‘Pincer’ dicarbene complexes of some early transition metals and uranium†
David
Pugh
,
Joseph A.
Wright
,
Sandra
Freeman
and
Andreas A.
Danopoulos
*
School of Chemistry, University of Southampton, Highfield, Southampton, UK SO17 1BJ. E-mail: ad1@soton.ac.uk
First published on 8th November 2005
Abstract
The complexes [(C–N–C)MXn(thf)m] with the ‘pincer’ 2,6-bis(imidazolylidene)pyridine, (C–N–C) = 2,6-bis(arylimidazol-2-ylidene)pyridine, aryl = 2,6-Pri2C6H3, M = V, X = Cl, n = 2, m = 1 1a; M = Cr, X = Cl, n = 2, m = 0, 2a, X = Br, 2b; M = Mn, X = Br, n = 2, m = 0, 3; M = Nb, X = Cl, n = 3, m = 0, 4; and M = U, X = Cl, n = 4, m = 0, 5, were synthesised by (a) substitution of labile tmed (1a), thf (2a, 3, 5) or dme (4) by free (C–N–C) or by (b) reaction of the bisimidazolium salt (CH–N–CH)Br2 with {Cr[N(SiMe3)2]2(thf)2} followed by amine elimination (2b). Attempted alkylation of 1a, 2, 3a and 4 with Grignard or alkyl lithiums gave intractable mixtures, and in one case [reaction of 1a with (mesityl)MgBr] resulted in exchange of Cl by Br (1b). Oxidation of 1a or [(C–N–C)VCl3] with 4-methylmorpholine N-oxide afforded the trans-V(C–N–C)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Cl2, 6, which by reaction with AgBF4 in MeCN gave trans-[V(C–N–C)(O)(MeCN)2][BF4]2, 7. Reaction of 1a with p-tolyl azide gave trans-V(C–N–C)(N-p-tolyl)Cl28. The complex trans-Ti(C–N–C)(NBut)Cl2, 9, was prepared by substitution of the pyridine ligands in Ti(NBut)Cl2(py)3 by C–N–C.
O)Cl2, 6, which by reaction with AgBF4 in MeCN gave trans-[V(C–N–C)(O)(MeCN)2][BF4]2, 7. Reaction of 1a with p-tolyl azide gave trans-V(C–N–C)(N-p-tolyl)Cl28. The complex trans-Ti(C–N–C)(NBut)Cl2, 9, was prepared by substitution of the pyridine ligands in Ti(NBut)Cl2(py)3 by C–N–C.
Introduction
The mer-coordinating ‘pincer’ ligands have been frequently used as versatile ancillary ligands in organometallic chemistry and catalysis.1 In particular, the ‘pincer’ dicarbene ligand C–N–C (I, Scheme 1) provides robust Pd, Ru and Rh complexes by virtue of the strong σ-donating NHC functional groups flanking the central pyridine donor.2 | ||
| Scheme 1 Metal complexes described in this paper: M = V, X = Cl, L = thf, n = 2, m = 1, 1a (method A); M = Cr, X = Cl, n = 2, m = 0, 2a (method A), X = Br, 2b, (method B); M = Mn, X = Br, n = 2, m = 0, 3; M = Nb, X = Cl, n = 3, m = 0, 4 (method A); M = U, X = Cl, n = 4, m = 0, 5, (method A); Ar = 2,6-Pri2C6H3. Reagents and conditions: (i) 4-methylmorpholine N-oxide–thf, X = Cl, Y = O, n = 2 or 3, m = 1; tolylN3, X = Cl, Y = N-tolyl, n = 2, m = 1; (ii) AgBF4, MeCN. | ||
Some catalytic applications in the Heck reaction (Pd) and transfer hydrogenation of ketones (Ru) have been reported.3 Furthermore, unusual and reactive alkyl and dinitrogen complexes of Co(I) and Fe(0) with the C–N–C ligand have been recently described.4 NHC complexes of high oxidation state and early transition metals have been poorly investigated.5 Functionalised NHC ligand designs have been introduced in this area.6 Recently, M(C–N–C)Cl3 complexes M = Ti(III), V(III), Cr(III) were synthesised and their ethylene polymerisation and oligomerisation activity studied.7
In this paper we report the synthesis and full characterisation of C–N–C complexes of Ti(IV), V(II), Cr(II), Mn(II), Nb(III) and U(IV) halides, and the oxidation of V(II) and V(III) complexes by oxo and imido transfer reagents. We also describe the structures of the previously prepared (C–N–C)MCl3 M = Ti and V.7
Results and discussion
The substitution of labile ligands by C–N–C served as a general method for the synthesis of the early transition metal and uranium halide complexes that are described in this paper. The reactions were carried out in thf and the products isolated by crystallisation from thf/petroleum. In addition, the aminolysis from {Cr[N(SiMe3)2]2(thf)2} was successfully used for the preparation of the chromium(II) carbene complex 3b. All new complexes are moderately sensitive to air and dissolve without decomposition in polar non-protic solvents. Due to their paramagnetic nature they were characterised by elemental analysis, magnetic susceptibility and in some cases single-crystal X-ray diffraction. The structure of the V(II) complex 1a is shown in Fig. 1. | ||
| Fig. 1 ORTEP representation of the molecular structure of 1a (50% probability ellipsoids). Hydrogen atoms and two thf molecules are omitted for clarity. Important bond lengths (Å) and angles (°): C(6)–V(1) 2.209(9), C(25)–V(1) 2.162(9), Cl(1)–V(1) 2.443(3), Cl(2)–V(1) 2.416(3), N(1)–V(1) 2.084(7), O(1)–V(1) 2.145(5); N(1)–V(1)–C(6) 75.2(3), N(1)–V(1)–C(25) 74.7(3), C(25)–V(1)–C(6) 149.6(3), Cl(2)–V(1)–Cl(1) 173.73(10). | ||
Complex 1a features a distorted octahedral metal centre with the ‘pincer’ ligand occupying meridional sites. The two chlorides are mutually trans and the sixth coordination site is filled by one thf molecule. The vanadium lies on the plane defined by the pyridine and the NHC rings; the bulky aromatic substituents of the NHC are virtually perpendicular to the coordination plane (80.5–84.5°). The N–C bond lengths and the N–C–N angles of the NHC are virtually identical to those observed for the free ligand. The two V–C carbene bond lengths are slightly different [2.209(9) and 2.162(9) Å] and within the range for V–C σ(sp3) single bonds [2.124–2.254 Å].8 The Cl–V–Cl angle is 173.73(10)° with the chlorine atoms pointing away from the pyridine ring; there is no obvious interaction of any of the chlorine atoms with the carbene carbons.
For comparison purposes the structures of the previously reported (C–N–C)VCl3 and (C–N–C)TiCl3 and of the niobium complex 4 were also determined; an ORTEP diagram of the former is given in Fig. 2; structural data for the latter two are included in the ESI.†
 | ||
| Fig. 2 ORTEP representation of the molecular structure of (C–N–C)VCl3 (50% probability ellipsoids). Hydrogen atoms are omitted for clarity. Important bond lengths (Å) and angles (°): C(2)–N(3) 1.348(3), C(2)–N(2) 1.368(3), C(2)–V(1) 2.144(3), C(21)–V(1) 2.147(3), N(1)–V(1) 2.160(2), Cl(3)–V(1) 2.2578(9); C(2)–V(1)–N(1) 72.77(10), C(2)–V(1)–Cl(1) 91.82(8), Cl(1)–V(1)–Cl(2) 167.17(3). | ||
In all three complexes the distorted octahedral geometry with occupation of the meridional sites by the ‘pincer’ is a common feature. The Ti–C bonds [2.211(5) and 2.191(5) Å] are slightly longer than the V(III)–C bonds [2.114(3) and 2.191(5) Å] and, surprisingly, shorter than other Ti(IV)–NHC bonds reported in the literature;5f they are longer than typical Ti (IV)–CH3 σ(sp3) bonds (ca. 2.175 Å)9a–c and some Ti–C σ(sp2) bonds [2.060–2.070 Å].9d,e The Nb–C bonds in 4 are within the range 2.203(6)–2.206(6) Å and are shorter than other Nb–C σ(sp3)10a–c and Nb–C σ(sp2)10d,e bonds, possibly due to some backbonding. In common with the other known metal (III) trihalide ‘pincer’ complexes,4a,7 the metal–chloride bond trans to the pyridine is significantly shorter than those to the mutually trans chlorides. The ligand bite angle increases in the order V(II) > V(III) > Ti(III) > Nb(III) [74.9(3), 72.89(10), 71.32(16), 69.6(2)°, respectively] showing an inverse correlation with the metal covalent radius; the trend can be rationalised by assuming that as the size of the metal increases it moves further out of pocket of the ‘pincer’ ligand.
The structures of the chromium and manganese bromide complexes 2b and 3 have also been determined by single-crystal X-ray diffraction; an ORTEP diagram of the manganese complex is shown in Fig. 3, while details of the structure of the chromium complex have been included in the ESI.†
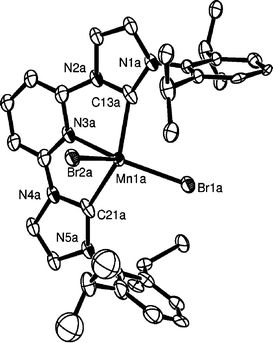 | ||
| Fig. 3 ORTEP representation of the molecular structure of 3 (50% probability ellipsoids) showing one refined orientation of the molecule. Hydrogen atoms and one thf molecule are omitted for clarity. Important bond lengths (Å) and angles (°): Mn(1A)–C(13A) 2.206(2), Mn(1A)–C(21A) 2.210(2), Mn(1A)–Br(1A) 2.5458(7), Mn(1A)–Br(2A) 2.5708(7), Mn(1A)–N(3A) 2.2574(16); C(13A)–Mn(1A)–C(21A) 143.58(8), C(13A)–Mn(1A)–N(3A) 74.99(7), C(21A)–Mn(1A)–N(3A) 71.60(6), Br(1A)–Mn(1A)–Br(2A) 124.57(3), N(3A)–Mn(1A)–Br(2A) 84.52(4), C(21A)–Mn(1A)–Br(2A) 93.48(6), C(13A)–Mn(1A)–Br(2A) 97.41(6). | ||
Both contain five-coordinate metal centres in distorted square-pyramidal geometries with apical bromides. There is an approximate Cs symmetry arising from the plane containing the metal, the two bromides and the pyridine nitrogen. Even though the position of the manganese atom in the structure of 3 is disordered, modelling of the disorder allowed the calculation of bond lengths and angles in the complex. The metal–C bond lengths are 2.206(2) and 2.210(2) Å for the Mn(II) and 2.122(10) and 2.125(10) Å for Cr(II). The apical M–Br is slightly longer than the basal. The bite angle of the ligand is in the range 71.6–74.9° (Mn) and 74.9–76.3° (Cr). The metals are positioned out of the basal plane (0.3873 Å for the Mn complex and 0.3114 Å for the Cr complex). Complex 3 constitutes a rare example of a structurally characterised Mn–NHC complex.11a,b The Mn–NHC bond lengths in 3 are in agreement with those previously reported; furthermore they are longer than Mn–C σ(sp3) [2.145–2.175 Å]11c,d and Mn–C σ(sp2) [2.039 Å].11e Tetrahedral chromium(II)–NHC complexes have been previously reported,12 with Cr–NHC bond length [2.175(3) Å] slightly longer than those in 2b within the observed esds. The Cr(II)–CH3 σ(sp3) bond lengths are within the range 2.150–2.168 Å.12b,c
The structure of the uranium complex is shown in Fig. 4.
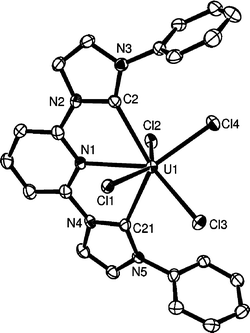 | ||
| Fig. 4 ORTEP representation of the molecular structure of 5 (50% probability ellipsoids). Hydrogen atoms, one thf molecule and Pri groups are omitted for clarity. Important bond lengths (Å) and angles (°): C(2)–U(1) 2.573(5), C(21)–U(1) 2.587(5), N(1)–U(1) 2.653(4), Cl(1)–U(1) 2.6486(12), Cl(2)–U(1) 2.6725(12), Cl(3)–U(1) 2.6314(13), Cl(4)–U(1) 2.5936(13); C(2)–U(1)–N(1) 62.47(14), C(21)–U(1)–N(1) 62.30(14), Cl(1)–U(1)–Cl(2) 145.55(4), Cl(4)–U(1)–Cl(3) 81.86(4), Cl(4)–U(1)–Cl(2) 83.44(4), Cl(3)–U(1)–Cl(2) 126.22(4). | ||
Complex 5 is only the second example of a U(IV) N-heterocyclic carbene complex13a even though U(III) and U(VI) complexes have been described recently.13b–d The metal adopts a distorted seven-coordinate geometry with an approximate C2 axis passing through the pyridine N atom and the uranium.14 The U–Cl bonds are within 2.587–2.673 Å. The U–C carbene bond lengths in 5 [2.573(5)–2.587(5) Å] are shorter than those observed previously for U(IV) [2.636(9) Å], U(III) [2.672(5)–2.789(14) Å] and U(VI) complexes (2.64 Å). However, they are longer than other known U–C σ(sp3)-alkyl [2.405–2.539 Å]13e–g and U–C σ(sp2)-vinyl (2.435 Å)13h bonds.
Oxidation reactions
Despite the fact that good σ-donors are expected to stabilise high oxidation state metal centres, only a few examples of NHC complexes in high oxidation states have been reported.5a–d The interaction of free NHCs with high oxidation state metal precursors was the method of choice for the synthesis of the majority of known high oxidation state NHC complexes,5 but it suffers from unwanted side reactions (metal reduction, ligand oxidation). Alternatively, the oxidation of lower oxidation state NHC complexes has not been explored. In this section we describe the application of the latter strategy to the synthesis of high oxidation state NHC complexes.The reaction of (C–N–C)VCl3 with 4-methylmorpholine N-oxide in thf at room temperature gave rise to the light green, paramagnetic V(IV)–oxo complex 6. The same product was also obtained, albeit in lower yields, by the reaction of 1a with the same oxidant. Complex 6 was characterised by analytical and spectroscopic methods. The VO absorption in the IR spectrum is found at 974 cm−1. Compared to other V(IV)–oxo complexes15 the position of this absorption is at the lower end of the observed range and is in agreement with decreased VO π-bonding due to the strong σ-donating NHCs. The structure of 6 was determined crystallographically and a diagram of the molecule is given in Fig. 5.
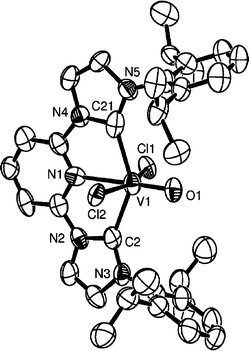 | ||
| Fig. 5 ORTEP representation of the molecular structure of 6 (50% probability ellipsoids). Only one out of two identical molecules in the asymmetric unit is shown. Hydrogen atoms are omitted for clarity. Important bond lengths (Å) and angles (°): C(2)–V(1) 2.169(10), C(21)–V(1) 2.182(11), O(1)–V(1) 1.600(5), N(1)–V(1) 2.275(7), Cl(1)–V(1) 2.398(3), Cl(2)–V(1) 2.421(3); Cl(1)–V(1)–Cl(2) 161.97(10), C(2)–V(1)–N(1) 71.6(3), C(21)–V(1)–N(1) 70.4(3). | ||
The geometry around the metal is octahedral with the ‘pincer’ ligand occupying mer sites and the chlorides being mutually trans. The V(IV)–C bond lengths are in the range 2.169(10)–2.182(11) Å, comparable to the corresponding V–C bond lengths of 1a and (C–N–C)VCl3; the VO bond [1.600(5) Å] is in the range reported for similar V(IV)O complexes.16 The trans chlorides deviate from linearity [161.97(10)°] in this case pointing away from the oxo-ligand, presumably in order to minimise interelectronic repulsions.
The chloride ligands in 6 can be easily abstracted with AgBF4 in acetonitrile, giving good yields of the salt 7 comprising a V(IV)–oxo pincer complex dication. Characterisation of 7 was accomplished by analytical and crystallographic methods. A diagram of the molecule is shown in Fig. 6.
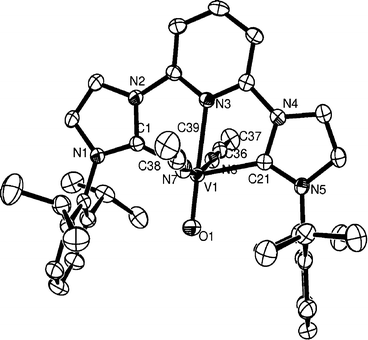 | ||
| Fig. 6 ORTEP representation of the cation in 7 (50% probability ellipsoids). Hydrogen atoms, BF4− anions and one MeCN molecule are omitted for clarity. Important bond lengths (Å) and angles (°): V(1)–C(1) 2.129(3), V(1)–C(21) 2.136(3), V(1)–N(3) 2.236(2), V(1)–O(1) 1.5913(19), V(1)–N(7) 2.091(2); C(1)–V(1)–N(3) 72.04(9), C(21)–V(1)–N(3) 71.59(9). | ||
The octahedral geometry is maintained in 7; the acetonitrile ligands occupy trans positions and the V-carbene bond lengths [2.129(3)–2.136(3) Å] are shorter than those observed in 6. The origin of this shortening is not clear but it may be due to increased ionic interaction of the carbene ligand with the metal. The VO bond length is comparable to the one observed in 6. As expected, the V–N bond length which is trans to the oxo ligand is longer than the V–acetonitrile bond lengths due to the increased trans influence of the oxo ligand.
The reaction of 1a with p-tolylazide gave the brown paramagnetic V(IV)–tolylimido complex 8 which was characterised by analytical and spectroscopic methods. The VN(o-tolyl) bond stretching was observed at 970 cm−1 in the infrared spectrum, while the magnetic susceptibility is almost equal to the values measured for 6 and 7, in agreement with one unpaired electron. All data obtained for 8 point to a monomeric six coordinate complex, most likely analogous to 6. We were unable to obtain the crystal structure of 8 due to facile solvent loss from the crystals.
The related Ti(IV) analogue of 8 was prepared by substitution of the labile pyridine ligands in Ti(NBut)Cl2(pyridine)3 with the free C–N–C. Complex 9 was characterised by spectroscopic, analytical and diffraction methods. The 1H NMR data support the presence of a plane of symmetry perpendicular to the plane of the ‘pincer’ and therefore the chloride ligands are mutually trans. The structure of the molecule was confirmed crystallographically and is shown in Fig. 7.
 | ||
| Fig. 7 ORTEP representation of the molecular structure of 9 (50% probability ellipsoids). Hydrogen atoms and disorder of the Pri and But groups are omitted for clarity. Important bond lengths (Å) and angles (°): Ti(1)–N(6) 1.677(5), Ti(1)–C(13) 2.281(6), Ti(1)–C(21) 2.286(6), Ti(1)–N(3) 2.332(5), Ti(1)–Cl(2) 2.415(2), Ti(1)–Cl(1) 2.433(2); C(21)–Ti(1)–N(3) 69.77(18), C(13)–Ti(1)–N(3) 70.82(19), Cl(2)–Ti(1)–Cl(1) 161.42(7). | ||
The octahedral titanium centre is coordinated by the ‘pincer’ ligand in the usual way, two trans-chlorides and one tert-butylimido group. The Ti–C bond lengths are in the range 2.281(6)–2.286(6) Å, significantly longer than in (C–N–C)TiCl3 [2.191(5)–2.211(5) Å]. The origin of this elongation is unclear. The Cl1–Ti1–Cl2 angle is 161.42(7)° with the chlorides pointing towards the metal, possibly in order to minimise interelectronic repulsions.
The Ti–NHC bond in 9 is relatively inert to a variety of reagents. For example the 1H NMR spectrum of 9 in C6D6 remains unchanged in the presence of excess tmed or PMe3, but quickly decomposes by the addition of water. In addition, 6 and 7 do not oxidise simple organic substrates under mild conditions.
In conclusion, we have shown that the rigid C–N–C dicarbene ligand is ideal for stabilising certain low and high oxidation state early transition metal complexes bearing metal–NHC bonds. In particular, the V–NHC bond is compatible with oxidising reagents but susceptible to hydrolysis. A comparison of the metal–carbene bond lengths observed in the series of the reported complexes does not help in clarifying the nature of the early metal–NHC bond (covalent with or without backbonding and varying degree of electrostatic interaction). However, first indications show that the coordinated NHC is neither susceptible to external nucleophilic attack nor showing any lability in the presence of other good donors (possibly due to the rigid nature of the tridentate ligand). Further work on the use of the C–N–C as a spectator ligand with electropositive metals and the reactivity of the complexes reported here is in progress.
Experimental
Elemental analyses were carried out by the London Metropolitan University microanalytical laboratory. NMR data were recorded on Bruker AMX-300, AM-300 and DPX-400 spectrometers, operating at 300 MHz and 400 MHz, respectively (1H). The spectra were referenced internally using the signal from the residual protio-solvent (1H) or the signals of the solvent (13C). Magnetic susceptibility measurements at room temperature were carried out using a Johnson-Matthey balance. All manipulations involving air sensitive materials were carried out under vacuum or N2 using standard Schlenkware techniques. All solvents used were dried by continuous reflux under N2 and distillation from suitable drying agents immediately prior to use; the light petroleum had bp 40–60 °C. Commercial chemicals were from Aldrich, Lancaster and Avocado. The following starting materials were prepared following literature methods: [Cr(N(SiMe3)2](thf)2],17 (CH–N–CH)Br2,18 (CHmes–N–CHmes)Br2,18 VCl2(tmed)2,19 VCl3(C–N–C),7 Ti(NBut)Cl2(py)3,20 FeBr2(C–N–C).21[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine, C–N–C 3a
The imidazolium salt (CH–N–CH)Br2 (6.86 g, 9.93 mmol) was suspended in thf (100 cm3) and cooled to −78 °C. A solution of KN(SiMe3)2 (4.32 g, 21.6 mmol) in thf (100 cm3) was cooled to −78 °C, and added to the vigorously stirred reaction solution. The mixture was allowed to warm slowly to room temperature overnight. After evaporation of the solvent at reduced pressure, the residue was dissolved in toluene (∼100 cm3) and filtered through Celite. The solvent volume was reduced to ∼20 cm3, and light petroleum (40 cm3) was added. Cooling to −30 °C overnight gave a cream solid and a red solution. The solution was filtered and the solid washed with light petroleum (∼20 cm3). The product was obtained as a cream solid (4.06 g, 77%). δH (C6D6) 1.09 (d, 12H, J = 6.9 Hz, Me), 1.18 (d, 12H, J = 6.8 Hz, Me), 2.90 [sept., 4H, J = 6.9 Hz, CH(CH3)2], 6.62 (d, 2H, J = 1.8 Hz, imidazole backbone CH), 7.05 (t, 1 H, J = 8.0 Hz, central pyridine CH), 7.14 (d, 4H, J = 7.4 Hz, aromatic CH), 7.25 (dd, 2H, J = 6.9, 8.4 Hz; central aromatic CH), 8.06 (d, 2H, J = 1.7 Hz, imidazole backbone CH), 8.43 (d, 2H, J = 8.0 Hz, pyridine CH). δC (C6D6) 24.03 (Me), 24.42 (Me), 28.60 [CH(CH3)2], 111.70 (aromatic CH), 116.25 (aromatic CH), 122.82 (aromatic CH), 128.53 (aromatic CH), 129.29 (aromatic CH), 138.72 (aromatic C), 140.69 (aromatic C), 146.20 (aromatic CH), 152.62 (aromatic C), 220.34 (carbene C).[2,6-Bis{3-(2,4,6-trimethylphenyl)imidazol-2-ylidene]pyridine, Cmes–N–Cmes
The reaction was carried out as described above, using the imidazolium salt (CHmes–N–CHmes)Br2 (2.78 g, 4.00 mmol) and KN(SiMe3)2 (1.68 g, 8.42 mmol). For solubility reasons, benzene was used in place of toluene in the work up, giving the carbene as a cream solid (1.07 g, 60%). δH (C6D6) 2.12 (s, 12H, Me), 2.14 (s, 6H, Me), 6.44 (d, 2H, J = 1.4 Hz, imidazole backbone CH), 6.79 (s, 4H, aromatic CH), 7.09 (t, 1H, J = 7.6 Hz, central pyridine CH), 8.11 (d, 2H, J = 1.4 Hz), 8.54 (d, 2H, J = 7.6 Hz. δC (C6D6) 18.07 (Me), 21.00 (Me), 111.48 (aromatic CH), 116.49 (aromatic CH), 119.66 (aromatic C), 121.47 (aromatic CH), 129.18 (aromatic CH), 135.29 (aromatic C), 137.61 (aromatic C), 140.68 (aromatic CH), 152.70 (aromatic C), 219.75 (carbene).Preparation of the complexes
[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene}]pyridine vanadium dichloride tetrahydrofuran 1a
This was prepared following method A from the carbene C–N–C (266 mg, 0.50 mmol) in thf (5 cm3) and [VCl2(tmeda)2] (0.177 g, 0.50 mmol) in thf (5 cm3) at −78 °C. The mixture was stirred for 10 min at −78 °C, then allowed to warm to room temperature, giving a dark green solution. Layering with light petroleum overnight led to the formation of crystals suitable for X-ray diffraction. Yield: 0.355 g, 98%. Mp 130–132 °C (decomp.). Found: C, 64.10; H, 6.63; N, 9.48%. Calc. for C39H49N5Cl2OV: C, 64.55; H, 6.81; N, 9.65%. μeff = 3.79 μB.[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene}]pyridine chromium dichloride 2a
To a suspension of CrCl2 (0.231 g, 1.88 mmol) in thf (25 cm3) was added a solution of C–N–C (1.0 g, 1.88 mmol) in thf (75 cm3) at −78 °C. The reaction mixture was stirred for 15 min at −78 °C and then allowed to reach room temperature and stirred for 16 h. Removal of the volatiles under reduced pressure gave 2a as a purple solid. Under prolonged exposure to vacuum the solid turns green. Yield: 1.256 g, 92%. Mp 101–102 °C. Found: C, 64.57, H, 6.75, N, 9.60. Calc. for C39H49N5OCrCl2: C, 64.45, H, 6.80, N, 9.64%. μeff = 4.99 μB.[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine chromium dibromide 2b
This was prepared following method B from (CH–N–CH)Br2 (0.385 g, 1 mmol) and {Cr[N(SiMe3)2]2(thf)2} (0.540 g, 1 mmol). The product was crystallised from a thf solution by slow diffusion of light petroleum. Found: C, 57.51; H, 6.05; N, 8.12%. Calc. for C35H41N5Br2Cr·1.5C4H8O: C, 57.82; H, 6.27; N, 8.22%. μeff = 4.97 μB.[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine manganese dibromide 3
This was prepared following method A from MnBr2 (0.607 g, 2.8 mmol) and C–N–C (1.50 g, 2.8 mmol). Yield: 2.15 g (yellow powder) 93%. Mp 106–108 °C (decomp.). Crystals were grown by slow vapour diffusion of light petroleum into a thf solution. Found: C, 57.11; H, 6.00; N, 8.47. Calc. for C39H49N5OMnBr2: C, 57.22; H, 6.03, N; 8.56%. μeff = 5.87 μB.[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine niobium trichloride 4
This was prepared following method A from NbCl3(dme) (0.65 g, 2.25 mmol) and C–N–C (1.98 g, 2.25 mmol). Filtration of the dark blue/purple solution followed by evaporation of the volatiles under reduced pressure gave 4 as a purple solid residue. X-Ray quality crystals were obtained by layering a thf solution with light petroleum. Yield: 1.56 g, 95%. Mp 260–263 °C (decomp.). Found: C, 57.58; H, 5.79; N, 9.42. Calc. for C39H49N5ONbCl3: C, 57.51; H, 5.65; N, 9.58%. μeff = 2.04 μB.[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine uranium tetrachloride 5
This was prepared following method A from UCl4 (0.300 g, 0.79 mmol) and C–N–C (0.42 g, 0.79 mmol). Yield: 0.36 g, 50% of yellow–green solid. X-Ray quality crystals were obtained by layering of a dilute thf solution with light petroleum. Found: C, 46.02; H, 4.86; N, 7.51. Calc. for C35H41N5UCl4: C, 46.12; H, 4.53; N, 7.68%.trans-[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine oxovanadium dichloride 6
To a stirred suspension of (C–N–C)VCl3 (1.10 g, 1.60 mmol) in THF (100 cm3) under a counterflow of dry N2 was added N-methylmorpholine N-oxide (0.206 g, 1.76 mmol) and the reaction was stirred for 16 h at room temperature. A green solution with a dark precipitate was obtained which was diluted with THF (50 cm3), filtered through Celite and concentrated in vacuo, to give 6 as a green solid. Yield: 0.965 g, 90%. X-Ray quality crystals were grown by layering a THF solution with light petroleum. Mp 286–288 °C (decomp.). Found: C, 62.54; H, 6.24; N, 10.54. Calc. for C35H41N5OCl2V: C, 62.78; H, 6.17; N, 10.46%. IR (Nujol mull): 1623 (w), 1595 (w), 1448 (s), 1291 (m), 1151 (w), 974 (m), 801 (m), 761 (m), 722 (s), 336 (m). MS (ES+, MeCN): 633.1 ([M − Cl]+, 100%).trans-[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine bis(acetonitrile) oxovanadium bis(tetrafluoroborate) 7
To a mixture of 6 (0.150 g, 0.22 mmol) and AgBF4 (0.092 g, 0.47 mmol) was added acetonitrile (15 cm3), and the reaction was stirred at room temperature for 4 h. The white precipitate that was formed was removed by filtration through Celite and the blue solution was evaporated to dryness. Yield: 0.191 g, 97%. X-Ray quality crystals were grown by slow diffusion of diethyl ether into an acetonitrile solution. Mp 145–146 °C. Found: C, 54.74, H, 5.41, N, 11.65. Calc. for C39H47N7B2F8V: C, 54.82; H, 5.54; 11.48%; IR (Nujol mull): 2361 (m), 2343 (w), 2292 (w), 1630 (s), 1594 (w), 1300 (s), 1261 (m), 1075 (m), 1020 (m), 976 (w), 803 (s), 778 (w) cm−1. μeff = 1.79 μB.trans-[2,6-Bis{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine vanadium (p-tolylimido) dichloride 8
To a stirred suspension of 1a (0.200 g, 0.28 mmol) in THF (20 cm3) under a counterflow of dry N2 was added p-tolyl azide (0.037 g, 0.28 mmol) and the reaction mixture was stirred for 16 h at room temperature. The brown solution was filtered through Celite and evaporated to dryness, affording 8 as a brown solid. Mp 169–170 °C. Yield: 0.150 g, 72%. Found: C, 66.32; H, 6.50; N, 10.97. Calc. for C42H48N6Cl2V: C, 66.49; H, 6.38; N, 11.08%. IR (Nujol mull): 1623 (w), 1592 (w), 1304 (m), 1169 (w), 1153 (w), 970 (w), 722 (s). MS (ES+, MeCN): 722.3 ([M − Cl]+, 100%). μeff = 1.66 μB.trans-[2,6-Bs{3-(2,6-diisopropylphenyl)imidazol-2-ylidene]pyridine titanium (tert-butylimido) dichloride 9
To a solution of Ti(NBut)Cl2(py)3 (0.161 g, 0.38 mmol) in thf (5 cm3) was added a solution of C–N–C (0.200 g, 0.38 mmol) in the same solvent (15 cm3). The mixture was stirred at room temperature for three days. Evaporation of the volatiles under reduced pressure, afforded 9 as an orange solid. Yield: 0.285 g, 95%. X-Ray quality crystals were grown by slow diffusion of light petroleum into a thf solution of 9. Found: C, 65.01, H, 7.05, N, 11.60%. Calc. for C39H50N6Cl2Ti: C, 64.91; H, 6.98; N, 11.65%. δH (C6D6) 7.03–7.23 (6H, m, aromatic), 6.80 (2H, br s, aromatic), 6.62 (1H, br t, J = 6.0 Hz, aromatic), 6.43 (2H, d, J = 7.5 Hz, imidazol-2-ylidene CH), 6.36 (2H, d, J = 1.5 Hz, imidazol-2-ylidene CH), 3.27 (4H, septet, J = 6.5 Hz, Pri CH), 1.68 and 1.02 (each 12H, d, J = 6.5 Hz, Pri CH3), 0.54 [9H, s, C(CH3)3]. δC (C6D6) 200.72 (C, carbene), 148.18, 146.76, 137.42 (C, Ar); 141.96, 130.29, 128.34, 124.88, 124.57, 123.53 (CH, Ar); 114.55, 107.79 (CH, imidazol-2-ylidene); 31.91 (CH3, But); 28.62, 25.87 (CH3, Pri); 25.47 (C, But); 24.16 (CH, Pri). IR (Nujol mull): 2360 (m), 2342 (w), 1621 (m), 1598 (m), 1481 (s), 1410 (s), 1377 (s), 1286 (m), 1250 (w), 1149 (w), 1107 (m), 1037 (m), 1016 (m), 799 (s), 778 (w), 769 (w), 740 cm−1Crystallography
A summary of the crystal data, data collection and refinement parameters for compounds 1a,(C–N–C)VCl3, 3, 5, 6, and 7 are given in Table 1; in addition, detailed data for the crystallographic characterisation of 1b, 2b, 4 and [(C–N–C)TiCl3 have been included in the ESI.†| 1a | (C–N–C)VCl3 | 3 | 5 | 6 | 7 | 9 | |
|---|---|---|---|---|---|---|---|
| Chemical formula | C47H65Cl2N5O3V | C39H49Cl3N5OV | C39H49Br2MnN5O | C39H49Cl4N5OU | C35H41Cl2N5OV | C39H47B2F8N7OV | C39H49Cl2N6Ti |
| M r | 869.88 | 761.12 | 818.59 | 983.66 | 669.57 | 895.45 | 720.64 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | Pna21 | P21/c | P21/c | P21/c | P21/n | P21/c | P212121 |
| a/Å | 10.306(3) | 10.8934(12) | 10.6948(8) | 10.7146(8) | 16.699(5) | 16.851(3) | 10.802(9) |
| b/Å | 32.019(11) | 18.7259(17) | 18.7188(14) | 18.384(2) | 19.125(5) | 12.707(4) | 13.630(13) |
| c/Å | 13.807(5) | 18.381(2) | 19.2039(19) | 20.229(4) | 22.226(6) | 21.564(4) | 32.45(2) |
| β/° | 90 | 94.051(8) | 93.796(9) | 93.628(10) | 97.012(4) | 100.152(13) | 90 |
| V/Å3 | 4556(3) | 3740.2(7) | 3836.1(6) | 3976.7(9) | 7045(3) | 4545.1(18) | 4778(7) |
| Z | 4 | 4 | 4 | 4 | 8 | 4 | 4 |
| T/K | 120(2) | 120(2) | 95(2) | 120(2) | 120(2) | 120(2) | 120(2) |
| μ/mm−1 | 0.380 | 0.518 | 2.464 | 4.388 | 0.467 | 0.292 | 0.319 |
| No. of data collected | 21996 | 19040 | 26916 | 37286 | 34114 | 63406 | 33304 |
| No. of unique data | 6436 | 8522 | 6754 | 7713 | 7369 | 10899 | 10871 |
| R int | 0.1892 | 0.0570 | 0.0661 | 0.0447 | 0.1383 | 0.073 | 0.1118 |
| Final R(|F|) for Fo > 2σ(Fo) | 0.0656 | 0.0571 | 0.0874 | 0.0370 | 0.0763 | 0.0604 | 0.0944 |
| Final R(F2) for all data | 0.0915 | 0.1282 | 0.2042 | 0.0715 | 0.2073 | 0.1382 | 0.1944 |
All data sets were collected on an Enraf Nonius KappaCCD area detector diffractometer with rotating anode FR591 and an Oxford Cryosystems low-temperature device operating in omega scanning mode with phi and omega scans to fill the Ewald sphere. The crystals were mounted on a glass fibre with silicon grease, from Fomblin vacuum oil. The programs used for control and integration were Collect, Scalepack and Denzo.22 All solutions and refinements were performed using the WinGX package and all software packages within.23 The SQUEEZE algorithm24 was used to remove highly disordered water molecule in 6 and thf molecules in 9 after attempts to satisfactorily model the disorder failed.
CCDC reference numbers 282396–282406.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512133g
References
- For recent reviews, see: (a) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS; (b) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- A. A. Danopoulos, A. A. D. Tulloch, S. Winston and G. Eastham, Dalton Trans., 2003, 1009–1015 RSC , and references therein.
- (a) A. A. Danopoulos, S. Winston and W. B. Motherwell, Chem. Commun., 2002, 1376–1377 RSC; (b) M. Poyatos, J. A. Mata, E. Falomir, R. H. Crabtree and E. Peris, Organometallics, 2003, 22, 1110–1114 CrossRef CAS.
- (a) A. A. Danopoulos, J. A. Wright, W. B. Motherwell and S. Ellwood, Organometallics, 2004, 23, 4807–4810 CAS; (b) A. A. Danopoulos, J. A. Wright and W. B. Motherwell, Chem. Commun., 2005, 784–786 RSC.
- (a) H. Braband and U. Abram, Chem. Commun., 2003, 2436–2437 RSC; (b) H. Braband, T. I. Zahn and U. Abram, Inorg. Chem., 2003, 42, 6160 CrossRef CAS; (c) T. I. Kuckmann and U. Abram, Inorg. Chem., 2004, 43, 7068 CrossRef; (d) C. D. Abernethy, G. M. Codd, M. D. Spicer and M. K. Taylor, J. Am. Chem. Soc., 2003, 125, 1128 CrossRef; (e) P. Shykla, J. A. Johnson, D. Vidovic, A. H. Cowley and C. Abernethy, Chem. Commun., 2004, 360 RSC; (f) M. Niehues, G. Erker, G. Kehr, P. Schwab, R. Frohlich, O. Blacque and H. Berke, Organometallics, 2002, 21, 2905 CrossRef CAS; (g) M. Niehues, G. Erker, G. Kehr, B. Wibbeling, R. Frohlich, O. Blacque and H. Berke, J. Organomet. Chem., 2002, 633, 192 CrossRef; (h) W. J. Oldham, Jr., S. M. Oldham, B. L. Scott, W. H. Smith and D. A. Costa, Chem. Commun., 2001, 1348 RSC; (i) W. A. Herrmann, F. C. Munck, G. R. J. Artus, O. Runte and R. Anwander, Organometallics, 1997, 16, 682 CrossRef CAS; (j) W. A. Herrmann, G. M. Lobmaier and M. Edison, J. Organomet. Chem., 1996, 520, 231 CrossRef CAS.
- P. L. Arnold, S. A. Mungur, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2003, 42, 5981 CrossRef CAS.
- D. S. McGuiness, V. C. Gibson and J. W. Steed, Organometallics, 2004, 23, 6288 CrossRef CAS.
- (a) T. W. Hayton, B. O. Patrick and P. Legzdins, Organometallics, 2004, 23, 657 CrossRef CAS; (b) B. Hessen, J. H. Teuben, T. H. Lemmen, J. C. Huffman and K. G. Caulton, Organometallics, 1985, 4, 946 CrossRef CAS; (c) R. J. Morris, S. R. Wilson and G. S. Girolami, J. Organomet. Chem., 1994, 480, 1 CrossRef CAS.
- (a) Z. Dawoodi, M. L. H. Green, V. S. B. Mtetwa, K. Prout, A. J. Schultz, J. M. Williams and T. F. Koetzle, J. Chem. Soc., Dalton Trans., 1986, 1629 RSC; (b) R. J. McKinney, T. H. Tulip, D. L. Thorn, T. S. Goolbaugh and F. N. Tebbe, J. Am. Chem. Soc., 1981, 103, 5584 CrossRef CAS; (c) J. Lee, P. E. Fanwick and I. P. Rothwell, Organometallics, 2003, 22, 1546 CrossRef CAS; (d) J. L. Polse, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 13405 CrossRef CAS.
- (a) S. I. Bailey, L. M. Engelhardt, W. P. Leung, C. L. Raston, I. M. Ritchie and A. H. White, J. Chem. Soc., Dalton Trans., 1985, 1747 RSC; (b) A. Spannenberg, H. Fuhrmann, P. Arndt, W. Baumann and R. Kempe, Angew. Chem., Int. Ed., 1998, 37, 3363 CrossRef CAS; (c) P.-F. Fu, M. A. Khan and K. M. Nicholas, J. Am. Chem. Soc., 1992, 114, 6579 CrossRef CAS; (d) G. E. Herberich and H. Mayer, J. Organomet. Chem., 1988, 347, 93 CrossRef CAS; (e) J. Sala-Pala and J. E. Guerchais, J. Organomet. Chem., 1990, 391, 61 CrossRef CAS.
- (a) J. Chai, H. Zhu, K. Most, H. W. Roesky, D. Vidovic, H.-G. Schmidt and M. Noltenmeyer, Eur. J. Inorg. Chem., 2003, 4332 Search PubMed; (b) J. Chai, H. Zhu, Y. Peng, H. W. Roesky, S. Singh, H.-G. Schmidt and M. Noltenmeyer, Eur. J. Inorg. Chem., 2004, 2673 CrossRef CAS; (c) H. G. Howard, G. S. Girolami, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1983, 2631 RSC; (d) E. Gallo, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1997, 36, 2178 CrossRef CAS; (e) C. A. Dullaghen, X. Zhang, D. Walther, G. B. Carpenter and D. A. Sweigart, Organometallics, 1997, 16, 5604 CrossRef.
- (a) A. A. Danopoulos, D. M. Hankin, G. Wilkinson, S. Cafferkey, T. K. N. Sweet and M. B. Hursthouse, Polyhedron, 1997, 16, 3879 CrossRef CAS; (b) G. S. Girolami, J. E. Salt and G. Wilkinson, J. Am. Chem. Soc., 1983, 105, 5954 CrossRef CAS; B. J. Thomas, S. K. Noh, G. K. Shulte, S. C. Sendlinger and K. H. Theopold, J. Am. Chem. Soc., 1991, 113, 893 CrossRef CAS.
- (a) W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689 CrossRef CAS; (b) H. Nakai, X. Hu, L. N. Zakharov, A. L. Rheingold and K. Meyer, Inorg. Chem., 2004, 43, 855 CrossRef CAS; (c) W. J. Oldham, S. M. Oldham, B. L. Scott, K. D. Abney, W. H. Smith and D. A. Costa, Chem. Commun., 2001, 1348 RSC; (d) S. A. Mungur, S. T. Liddle, C. Wilson, M. J. Sarsfield and P. L. Arnold, Chem. Commun., 2004, 2738 RSC; (e) P. G. Edwards, R. A. Andersen and A. Zalkin, Organometallics, 1984, 3, 293 CrossRef CAS; (f) P. G. Edwards, R. A. Andersen and A. Zalkin, J. Chem. Soc., Chem. Commun., 1987, 1846 RSC; (g) W. W. Lukens, Jr., S. M. Beshouri, L. L. Blosch, A. L. Stuart and R. A. Andersen, Organometallics, 1999, 18, 1235 CrossRef CAS.
- D. L. Kepert The Early Transition Metals, Academic Press, London, 1972, pp. 8–11 Search PubMed.
- W. A. Nugent and J. M. Mayer, in Metal–Ligand Multiple Bonds, Wiley, N.Y., 1988, p. 116 Search PubMed.
- W. A. Nugent and J. M. Mayer, in Metal–Ligand Multiple Bonds, Wiley, N.Y., 1988, p. 159 Search PubMed.
- B. Horvarh, J. Strutz and E. G. Horvath, Z. Anorg. Allg. Chem., 1979, 457, 38–50 CrossRef CAS.
- A. A. Danopoulos, A. A. D. Tulloch, S. Winston, G. Eastham and M. B. Hursthouse, Dalton Trans., 2003, 1009 RSC.
- The Synthesis of Organometallic Compounds. A Practical Guide, ed. S. Komiya, J. Wiley and Sons, Chichester, 1997, p. 106 Search PubMed.
- A. J. Blake, P. E. Collier, S. C. Dunn, W. S. Li, P. Mountford and O. V. Shiskhin, J. Chem. Soc., Dalton Trans., 1997, 1549 RSC.
- A. A. Danopoulos, N. Tsoureas, J. A. Wright and M. E. Light, Organometallics, 2004, 23, 166 CrossRef CAS.
- R. Hooft, COLLECT, Nonius BV, 1997-2000 Search PubMed; Z. Otwinowski and W. Minor, SCALEPACK, DENZO, Methods Enzymol., 1997, 276, 307 Search PubMed.
- L. J. J. Farrugia, Appl. Crystallogr., 1999, 32, 83 Search PubMed.
- P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, 194 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full crystallographic data and ORTEP diagrams for the structurally characterised species. See DOI: 10.1039/b512133g |
| This journal is © The Royal Society of Chemistry 2006 |