm is highest pressure we are able to obtain at 22 °C using our standard copolymerization setup and was therefore the highest pressure applied. Decreasing the CO2 pressure from 54.4 atm to 47.6 atm caused a dramatic loss in catalyst activity, consistent with the results we observe with (salen)CoX catalysts for PO/CO2 copolymerization.5 Further reductions in CO2 pressure resulted in a slow but steady decrease in catalyst activity. The syndiospecificity of catalyst rac-(salen-6)CoOBzF5 is optimized at 54.4 atm of CO2 and parallels the catalyst activity at all pressures employed.
| Entry | Catalyst | Time/h | Yieldb (%) | TOFc/h−1 | Carbonate linkagesd (%) | M n e/kg mol−1 | M w/Mne | r-Centered tetradsf (%) |
|---|---|---|---|---|---|---|---|---|
| a Copolymerizations run in neat cyclohexene oxide (CHO) at 22 °C with [CHO] : [Co] = 500 : 1 at 6.8 atm of CO2. In all cases, cyclohexene carbonate byproduct is not observed as determined by 1H NMR spectroscopy (CDCl3, 300 MHz). b Based on isolated poly(cyclohexene carbonate) yield. c Turnover frequency = mol CHO (mol Co)−1 h−1. d Determined by 1H NMR spectroscopy (CDCl3, 300 MHz). e Determined by gel permeation chromatography calibrated with polystyrene standards in tetrahydrofuran at 40 °C. f Determined by 13C NMR spectroscopy (CDCl3, 125 MHz). | ||||||||
| 1 | (R,R)-(salen-1)CoBr | 48 | 35 | 4 | 90 | 12.6 | 1.23 | 50 |
| 2 | (R,R)-(salen-11)CoBr | 72 | 32 | 2 | 99 | 8.9 | 1.19 | 35 |
| 3 | (R,R)-(salen-2)CoBr | 72 | 10 | <1 | 20 | 10.8 | 1.83 | NA |
| 4 | rac-(salen-6)CoOBzF5 | 48 | 35 | 4 | 88 | 9.3 | 1.27 | 60 |
| 5 | rac-(salen-6)CoBr | 72 | 17 | 1 | 86 | 6.5 | 1.33 | 51 |
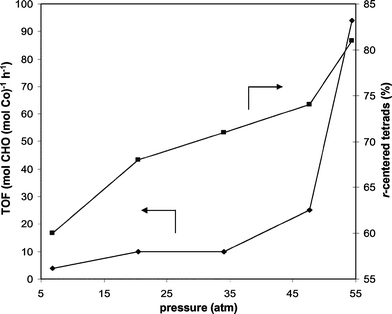 | ||
| Fig. 7 Effect of CO2 pressure on catalyst activity and syndiospecificity of CHO/CO2 copolymerization with rac-(salen-6)CoOBzF5. | ||
| Entry | Catalyst | CO2 Pressure/atm | Time/h | Yieldb (%) | TOFc/h−1 | M n d/kg mol−1 | M w/Mnd | r-Centered tetradse (%) |
|---|---|---|---|---|---|---|---|---|
| a Copolymerizations run in neat cyclohexene oxide (CHO) at 22 °C with [CHO] : [Co] : [[PPN]Cl] = 1000 : 1 : 1. In all cases, cyclohexene carbonate byproduct is not observed and product poly(cyclohexene carbonate)s have ≥97% carbonate linkages as determined by 1H NMR spectroscopy (CDCl3, 300 MHz). b Based on isolated PCHC yield. c Turnover frequency = mol CHO (mol Co)−1 h−1. d Determined by gel permeation chromatography calibrated with polystyrene standards in tetrahydrofuran at 40 °C. e Determined by 13C NMR spectroscopy (CDCl3, 125 MHz). f 70 °C. | ||||||||
| 1 | (R,R)-(salen-1)CoBr | 54.4 | 6 | 34 | 57 | 14.8 | 1.38 | 62 |
| 2 | (R,R)-(salen-1)CoOBzF5 | 54.4 | 6 | 40 | 67 | 15.0 | 1.36 | 59 |
| 3 | rac-(salen-6)CoOBzF5 | 54.4 | 6 | 18 | 30 | 10.1 | 1.44 | 62 |
| 4 | (R,R)-(salen-2)CoOBzF5 | 54.4 | 6 | 36 | 60 | 11.8 | 1.38 | 50 |
| 5 | (R,R)-(salen-11)CoOBzF5 | 54.4 | 6 | 27 | 45 | 10.6 | 1.41 | 51 |
| 6 | (R,R)-(salen-1)CoBr | 6.8 | 2 | 22 | 110 | 8.2 | 1.24 | 47 |
| 7 | (R,R)-(salen-1)CoOBzF5 | 6.8 | 2 | 32 | 160 | 14.9 | 1.14 | 49 |
| 8 | rac-(salen-6)CoOBzF5 | 6.8 | 2 | 18 | 90 | 7.4 | 1.22 | 56 |
| 9 | (R,R)-(salen-2)CoOBzF5 | 6.8 | 2 | 13 | 65 | 6.6 | 1.18 | 42 |
| 10 | (R,R)-(salen-11)CoOBzF5 | 6.8 | 14 | 44 | 31 | 15.3 | 1.17 | 44 |
| 11f | (R,R)-(salen-1)CoOBzF5 | 6.8 | 1 | 44 | 440 | 11.9 | 1.23 | 47 |
We initially applied [PPN]Cl cocatalysts to the (salen)CoX catalyzed CHO/CO2 copolymerization at high CO2 pressures in an effort to maximize the catalyst syndiospecificity. Interestingly, catalyst system (R,R)-(salen-1)CoBr/[PPN]Cl showed a loss in syndiospecificity from use of (R,R)-(salen-1)CoBr alone. Additionally, only a small increase in activity was observed upon the addition of the [PPN]Cl cocatalyst. By varying the catalyst axial ligand in (R,R)-(salen-1)CoX/[PPN]Cl from Br to OBzF5 we observe little change in activity and syndiospecificity (entries 1 and 2). Surprisingly, catalyst rac-(salen-6)CoOBzF5, one of the fastest catalysts without an additive, was less active for CHO/CO2 copolymerization upon the inclusion of [PPN]Cl (entry 3). Similar to the (R,R)-(salen)CoX/[PPN]Cl (X = Br, OBzF5) catalyst systems, little syndiospecificity was realized in this case.
The loss of syndiospecificity upon the addition of [PPN]Cl to the (salen)CoX catalyzed CHO/CO2 copolymerization indicates that a chain-end control mechanism exerts less control on the stereospecificity of the polymerization. We therefore explored (salen)CoOBzF5 catalysts with sterically bulky salen ligands in an effort to invoke a site-control mechanism for isotactic PCHC. Application of (R,R)-(salen-2)CoOBzF5/[PPN]Cl, with a more sterically bulky diimine backbone, had comparable activity to (R,R)-(salen-1)CoOBzF5/[PPN]Cl however the resultant PCHC was atactic (entry 4). Additionally, employing a more sterically bulky phenolate moiety ((R,R)-(salen-11)CoOBzF5/[PPN]Cl) also generated atactic PCHC with slightly lower activity (entry 5). In general, addition of [PPN]Cl cocatalysts to (salen)CoX catalyzed CHO/CO2 copolymerization at high CO2 pressures results in a loss in stereospecificity with little or no increase in activity.
At low CO2 pressures (6.8 atm), catalyst activities increase dramatically upon the addition of [PPN]Cl to the copolymerization, although product PCHCs become virtually atactic. Catalyst systems (R,R)-(salen-1)CoX/[PPN]Cl (X = Br, OBzF5) are highly active with TOFs of 110 and 160 h−1, respectively, producing atactic PCHC (entries 6 and 7). rac-(salen-6)CoOBzF5/[PPN]Cl was less active for CHO/CO2 copolymerization with a TOF of 90 h−1 (entry 8), while use of the more sterically bulky (R,R)-(salen-2)CoOBzF5/[PPN]Cl or (R,R)-(salen-11)CoOBzF5/[PPN]Cl resulted in even lower TOFs of 65 and 31 h−1 (entries 9 and 10). Overall, when using the cocatalyst [PPN]Cl, (R,R)-(salen-1)CoOBzF5 is the most active catalyst for CHO/CO2 copolymerization at low CO2 pressures.
Although use of elevated temperatures for (R,R)-(salen-1)CoOBzF5/[PPN]Cl catalyzed PO/CO2 copolymerization predominantly forms propylene carbonate, we did not observe the analogous cyclohexene carbonate upon heating the CHO/CO2 copolymerization. Furthermore, at the optimized temperature of 70 °C, catalyst system (R,R)-(salen-1)CoOBzF5/[PPN]Cl generates PCHC with a TOF of 440 h−1 (entry 11). Overall, this is the fastest catalyst activity we observe with the (salen)CoX/[PPN]Cl systems, although it still is inferior to zinc- and chromium-based alternatives.1
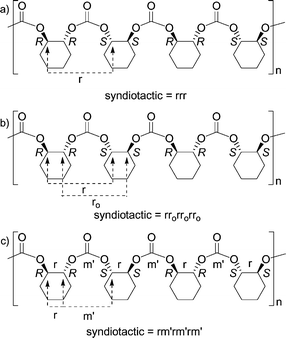 | ||
| Fig. 8 Classification of poly(cyclohexene carbonate) according to the relative stereochemistry of the carbon at which the main chain enters the cyclohexane unit (a), the carbon at which the main chain enters and leaves the cyclohexane unit (b) and adjacent carbons (c). | ||
In earlier reports, we and Nozaki assigned the carbonyl region of the 13C NMR spectrum for isotactic PCHC,13,14 which was further corroborated using statistical methods.14 Additionally, Nozaki has assigned the resonances corresponding to [mmm] and [rrr] tetrad sequences by synthesizing model polycyclohexene carbonate oligomers.19 In each case, all [mmm], [mmr] and [rmr] tetrads were correlated to one central resonance at 153.7 ppm and the remaining r-centered tetrads resided in the 153.3–153.1 ppm range. Isotactic PCHC was therefore characterized by a strong resonance at 153.7 ppm with a smaller series of resonances from 153.3–153.1 ppm due to stereoerrors. Statistically (enantiomorphic-site control), the most prominent stereoerrors in isotactic PCHC are [mrr] and [mmr] tetrads, with much smaller contributions from the remaining [rrr], [rmr] and [mrm] tetrads. In contrast, the PCHC that we obtain with catalyst rac-(salen-6)CoOBzF5 revealed a 13C NMR spectrum with four distinct resonances at 152.94, 153.06, 153.12 and 153.70 ppm integrating to 9, 56, 24 and 11%, respectively (Fig. 9(a)).22 The peak at 152.94 ppm is unique to this polymer and was unobserved in isotactic PCHC.14,19 Based on the previous assignments, we correlated the most intense resonance at 153.06 ppm to the [rrr] tetrad, with a relative integration of 56%. Using Bernoullian (chain-end control) statistical methods, such that the calculated probability of a racemo diad (Pr) was determined by the maximized weighted fit between calculated and observed (obs) tetrad concentrations we calculate 56% [rrr] tetrads, 24% [rrm] tetrads and 12% [rmr] tetrads, with the remaining tetrads: [mmr] = 5%, [mrm] = 2% and [mmm] = 1% (Table 6).25 The previous reports assigning all m-centered tetrads to a central resonance at 153.7 ppm do not agree with these statistical calculations. We therefore propose that the [rmr] tetrad corresponds to the most upfield resonance at 152.94 ppm in our spectrum, an unobservable stereoerror in isotactic PCHC samples, due to its low probability, leading to a previously incorrect assignment. It is also important to note that the [rmr] tetrad was not investigated in previous syntheses of model oligomers,19 precluding its direct assignment through chemical means. Finally, we attribute the resonance at 153.11 ppm to the [rrm] sequence, leaving the remaining [mmr] and [mmm] tetrads collectively at 153.70 ppm and the negligible [mrm] tetrad absent. It is notable that the [rrm] resonance in the 13C NMR spectrum of isotactic PCHC (Fig. 9(d)) is shifted 0.1 ppm upfield from that of syndiotactic PCHC (Fig. 9(a) and (b)), which we attribute to the minor influence of the long-range polymer tacticity on this shift.
| P r | [mmm] + [mmr] | [rmr] | [rrr] | [rrm] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Table/entry | Calc. (%) | Obs. (%) | Calc. (%) | Obs. (%) | Calc. (%) | Obs. (%) | Calc. (%) | Obs. (%) | Calc. (%) | Obs. (%) |
| a Calculated (calc.) statistical occurrences for each tetrad were determined through application of Bernoullian (chain-end control) statistical methods where the calculated probability of a racemo diad (Pr) was determined by the maximized weighted fit between calculated and observed (obs.) tetrad concentrations. All observed values determined by quantitative 13C NMR spectroscopy (CDCl3, 125 MHz) with deconvolution of 13C NMR resonances. In all cases, the [mrm] tetrad concentrations (calc values between 2–7%) were too small for analysis by the deconvolution methods used and were therefore omitted. | ||||||||||
| 2/7 | 82 | 80 | 6 | 10 | 12 | 12 | 55 | 55 | 24 | 24 |
| 3/1 | 82 | 76 | 6 | 9 | 12 | 11 | 56 | 56 | 24 | 24 |
| 3/5 | 81 | 75 | 7 | 11 | 13 | 10 | 53 | 53 | 25 | 26 |
| 2/2 | 79 | 78 | 8 | 11 | 13 | 11 | 49 | 49 | 26 | 29 |
| 2/6 | 77 | 75 | 9 | 15 | 14 | 14 | 46 | 46 | 27 | 26 |
| 1/5 | 71 | 66 | 15 | 25 | 15 | 13 | 35 | 35 | 29 | 26 |
| 1/2 | 68 | 66 | 17 | 24 | 15 | 16 | 32 | 32 | 30 | 28 |
| 3/2 | 66 | 65 | 19 | 26 | 15 | 15 | 29 | 29 | 30 | 31 |
 | ||
| Fig. 9 Carbonyl region of the 13C NMR spectra (125 MHz, CDCl3) of poly(cyclohexene carbonate)s generated using CHO/CO2 (54.4 atm) with catalyst rac-(salen-6)CoOBzF5 (a), CHO/CO2 (54.4 atm) with catalyst (R,R)-(salen-1)CoOBzF5 (b), CHO/CO2 (54.4 atm) with catalyst rac-(salen-8)CoOBzF5 (c) and CHO/CO2 (6.8 atm) with catalyst (R,R)-(salen-11)CoBr (d). | ||
To further support the PCHC 13C NMR assignments that we propose, we applied Bernoullian statistical methods and compared the observed concentration of each tetrad with its corresponding calculated amount. Table 6 compares observed tetrad concentrations to their calculated statistical occurrences for a series of syndiotactic PCHCs. In most cases, the observed and calculated concentrations are in close agreement, and are within the expected error of the deconvolution methods used. Notably, the PCHCs with the highest observed % r-centered tetrads overall have relative tetrad concentrations that best agree with the corresponding calculated values.
The 13C NMR spectrum of less syndiotactic PCHC, as generated from catalyst (R,R)-(salen-1)CoOBzF5 shows the [rrr] and [rrm] tetrad resonances approach each other in magnitude with a more significant contribution from the [mmm] and [mmr] tetrads (Fig. 9(b)). Using Bernoullian statistical methods, we calculate relative tetrad occurrences of [mmm] + [mmr] = 15%, [rmr] = 15%, [rrr] = 35%, [rrm] = 29% and [mrm] = 6% (Table 6). This is indeed consistent with what we observe, where a previously minor resonance at 153.18 ppm is now more prominent, which we assign to the [mrm] tetrad. In this spectrum we detect fine splitting of various resonances in the 153.2–152.9 ppm region and hypothesize that due to the increase of PCHC stereoerrors we are able to observe signals at greater than tetrad resolution. Furthermore, the 13C NMR spectrum of the same PCHC sample using a lower resolution spectrometer showed only the five resonances at 152.95, 153.06, 153.12, 153.17 and 153.70 ppm.
The 13C NMR spectrum of PCHC, as generated from catalyst rac-(salen-8)CoOBzF5, reveals that the [mmm] + [mmr] tetrads integrate equally to those of the remaining tetrads, indicative of the formation of atactic polymer (Fig. 9(c)). Finally, isoenriched PCHC, as generated from catalyst (R,R)-(salen-11)CoBr while carrying out the CHO/CO2 copolymerization at low CO2 pressures (6.8 atm), has a 13C NMR spectrum similar to that of previously reported isotactic PCHC.13,14 In this case we observe a large resonance correlating to the combined [mmm] + [mmr] tetrads at 153.70 ppm and additional resonances representing the [rrm] and [mrm] stereoerrors with no detectable [rrr] or [rmr] tetrads (Fig. 9(d)).
The methylene regions in the PCHC 13C NMR spectra also vary with PCHC stereochemistry (Fig. 10). In the case of syndiotactic PCHC, two series of two adjacent resonances are observed which we assign to the [rr] and [mr] triads corresponding to the non-equivalent methylene carbons in the PCHC backbone (Fig. 10(a)). In each case, the [mr] and [rm] triad resonances cannot be distinguished. Furthermore, we suspect that one of these two triads has the same 13C NMR shift as the [rr] triad. The 13C NMR spectrum of less syndiotactic PCHC shows [rr] triads of decreased intensity relative to the [mr] triad resonances and additional downfield resonances are detected which we assign to the [mm] triads (Fig. 10(b)). Alternatively, the 13C NMR spectrum of atactic PCHC reveals more intense [mm] triad resonances compared to the syndiotactic PCHC samples, with the [mr] and [rr] triad resonances also present (Fig. 10(c)). In this case, we observe some tetrad resolution where the [rr] triad resonances are split into their [rrm] and [rrr] components and the [mr] triad resonances are broadened. Finally, the 13C NMR spectrum of isoenriched PCHC shows that the downfield [mm] triad resonances in the methylene region have the greatest intensities relative to the [mr] and [rr] triad signals (Fig. 10(d)).
 | ||
| Fig. 10 Methylene region of the 13C NMR spectra (125 MHz, CDCl3) of poly(cyclohexene carbonate)s generated using CHO/CO2 (54.4 atm) with catalyst rac-(salen-6)CoOBzF5 (a), CHO/CO2 (54.4 atm) with catalyst (R,R)-(salen-1)CoOBzF5 (b), CHO/CO2 (54.4 atm) with catalyst rac-(salen-8)CoOBzF5 (c) and CHO/CO2 (6.8 atm) with catalyst (R,R)-(salen-11)CoBr (d). | ||
Conclusions
We have explored a series of (salen)CoX catalysts for the alternating copolymerization of CHO and CO2. These catalysts provide a viable route to syndiotactic PCHC, a previously unreported PCHC microstructure. The structural properties of the salen ligand in these catalysts dictate the syndiotacticity of the resultant PCHC, a correlation which we attribute to a competition of chain-end and enantiomorphic-site control mechanisms. Additionally, catalyst syndiospecificity is pressure dependent, with the best selectivity realized employing high CO2 pressures. PCHC with as high as 81% r-centered tetrads was obtained using this class of catalyst. Inclusion of [PPN]Cl cocatalysts to the (salen)CoX catalyzed CHO/CO2 copolymerization results in an increase in catalyst activity in most cases with an overall loss in syndiospecificity. Under optimized conditions, these systems achieve a TOF of 440 h−1 for the generation of PCHC.Experimental
Materials and physical measurements
All air- or water-sensitive reactions were carried out under dry nitrogen using an MBraun Labmaster drybox or standard Schlenk-line techniques. Methylene chloride and diethyl ether were dried and degassed by passing through a column of activated alumina and by sparging with dry nitrogen. (S,S)- and (R,R)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-diaminocyclohexane cobalt ((S,S)-(salen-1)Co and (R,R)-(salen-1)Co) were purchased from Aldrich and recrystallized from methylene chloride and methanol. Bis(triphenylphosphine)iminium chloride ([PPN]Cl) was purchased from Strem and recrystallized from dry methylene chloride and diethyl ether under nitrogen before use. CHO was dried over calcium hydride and vacuum transferred before use. CO2 (99.998% purity) was purchased from Airgas and passed over a column of 4 Å molecular sieves. All other reagents were purchased from commercial sources and used as received. Varian Mercury (300 MHz) and Inova (500 MHz) spectrometers were used to record 1H NMR spectra, which were referenced vs. residual nondeuterated solvent shifts. An Inova (500 MHz) spectrometer was used to record 13C NMR spectra (at 125 MHz), which were referenced vs. residual deuterated solvent shifts. C6F6 (−162.90 ppm) was used as an internal reference for all 19F NMR spectra. GPC analyses were carried out using a Waters instrument (M515 pump, U6 K injector) equipped with Waters UV486 and Waters 2410 detectors, and four 5-µm PL Gel columns (Polymer Laboratories; 100, 500, 1000 Å and Mixed C porosities) in series. The GPC columns were eluted with THF at 40 °C at 1 mL min−1 and were calibrated using 23 monodisperse polystyrene standards. IR spectra were measured on a Mattson Research Series FTIR. High resolution mass spectra were obtained from the Mass Spectrometry Laboratory, School of Chemical Sciences, University of Illinois. All (salen)CoX (X = Br or OBzF5) complexes reveal axial ligand (X) loss in the (EI) mass spectra attributable to the poor stability of this complex under the applied conditions. In the case of all (salen)CoOBzF5 complexes, carbons on the phenyl group of pentafluorobenzoate were not assigned in the 13C NMR spectra due to complex carbon fluorine splitting patterns.Ligand preparation
Schiff-base condensation of salicylaldehydes with 1,2-diamines to generate salen-type ligands is readily achieved by refluxing 2 eq of the parent aldehyde and 1 equiv of the parent diamine in absolute ethyl alcohol.26 In most cases, 3 h reaction time is sufficient for obtaining high product yields. Following the removal of solvent in vacuo, the crude yellow solid is recrystalized from ethyl alcohol yielding the product salen ligand in most cases. Alternatively, the tartrate salt of the parent diamine can be employed in the synthesis, where the diamine tartrate is stirred with K2CO3 in water until dissolution prior to reaction with the aldehyde.27 The analytical data and synthetic modifications for new salen ligands are listed below:Preparation of (salen)Co complexes
Complexes (R,R)-(salen-1)Co and (S,S)-(salen-1)Co are commercially available. All other (salen)Co complexes were prepared according to the general procedure for the synthesis of (salen-4)Co. Complexes (R,R)-(salen-2)Co,30 (salen-3)Co,31 and (salen-5)Co32 prepared by this method are in agreement with literature characterization. The analytical data and synthetic modifications for new (salen)Co complexes are listed below:Preparation of (salen)CoX (X = Cl, Br, I, OAc, OBzF5) complexes
The synthesis of (salen-1)CoX (X = Cl, Br, I, OAc, OBzF5) have been described previously.6,20,21 A modified procedure as reported by Jacobsen and coworkers21 for the synthesis of (R,R)-(salen-1)CoCl was applied to the synthesis of all (salen)CoBr complexes with the substitution of NaBr for NaCl.Representative copolymerization procedure
A 100 mL Parr autoclave was heated to 120 °C under vacuum for 4 h, then cooled under vacuum to 22 °C and moved to a drybox. Complex rac-(salen-6)CoOBzF5 (15.6 mg, 0.0201 mmol) and CHO (1.00 mL, 9.88 mmol) were placed in a glass sleeve with a Teflon stir bar inside the Parr autoclave. The autoclave was pressurized to 54.4 atm of CO2 and was left to stir at 22 °C for 3 h. The reactor was vented at 22 °C. A small aliquot of the resultant polymerization mixture was removed from the reactor for 1H NMR and GPC analysis. The remaining polymerization mixture was then dissolved in methylene chloride (5 mL), quenched with 5% HCl solution in methanol (0.2 mL), and precipitated from methanol (30 mL). The polymer was collected and dried in vacuo to constant weight, and the polymer yield was determined (0.799 g, 56.9%).Crystallographic data for structures rac-(salen-1)CoI and (R,R)-(salen-1)CoCl
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2); a = 10.969(4), b = 12.522(5), c = 16.694(7) Å, α = 97.299(9), β = 105.834(9), γ = 106.802(9)°, V = 2058.3(14) Å3, Z = 2, μ = 1.331 mm−1; 10442 reflections collected and 6822 independent reflections (Rint = 0.0493). Final R indices [I > 2σ(I)] R1 = 0.0562, wR2 = 0.1100; R indices (all data) R1 = 0.0996, wR2 = 0.1232.
(no. 2); a = 10.969(4), b = 12.522(5), c = 16.694(7) Å, α = 97.299(9), β = 105.834(9), γ = 106.802(9)°, V = 2058.3(14) Å3, Z = 2, μ = 1.331 mm−1; 10442 reflections collected and 6822 independent reflections (Rint = 0.0493). Final R indices [I > 2σ(I)] R1 = 0.0562, wR2 = 0.1100; R indices (all data) R1 = 0.0996, wR2 = 0.1232.
Crystallographic details for (salen-1)CoX (X = Cl, I) are summamised in the ESI.†
CCDC reference numbers 289855 and 289856.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513107c
Acknowledgements
G. W. C. gratefully acknowledges a Packard Foundation Fellowship in Science and Engineering, and funding from the NSF (CHE-0243605), the Swiss National Science Foundation Fellowship to C. M. T., the Cornell Center for Materials Research (supported through the NSF MRSEC program, DMR-0079992), Sumitomo Chemicals, the Cornell University Nanobiotechnology Center, and the Cornell University Center for Biotechnology, a New York State Center for Advanced Technology.References
- For reviews on epoxide/CO2 copolymerizations, see: (a) B. Ochiai and T. Endo, Prog. Polym. Sci., 2005, 30, 183–215 CrossRef CAS; (b) D. R. Moore and G. W. Coates, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS; (c) H. Sugimoto and S. Inoue, J. Polym. Sci., Part A, 2004, 42, 5561–5573 CrossRef CAS; (d) D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS; (e) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS; (f) M. S. Super and E. J. Beckman, J. Trends Polym. Sci., 1997, 5, 236–240 Search PubMed; (g) W. Kuran, Prog. Polym. Sci., 1998, 23, 919–992 CrossRef CAS.
- (a) S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B, 1969, 7, 287–292 Search PubMed; (b) S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS.
- T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8–13 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831–1833 CAS.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS.
- T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39–48 CrossRef CAS.
- D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS.
- (a) D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144–148 CrossRef CAS; (b) D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622–4629 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024–6034 CrossRef CAS.
- K. Nakano, T. Hiyama and K. Nozaki, Chem. Commun., 2005, 1871–1873 RSC.
- K. Nakano, K. Nozaki and T. Hiyama, J. Am. Chem. Soc., 2003, 125, 5501–5510 CrossRef CAS.
- K. Nozaki, K. Nakano and T. Hiyama, J. Am. Chem. Soc., 1999, 121, 11008–11009 CrossRef CAS.
- M. Cheng, N. A. Darling, E. B. Lobkovsky and G. W. Coates, Chem. Commun., 2000, 2007–2008 RSC.
- D. J. Darensbourg, J. L. Rodgers and C. C. Fang, Inorg. Chem., 2003, 42, 4498–4500 CrossRef CAS.
- R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194–196 CrossRef CAS.
- For further examples of cobalt catalyzed PO/CO2 copolymerization, see: (a) R. L. Paddock and S. T. Nguyen, Macromolecules, 2005, 38, 6251–6253 CrossRef CAS; (b) X. B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS.
- K. Nakano, N. Kosaka, T. Hiyama and K. Nozaki, Dalton Trans., 2003, 4039–4050 RSC.
- K. Nakano, K. Nozaki and T. Hiyama, Macromolecules, 2001, 34, 6325–6332 CrossRef CAS.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef CAS.
- L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360–1362 CrossRef CAS.
- Quantitative 13C NMR spectra of PCHC were deconvoluted prior to integration in order to subtract out the [rmr] resonance from the remaining resonances in the 153.2–152.9 ppm regime. This provided for an approximation of the % r-centered tetrads in the PCHC.
- K. L. Peretti, H. Ajiro, C. T. Cohen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11566–11567 CrossRef CAS.
- M. L. Huggins, G. Natta, V. Desreux and H. Mark, Pure Appl. Chem., 1966, 645–656.
- The Bernoullian (chain-end control) expressions in the Bovey formalism (Pr is the probability of a racemic diad) for tetrad sequences are the following: [rrr] = Pr3; [rrm] = 2[Pr2(1 − Pr)]; [mrm] = Pr(1 − Pr)2; [mmm] = (1 − Pr)3; [mmr] = 2[Pr(1 − Pr)2]; [rmr] = Pr2(1 − Pr).
- W. Zhang and E. N. Jacobsen, J. Org. Chem., 1991, 56, 2296–2298 CrossRef CAS.
- P. J. Pospisil, D. H. Carsten and E. N. Jacobsen, Chem. Eur. J., 1996, 2, 974–980 CrossRef CAS.
- A. E. Cherian, E. B. Lobkovsky and G. W. Coates, Macromolecules, 2005, 38, 6259–6268 CrossRef CAS.
- F. Lam, J. X. Xu and K. S. Chan, J. Org. Chem., 1996, 61, 8414–8418 CrossRef CAS.
- T. Fukuda and T. Katsuki, Tetrahedron, 1997, 53, 7201–7208 CrossRef CAS.
- H. Shimakoshi, H. Takemoto, I. Aritome and Y. Hisaeda, Tetrahedron Lett., 2002, 43, 4809–4812 CrossRef CAS.
- B. Rhodes, S. Rowling, P. Tidswell, S. Woodward and S. M. Brown, J. Mol. Catal. A: Chem., 1997, 116, 375–384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details for (salen-1)CoX (X = Cl, I). See DOI: 10.1039/b513107c |
| This journal is © The Royal Society of Chemistry 2006 |