Organic chemistry in water
Chao-Jun
Li
*ab and
Liang
Chen
b
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 2K6, Canada
bDepartment of Chemistry, Tulane University, New Orleans, Louisiana 70118, USA
First published on 21st November 2005
Abstract
Water has emerged as a versatile solvent for organic chemistry in recent years. Water as a solvent is not only inexpensive and environmentally benign, but also gives completely new reactivity. The types of organic reactions in water are broad including pericyclic reactions, reactions of carbanion equivalent, reactions of carbocation equivalent, reactions of radicals and carbenes, transition-metal catalysis, oxidations-reductions, which we discuss in this tutorial review. Aqueous organic reactions have broad applications such as synthesis of biological compounds from carbohydrates and chemical modification of biomolecules.
 Chao-Jun Li | Chao-Jun Li received his PhD at McGill University (1992). He spent 1992–94 as a NSERC Postdoctoral Fellow at Stanford University and became assistant professor (1994), associate professor (1998), and full professor (2000) at Tulane University (US). In 2003, he became a Professor and Canada Research Chair (Tier I) in Organic/Green Chemistry at McGill University in Canada. His widely recognized researches include the development of Grignard-type reactions in water, transition-metal catalysis in air and water, Alkyne-Aldehyde-Amine coupling (A3-coupling), Asymmetric Alkyne-Aldehyde-Amine coupling (AA3-coupling), and Cross-Dehydrogenative-Coupling (CDC) reactions. |
 Liang Chen | Liang Chen was born in 1977 in Fujian, China. He received his Bachelor degree from Nanjing University, China. Thereafter, he spent two years working with Professor Xiao-rong Wang at the State Key Laboratory of Pollution Control & Resource Reuse in Nanjing, China. In 2005, he obtained his PhD under the supervision of Professor C.-J. Li at Tulane University, New Orleans, for the development of water-compatible catalytic reactions of sp-C–H bonds. Currently, he is a postdoctoral fellow with Professor Yandulov at Stanford University, focusing on catalytic fluorinations. |
1. Introduction
Life, the most complex form of organic compounds on Earth, requires the construction of chemical bonds in an aqueous environment. Following nature's lead, we should think of water as a versatile solvent for organic chemistry. Since it was reported that Diels–Alder reactions could be greatly accelerated by using water as a solvent instead of organic solvents,1 there has been considerable attention directed toward the development of organic reactions in water. Besides Diels–Alder reactions, other examples cover almost all of the most useful organic reactions, even reactions involving water-sensitive compounds.2 It is obvious that water is the most inexpensive and environmentally benign solvent. In many cases, due to hydrophobic effects, using water as a solvent not only accelerates reaction rates but also enhances reaction selectivities, even when the reactants are sparingly soluble or insoluble in this medium. Furthermore, the low solubility of oxygen gas in water, an important property in the early development of life in an anaerobic environment, can facilitate air-sensitive transition-metal catalysis in open air. The use of water as a solvent also implies the elimination of tedious protection–deprotection processes for certain acidic-hydrogen-containing functional groups, which contributes to the overall synthetic efficiency. Water soluble compounds such as carbohydrates, can be used directly without the need for laborious derivatization and water-soluble catalysts can be re-used after separation from water-insoluble organic products. Aqueous organic chemistry is also essential for the emerging field of chemical biology which uses chemical tools to study biological systems. Although aqueous organic chemistry has broad applications and a bright future, current organic chemistry textbooks do not touch on this subject. In order to compensate for this omission in organic chemistry textbooks, this introductory review will provide non-specialists with some insight into this field.2. Pericyclic reactions
Pericyclic reactions are important processes in organic synthesis for constructing cyclic structures and for controlling stereochemistry. Since Breslow's discovery that Diels–Alder reactions proceed faster (as high as 700-fold) and have a higher endo/exo selectivity in water than in organic solvents, extensive experimental and theoretical studies have been carried out on this topic. The small size and high polarity of a water molecule, as well as a three-dimensional hydrogen bonded network system of bulk water, provide some unique properties which include a large cohesive energy density, a high surface tension, and a hydrophobic effect. These unique properties are believed to be responsible for the rate and selectivity enhancements of pericyclic reactions and the “on-water” effect. The hydrogen bonding between water and organic molecules is also believed to play an important role in the rate accelerations of some organic reactions in water.It is worth mentioning that, in many cases, both water-soluble and water-insoluble reactants can be accelerated in water and, sometimes, larger rate increases are found with water-insoluble substrates (denoted by Sharpless and co-workers as the “on-water effect”).3 For instance, the Diels–Alder reaction (Scheme 1) of the water-insoluble trans, trans-2,4-hexadienyl acetate (1) and N-propylmaleimide (2) showed substantial rate acceleration in aqueous suspension over homogeneous solution. The reaction rate is faster in a protic solvent such as methanol than in nonprotic solvents such as acetonitrile and toluene.3 It suggests that hydrogen bonding and hydrophobic effects may both be important for the observed rate acceleration.
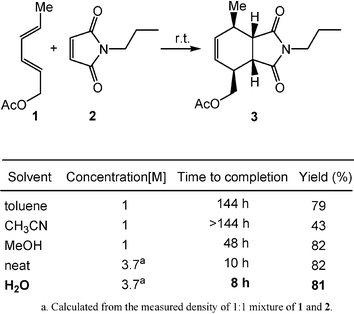 | ||
| Scheme 1 Rate acceleration of Diels–Alder reactions by water solvent. (Reprinted with permission from ref. 3. Copyright 2005, Wiley-VCH.) | ||
Claisen rearrangements, which have negative volume changes of activation as compared to the Diels–Alder reactions, are also accelerated in water. The rearrangement of naphthyl ether (4) to 5 in aqueous suspension is completed within five days at room temperature, whereas it is much slower in organic solvents (Scheme 2). The neat reaction is also slower than the aqueous reaction.3
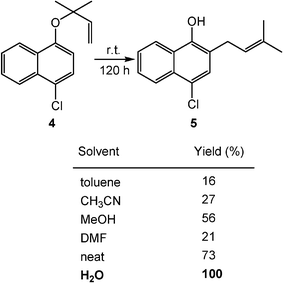 | ||
| Scheme 2 Rate acceleration of Claisen rearrangements by water solvent. (Reprinted with permission from ref. 3. Copyright 2005, Wiley-VCH.) | ||
Beside Diels–Alder cycloadditions and Claisen rearrangements, other pericyclic reactions also show rate enhancements in water. For instance, 2σ + 2σ + 2π cycloaddition (Scheme 3) of quadricyclane (6) with azodicarboxylates (7) at room temperature occurs much faster by using water as a solvent than organic solvents.3 The unusual reactivity described here is possibly due to the greater impact of the hydrophobic effect on the ground state than the transition state.
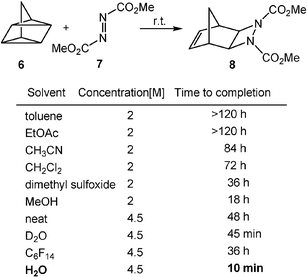 | ||
| Scheme 3 Rate acceleration of 2σ + 2σ + 2π cycloadditions by water solvent. (Reprinted with permission from ref. 3. Copyright 2005, Wiley-VCH.) | ||
The rate accelerations of pericyclic reactions in aqueous solution have been applied to the total synthesis of a variety of biologically active compounds. Total synthesis of gambogin (14), a biologically active compound against the Hela and HEL cell lines, includes two key steps of a Claisen rearrangement and a Claisen/Diels–Alder cascade reaction (Scheme 4). The Claisen/Diels–Alder cascade reaction from 10 to 11 showed dramatic rate accelerations in aqueous solutions (Table 1). The great rate enhancement of Claisen rearrangement from 12 to 13 is also observed in aqueous solutions (Table 2).4
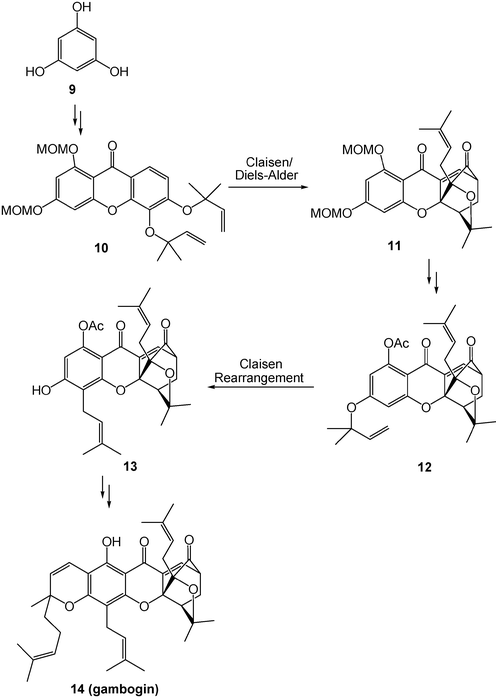 | ||
| Scheme 4 Solvent effects for total synthesis of gambogin. (Reprinted with permission from ref. 4. Copyright 2005, Wiley-VCH.) | ||
Various experiments indicate that the hydrogen-bonding contributes, to a certain degree, to the rate enhancements of pericyclic reactions in water as well as the hydrophobic effect.5 For example, the rate of the cycloaddition reactions of pyridazinium-dicyanomethanide 1,3-dipole (15) with the dipolarophile ethyl vinyl ketone (16) is enhanced by gradually increasing the mole fraction of water in the organic solvents (from 0 to 1) of acetonitrile, acetone, methanol, ethanol, and tert-butyl alcohol at 37 °C (Scheme 5). In each case, exponential rate enhancements are triggered as the mole fraction of water surpasses ca. 0.9. When methanol replaces water, no triggering effect is observed. These results suggest that hydrogen-bonding and hydrophobic effects dominate the rate enhancements. The theoretical calculation of the transition state structure strongly supports this conclusion.5
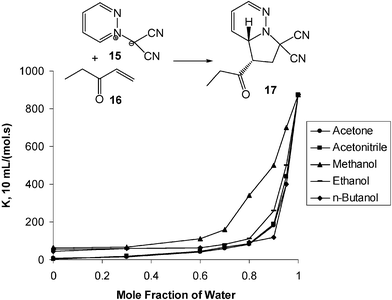 | ||
| Scheme 5 Triggering effect for the introduction of water to organic solvents in cycloaddition reactions. | ||
Beside the rate enhancement, the enhancement of endo/exo selectivity of aqueous reactions is observed not only in non-catalyzed Diels–Alder reactions but also in Lewis acid catalyzed-reactions. For instance, compared to organic solvents, the enantioselectivity of copper-catalyzed Diels–Alder reaction of 3-phenyl-1-(2-pyridyl)-2-propen-1-one (18) with cyclopentadiene (19) is greatly enhanced by the use of water as the solvent (Scheme 6).6
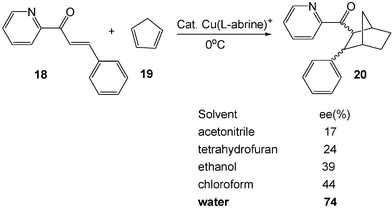 | ||
| Scheme 6 The enhancement of selectivity for Diels–Alder reaction by water. | ||
In agreement with experiments, density functional theory studies show that the computed endo preference is enhanced to 2.4 kcal mol−1 in an aqueous solution. The endo/exo selectivity partly arises from hydrogen bonding, and partly from the bulk-phase effects, which includes enforced hydrophobic interactions and anti-hydrophobic co-solvent effects.7
3. Reactions of carbanion equivalent
The metal-mediated carbanion based reaction is one of the most important methods used to form carbon–carbon bonds. Traditionally, many metal-mediated carbanion based reactions involving organomagnesium and organolithium reagents are often very hard to handle because these reactions require strict exclusion of moisture and oxygen and often require low temperature (e.g. −78 °C) conditions. Recently, they have been modified to work successfully in water by using metals such as indium, tin and zinc as the metal mediators for reactions of carbonyl compounds and imines with allyl halides.42Miyaura reported a rhodium-catalyzed addition of aryl or alkenylboronic acids to α,β-unsaturated carbonyl compounds (Scheme 7)8 and 1,2-addition to aldehydes in aqueous organic mixtures.9 Related asymmetric conjugate additions have been (most noticeably) reported by Hayashi and co-workers.10
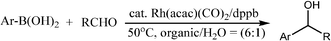 | ||
| Scheme 7 Addition of boronic acid derivative to aldehydes in aqueous conditions. | ||
The Rh(I) [Rh(COD)2Cl or Rh(COD)2BF4]-catalyzed Grignard-type phenylation of carbonyl compounds from trimethylphenylstannane with aldehydes 21 occurs smoothly in water and under an atmosphere of air.11 When methyl groups of organotin compounds are replaced by halogens, the reaction requires the addition of a base, which will generate PhSn(OH)nX3−n species in situ and enhance its reactivity (Scheme 8A).12 Barbier-Grignard type carbonyl alkylation can be carried out in water as well. In the presence of Zn/CuI and a catalytic amount of InCl, the Barbier–Grignard type reaction of various aldehydes 24 with alkyl iodides 25 yields the desired alcohol products 26 in water (Scheme 8B).13 It is important to note that the aldehyde bearing hydroxyl group does not have to be protected prior to the reaction in water.
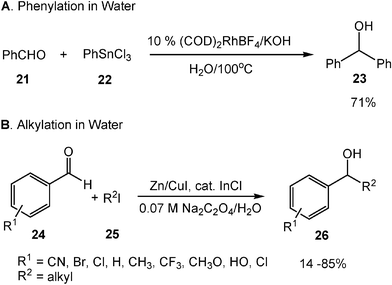 | ||
| Scheme 8 Barbier–Grignard type reaction in water. | ||
Although metal-mediated Barbier-type allylation reactions are highly regio- and stereoselective, α-adduct products are seldom to be obtained (Scheme 9A). With water as a solvent, regioselective allylation (α-adduct versus γ-adduct) becomes possible. In the presence of 2 mL of water solvent, reaction of 1 mmol of aldehyde 27 with 1.2 mmol allyl bromide (28) gives 100% of γ-adduct 29 in high yield; while in the presence of 6 equiv. of water solvent, 99% of α-adduct 30 is obtained (Scheme 9B).14 Here, water is not only a good solvent for the allylation reaction, but also is necessary for the formation of an oxonium ion intermediate which furnishes α-adduct products. Too much water solvent is believed to completely suppress the formation of the oxonium intermediate.
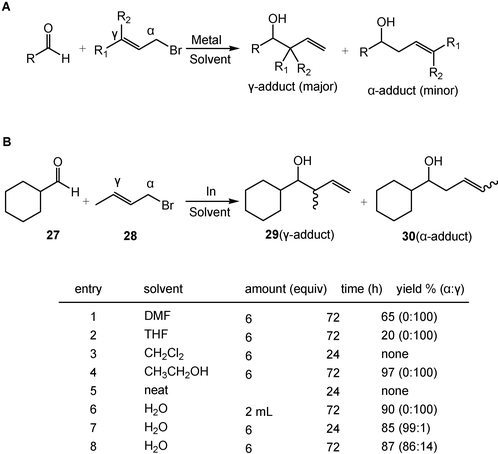 | ||
| Scheme 9 Water-induced selective allylation. (Reprinted with permission from ref. 14. Copyright 2003, American Chemical Society.) | ||
Conjugate addition of unsaturated carbonyl compounds with arylmetallic reagents also proceeds well in water. In the presence of a Rh(I) catalyst, the reaction of phenyl tin chloride 32 with conjugated enone 31 successfully gives the desired addition product 33 in high yield in water under basic conditions (Scheme 10).12
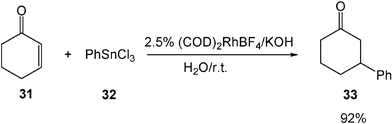 | ||
| Scheme 10 Conjugate addition of unsaturated carbonyl compounds with arylmetallic reagents in water. | ||
4. Reactions of carbocation equivalent
In organic reactions, carbocations are often initiated by the coordination/reaction of protonic and Lewis acids. A variety of Lewis and protonic acids-catalyzed reactions of carbocation equivalent have been developed and many have been applied in industry. However, these reactions generally are moisture sensitive and even a small amount of water stops the reactions because most acids react with water instead of the substrates. Recently, various kinds of protonic and various Lewis acids have been found to retain catalytic activities in aqueous media. Particularly, many of them are not only compatible with water, but also activated by water. Rare earth metal salts such as Sc(OTf)3 and Yb(OTf)3 can be used for aqueous organic reactions. For example, Yb(OTf)3 is found to be highly effective for catalyzing the Mukaiyama aldol reaction of benzaldehyde (34) with silyl enol ether 35 in aqueous solution which is much less effective in dry organic solvents (Scheme 11).15 The enhancement of the reaction rate by water is very likely due to the change of the acid catalyst's structure and activity caused by water, or to the regeneration of the active catalyst from the corresponding intermediate which is faster in water than in dry organic solvents.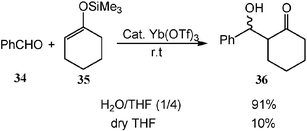 | ||
| Scheme 11 The enhancement of the reaction rate for Mukaiyama aldol reaction by water. | ||
Most catalytic asymmetric reactions of carbocation are conducted at low reaction temperatures (e.g., −78 °C) in aprotic anhydrous solvents such as dry toluene, dichloromethane, and propionitrile. Recently, Cu(II) together with a chiral ligand has been found to be effective for asymmetric aldol reactions in an aqueous solution. In the presence of a catalytic amount of Cu(OTf)2 and bis(oxazoline) ligand 37, the reaction of benzaldehyde (38) with silyl enol ether 39 furnishes the desired aldol product 40 in a high yield with good enantioselectivity (Scheme 12). In contrast, dry organic solvents such as dry ethanol and dry dichloromethane, give much lower yields and selectivities.15 The possible reason for the higher enantioselectivity in water is that water rapidly hydrolyzes the silyl cation equivalent, which is generated during the aldol reactions and is responsible for the decrease of enantioselectivity by catalyzing aldol reactions to give racemic products.
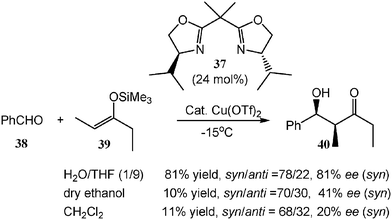 | ||
| Scheme 12 Lewis acid-catalyzed asymmetric reaction in aqueous solution. | ||
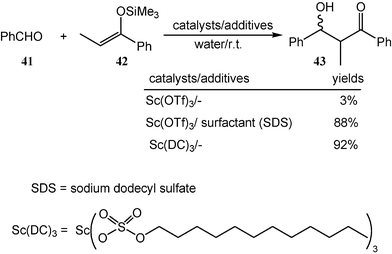
One of the major drawbacks of using water as a solvent is the low solubility of most organic substrates in water. It could be overcome by the use of surfactants, which solubilize organic substances in water. While the reaction of benzaldehyde (41) with silyl enol ether 42 gives almost no desired aldol product 43 in the presence of a catalytic amount of Sc(OTf)3 in water, a high yield of the corresponding aldol adduct is obtained when the reaction is carried out in the presence of a catalytic amount of Sc(OTf)3 and sodium dodecyl sulfate (SDS, a surfactant) in water (Scheme 13).15 Recently, a new type of catalyst, “Lewis acid-surfactant combined catalyst (LASC)” has been developed. It acts both as a Lewis acid to catalyze the reaction and as a surfactant to solubilize organic substrates in water. Scandium tris(dodecyl sulfate) (Sc(DS)3) is a good example because it is very effective for catalyzing the reaction of benzaldehyde (41) with silyl enol ether 42 in water (Scheme 13).15
 | ||
| Scheme 13 LASC-catalyzed organic reaction in water. | ||
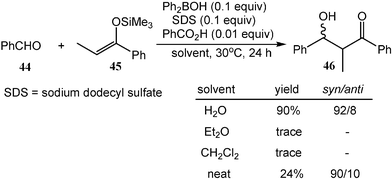
Boron compounds have also been developed as catalysts for the reaction of carbocation equivalents in water. In the presence of a catalytic amount of a boron source, SDS and benzoic acid, the reaction of benzaldehyde (44) with silyl enol ether (45) gives a highly diastereoselective aldol adduct (46) in water at ambient temperature (Scheme 14).16 In contrast, traditional boron aldol reactions are performed at a lower temperature and need strictly anhydrous conditions. It is important to note that the use of water as a solvent is essential in this reaction. Almost no corresponding products are obtained in organic solvents, such as ethyl ether and dichloromethane, and a very low yield of product is obtained under the neat condition (Scheme 14).
 | ||
| Scheme 14 Boron compound-catalyzed organic reactions in water. (Reprinted with permission from ref. 16. Copyright 2001, Wiely-VCH.) | ||
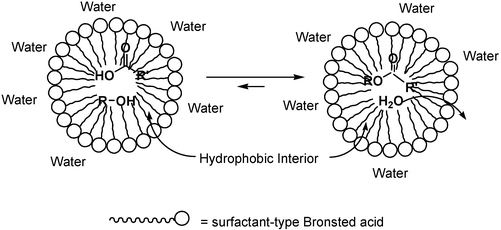
Dehydration is a very common reaction in organic chemistry. It is difficult to carry out in water because water molecules generated during the reaction must be removed to shift equilibrium toward the side of the dehydrated product. With a surfactant-type acid catalyst, dehydration has been successfully developed in water. Emulsion droplets are formed in water in the presence of a surfactant-type catalyst and organic substrates. The emulsion droplets have a hydrophobic interior which would concentrate acid catalysts onto the surface of droplets and enhance the reaction rate toward the side of the dehydrated product. On the other hand, water molecules generated during the reaction are removed from droplets due to the hydrophobic nature of their interior, which shift the equilibrium toward the desired side (Scheme 15).
 | ||
| Scheme 15 Dehydration in water with a surfactant-type catalyst. | ||
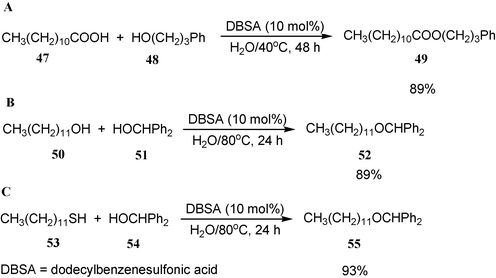
A representative example of dehydration in water is protonic acid-catalyzed dehydrative esterification. Dodecylbenzenesulfonic acid (DBSA), which acts as a protonic acid–surfactant–combined catalyst (BASC), is very effective for catalyzing esterification of acids with alcohols in water (Scheme 16A).17 Beside the esterification, etherification (Scheme 16B) and thioetherification (Scheme 16C) also proceed smoothly in water by the catalyst DBSA.
 | ||
| Scheme 16 Esterification, etherification and thioetherification in water. | ||
Dithioacetals are very useful in organic synthesis as powerful protective groups for carbonyl compounds. The DBSA-catalyzed system is also applicable to dithioacetalization in water which yields dithioacetal products (Scheme 17). The reaction proceeds very well not only for ketones but also for aldehydes.17
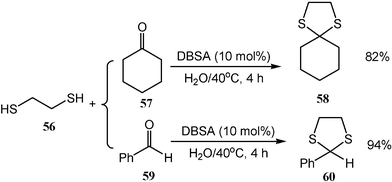 | ||
| Scheme 17 Dithioacetalization in water. | ||
The use of water as a solvent is the key to obtaining high yield and selectivity for the asymmetric Mannich-type reaction. With the combination of catalyst ZnF2 and a chiral diamine 61, the reaction of hydrazono ester 62 and silyl enol ether 63 gives the corresponding product (R)-64 in a high yield with a high enantioselectivity in an aqueous solution. However, in dry organic solvents, such as tetrahydrofuran, no product is obtained in this reaction. This asymmetric reaction can be used for a four-step enantioselective synthesis of HPA-12 (65), the first compound of a specific inhibitor for sphingomyelin synthesis in mammalian cells (Scheme 18).18
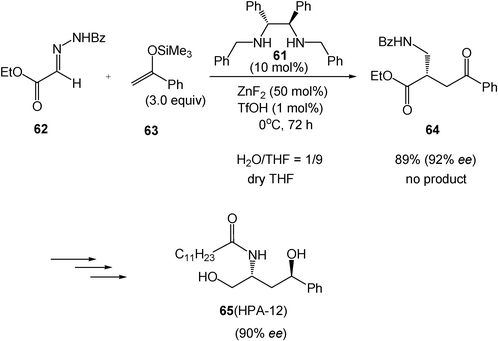 | ||
| Scheme 18 Asymmetric Mannich-type reaction in water and its application for total synthesis of HPA-12. | ||
5. Reactions of radicals and carbenes
Water is the solvent of choice for reactions of radicals and carbenes since strong O–H bonds (enthalpy 436 kJ mol−1) are not easily attacked. Recently, water has been examined as a solvent for these reactions. Significant solvent effects have been reported in atom-transfer radical cyclizations. For example, treatment of allyl iodoacetate 66 in water with triethylborane, a radical initiator, provides the corresponding lactone product 67 in 78% yield. In contrast, in organic solvents such as hexane, benzene, methanol, tetrahydrofuran (THF), DMSO, etc., the reaction is sluggish, and yields no or little lactone product (Scheme 19).19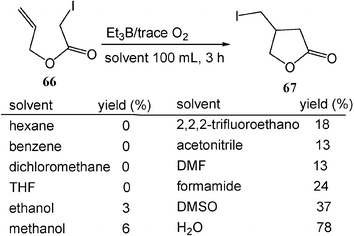 | ||
| Scheme 19 Atom-transfer radical cyclization in water. (Reprinted with permission from ref. 19. Copyright 2000, American Chemical Society.) | ||
The radical cyclization which forms medium- and large-ring lactones also proceeds much more efficiently in water than in organic solvents. The reaction of α-iodo ester 68 with triethylborane in water gives nine-membered lactone 69 in 69% yield. In contrast, in organic solvents such as hexane, benzene, and methanol, the atom-transfer radical cyclization provides no or little lactone (Scheme 20A).19 Similarly, the eighteen-membered lactone is formed by radical cyclization much more efficiently (70% yield) in water than in benzene (14% yield, Scheme 20B). Ab initio calculations suggest that both the large dielectric constant of water and the high cohesive energy density of water are responsible for the acceleration of the cyclization.19
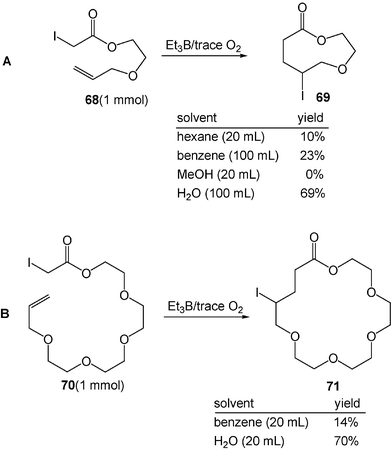 | ||
| Scheme 20 Medium- and large-ring formation by radical cyclization in water. (Reprinted with permission from ref. 19. Copyright 2000, American Chemical Society.) | ||
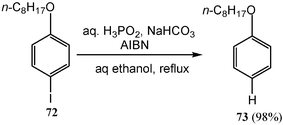
The radical reaction of organic halides in water proceeds smoothly to give the corresponding reduced products. For instance, reactions of various organic halides 72 with phosphinic acid (hypophosphorous acid) in aqueous ethanol in the presence of a radical initiator azo-bis-isobutryonitrile (AIBN) and a base NaHCO3 give the reduced products 73 in high yields (Scheme 21).20
Similarly, D3PO2 is excellent for radical deuteration of organic halides (Scheme 22). The reduction of organic halides 74 with D3PO2 in D2O provides labelled compounds 75 which can be assayed in vivo.20
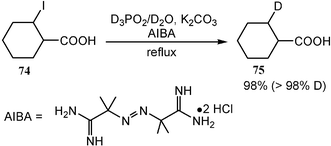 | ||
| Scheme 22 Isotope labelling via an aqueous radical reaction. | ||
Water is also a choice of solvent for free-radical polymerization. The high heat capacity of water allows effective transfer of the heat from polymerization. Compared to organic solvents, the high polarity of water distinguishes remarkably the miscibility of many monomers from polymers. Today, aqueous free-radical polymerization is applied in industries.21
Reactions of carbenes take place in water too. An example is that water-soluble ruthenium alkylidenes 76 bearing cationically functionalized phosphine ligands are stable and initiate olefin metathesis reactions in water. In the presence of 76, monomer 77 is quantitatively polymerized in aqueous environments (Scheme 23).22
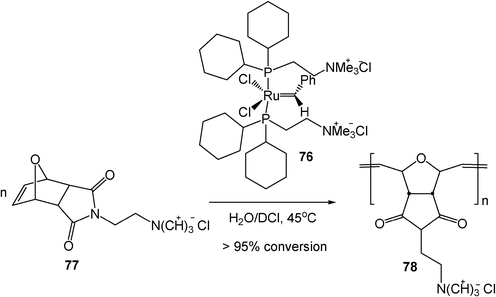 | ||
| Scheme 23 Ruthenium carbene-catalyzed aqueous polymerization. (Reprinted with permission from ref. 22. Copyright 2000, American Chemical Society.) | ||
6. Transition-metal catalysis
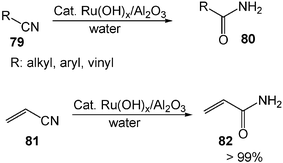
The use of late transition metals for catalyzing reactions is of growing importance in modern organic chemistry.23 Conventionally, organometallic chemistry and transition-metal catalysis are carried out under an inert gas atmosphere and the exclusion of moisture has been essential.24 The reason could be that water as a ligand often occupies metals' coordination sites. On the other hand, metal–carbon bonds are easily attacked by water either via proton transfer (electrophilic reaction) or via the oxygen (nucleophilic reaction). However, recent studies have shown that in water such catalyses are both facilitated and can be operated in open air.25Hydration of nitriles 79 to generate the corresponding amides 80 has wide application, such as as intermediates for organic synthesis and as raw materials for plastics, detergents and lubricants etc. Hydration of nitriles traditionally is carried out with acids or bases such as H2SO4 and NaOH. However, many functional groups such as carbon–carbon double bonds cannot survive this process. In addition, hydrolysis of amide products usually occurs to produce carboxylic acid by-products, especially in acidic conditions. Carefully controlled conditions with one equivalent of water with respect to starting material nitriles are necessary for the traditional hydration of nitriles to amides. However, with the easily prepared, inexpensive, and supported ruthenium hydroxide catalyst Ru(OH)x/Al2O3, the hydration of various nitriles gives quantitatively the corresponding amide products in water.26 Various functional groups such as carbon–carbon double bonds tolerate this hydration process. For example, the hydration of acrylonitrile (81), which is important for industry, provides acrylamide (82) quantitatively (Scheme 24). No side reactions, such as hydration of carbon–carbon double bonds and polymerization of acrylonitriles, occur. The water solvent also provides a way to recycle the transition metal catalyst. The Ru(OH)x/Al2O3 catalyst is easily separated from the reaction mixture by filtration. After separation of the catalyst, the catalyst can be reused at least twice with no considerable loss of activity.
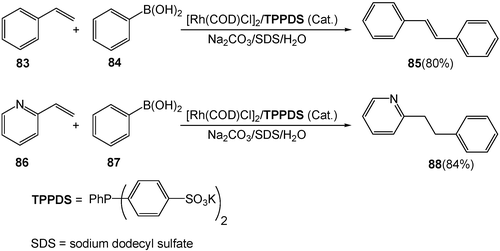
Rhodium-catalyzed addition of arylboronic acids to activated olefins (i.e. olefins with electron-withdrawing groups) has been significantly studied. However, a common requirement of this process is the activation of olefins with electron-withdrawing groups. The reaction substrates are expanded from activated olefins to general un-activated ones by switching organic solvents to water. With a combination of rhodium catalyst and water-soluble phosphine ligand TPPDS, a variety of un-activated styrenyl olefins 83 are reacted with arylboronic acids 84 in water to produce “Heck-type” products 85, while the reaction of vinyl heteroaromatic compounds 86 with arylboronic acids 87 gives addition–hydrolysis products 88 (Scheme 25).27 Notably, in each case, either no reaction or only sluggish reaction is observed in organic solvents with water-soluble sulfonated ligands or with triphenylphosphines.
Palladium catalyzed C–N bond formation can be carried out in water too. With a combination of a phosphine ligand 89 and a palladium catalyst, un-reactive aryl chlorides 90 are reacted with amines 91 in water smoothly to afford the corresponding amination products 92 (Scheme 26).28
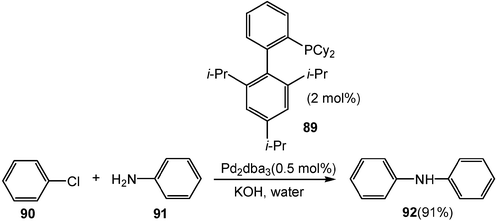 | ||
| Scheme 26 Pd-catalyzed C–N bond formations in water. | ||
Compared to other late transition metals, gold has not been extensively explored as a catalyst. However, recent studies show gold has unique catalytic properties in several organic reactions. Gold-catalyzed organic reactions can be carried out in water too. A representative example is that the gold-catalyzed three-component reaction of aldehydes 93, amines 94 and terminal alkynes 95 efficiently affords propargyl amine products 96 in water (Scheme 27).29 Transition metal-catalyzed asymmetric organic reactions are also possible with the use of water as a solvent. With Cu(I) as a catalyst and chiral pybox (100) as a ligand, the addition of terminal alkynes 97 to imines 98 in water at room temperature affords the corresponding propargyl amine products 99 with a good yield and enantioselectivity (Scheme 27).30
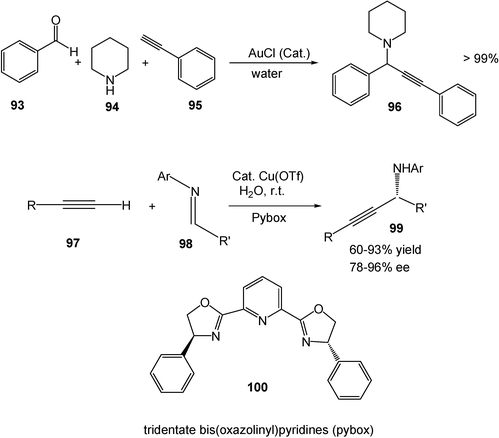 | ||
| Scheme 27 Gold- and copper-catalyzed reactions in water. | ||
7. Oxidations–reductions
Oxidation reactions have often been conducted using stoichiometric amounts of heavy metal reagents (e.g. potassium permanganate, V2O5) or moisture-sensitive oxidants (e.g. oxalyl chloride, N,N′-dicyclohexylcarbodiimide (DCC)). Recently, new oxidative reactions have been developed in aqueous media by water-compatible oxidants, such as O2 and H2O2.Using clean, safe, and inexpensive molecular oxygen as a sole oxidant, a variety of 2-naphthols and substituted phenols can be converted to the corresponding biaryl compounds by ruthenium catalyst Ru(OH)x/Al2O3 in moderate to excellent yields in water. Thus, in the presence of 5 mol% of Ru(OH)x/Al2O3 and 1 atm O2, the oxidative coupling of 2-naphthol (101) in water quantitatively produces 1,1′-binaphthalene-2,2′-diol (BINOL) (102), which is a very important building block and ligand. It is important to note, Ru(OH)x/Al2O3 catalyst can be reused without the loss of any catalytic activity even after the seventh recycling run (Scheme 28).31
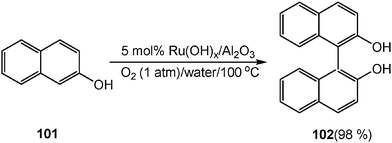 | ||
| Scheme 28 Oxidative coupling of 2-naphthol in water. | ||
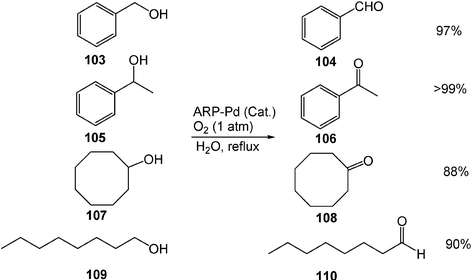
Catalytic oxidation of alcohols can be achieved in water by molecular oxygen by using a novel amphiphilic resin-dispersion of palladium nanoparticles (ARP-Pd). In the presence of the nano-palladium catalyst, primary alcohols give the corresponding aldehydes products, and secondary alcohols give the corresponding ketones in water (Scheme 29). ARP-Pd can be reused with negligible loss of catalytic activity after several runs.32
Aqueous oxidative polymerization provides a convenient way for product separation. Using molecular oxygen as an oxidant and K3[Fe(CN)6] as a catalyst, the oxidative polymerization of 2,6-dimethylphenol (DMP) 111 in aqueous NaOH solution gives poly(2,6-dimethyl-1,4-phenyleneoxide) (PPO) 112, an important plastic in engineering (Scheme 30).33 The DMP monomer is dissolved in aqueous NaOH solution, while the PPO polymer product is insoluble in it. The precipitated product is separated from the reaction mixture by simple distillation after the reaction and the aqueous solution can be reused for the polymerization.
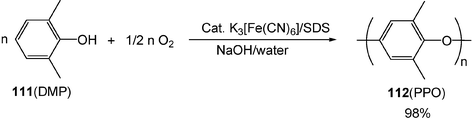 | ||
| Scheme 30 Polymerization of DMP in water. | ||
Hydrogen peroxide, another water-compatible oxidant, is also often used in aqueous oxidative reactions. In the presence of polyoxometalate, Na12[WZnZn2(H2O)2(ZnW9O34)2], which is easily prepared in one step in water from simple salts, various alcohols are converted into the corresponding aldehyde or ketone products in water by hydrogen peroxide. The in situ prepared catalyst polyoxometalate could be recycled in water.34
Water is rarely used as the solvent in a reduction reaction because most reducing agents are incompatible in water. However, water-compatible sodium/lithium borohydride is frequently used in water to reduce ketones or aldehydes. When lithium borohydride (LiBH4) is modified to the more hydrophobic reducing agent lithium pentafluorophenyl borohydride (LiC6F5BH3), a hydrophobically directed selective reduction of ketones is observed in water. As an example, quaternized, β-keto diamines (113) are reduced by LiC6F5BH3 to 114 almost exclusively in water. In contrast, the use of methanol as a solvent gives equal amounts of products 114 and 115 (Scheme 31).35
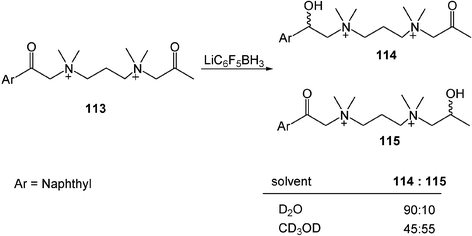 | ||
| Scheme 31 Hydrophobically directed selective reduction of ketones. (Reprinted with permission from ref. 35. Copyright 2003, American Chemical Society.) | ||
A new water-soluble ruthenium complex of β-cyclodextrin-modified amino alcohols 116 serves as a supramolecular catalyst in the selective reduction of ketones. In the presence of NaCOOH, this Ru-base catalyst reduces conjugated (117) and non-activated unconjugated ketones (118) to the corresponding alcohols with good yields and enantioselectivities in water (Scheme 32).36
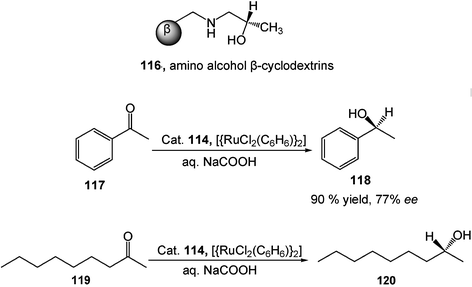 | ||
| Scheme 32 Enantioselective reduction of ketones in water. | ||
8. Synthetic applications
Because of its unique properties, aqueous organic chemistry has found wide applications in organic synthesis, life science and chemical biology.8.1. Carbohydrate chemistry
One of the most important advantages of aqueous organic reactions in organic synthesis is that water-soluble hydroxyl-containing molecules can be used directly without the need of tedious protection–deprotection processes. Thus, the most important application for carrying out organic reactions in water is carbohydrate chemistry where the protection–deprotection processes are involved most extensively. C-Glycosides have wide applications used as a variety of building blocks for total synthesis of natural products and as potential enzyme inhibitors. Traditionally, C-Glycosidic ketones are prepared from a protected sugar by addition of a suitable nucleophile in anhydrous conditions. A new protocol using water as a solvent eliminates tedious protection–deprotection processes, and thus it greatly increases the overall yields. With commercially available D-glucose (121) and pentane-2,4-dione (122) as starting substrates, β-C-glycosidic ketone 123 is obtained in one step with almost a quantitative yield in aqueous NaHCO3 solution (Scheme 33). In a sharp contrast, in organic solvents the β-C-glycosidic ketone 123 is prepared from benzylated D-glucose, a protected sugar, in seven steps in an overall low yield.37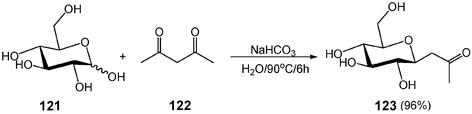 | ||
| Scheme 33 One-step synthesis of β-C-glycosidic ketone in water. | ||
Polyhydroxylated N-heterocycles, or azasugars, which show broad biological activities, are among the most attractive synthetic targets. The most commonly employed methods for the synthesis of azasugars have involved starting from commercially available carbohydrates because their structures are similar to those of sugars. However, their asymmetric synthesis usually requires lengthy procedures and produces low yields. With the use of water as a solvent for all the steps, an extremely efficient asymmetric synthesis of a pyrrolidine azasugar has been developed in only four steps, without any protection–deprotection process, and in 60% overall yield. At first, a modified Sharpless asymmetric dihydroxylation of achiral and cheap 1,6-dibromodiene 124 gives the chiral bis-electrophilic diol 125 in a good yield and enantioselectivity (70% yield, 97% ee). Notably, it has unique and useful reactivity in water (Scheme 34) because of the instability of 124 and 125 in organic solvents. The following selective hydrolysis of 125 affords triol 126 in 98% yield. Then, with dinuclear peroxotungstate K2[W2O3(O2)4(H2O)2] as a catalyst and H2O2 as an oxidant, a novel water-compatible epoxidation of 126 affords the desired epoxy alcohol 127 in 99% yield (92% ds). Finally, following the nucleophilic displacement of the bromide by ammonia, an intramolecular ring opening of the epoxide gives azasugar 128 in 88% yield. The overall yield of 128 is thus 60%.38
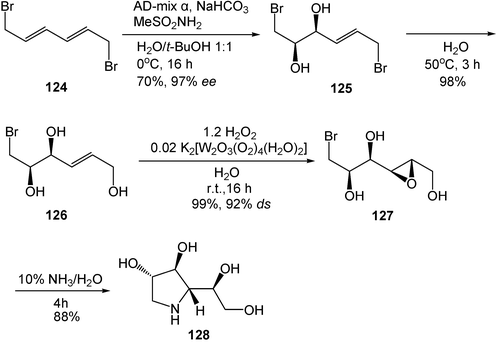 | ||
| Scheme 34 Total synthesis of azasugar in water. (Reprinted with permission from ref. 38. Copyright 2005, The Royal Society of Chemistry.) | ||
8.2. Chemical modification of biomolecules
Aqueous organic chemistry is essential for the emerging field of chemical biology, which uses chemical tools to study biology. Since life constructs chemical bonds in aqueous environments, selective chemical reactions designed to modify biomolecules are now recognized as powerful tools in chemical biology. They provide insight into cellular processes and inspire new strategies for protein engineering. To achieve this goal, the participating functional groups must have a narrow distribution of reactivity and must be inert toward biological molecules. In addition, the selective chemical reactions must occur at room temperature and in aqueous physiological environments.Azides and alkynes are highly energetic functional groups and nearly inert to biomolecules. The Huisgen cycloaddition of azides 129 and alkynes 130 is irreversible and thermodynamically more favorable by approximately 30–35 kcal mol−1. However, the reaction requires elevated temperature. A copper(I)-catalyzed version, recently developed by Sharpless and co-workers, occurs readily at physiological temperatures and in aqueous solutions (Scheme 35A). This reaction enables the selective modification of various biological molecules such as virus particles, nucleic acids, and proteins. An example is the cowpea mosaic virus (CPMV) which has 60 identical copies of a two-protein unit around a single-stranded RNA genome. In the presence of Cu(I) catalyst, the virus CPMV (132), decorated with azides, is efficiently coupled with the dye-alkyne 133 (Scheme 35B).39
![Chemical modification of CPMV by azide-alkyne [3 + 2] cycloaddition.](/image/article/2006/CS/b507207g/b507207g-s35.gif) | ||
| Scheme 35 Chemical modification of CPMV by azide-alkyne [3 + 2] cycloaddition. | ||
The modified Staudinger reaction is another chemoselective reaction designed to modify biomolecules. The Staudinger reaction occurs between phosphines and azides to produce aza-ylides. In the presence of water, the phosphines and azides react efficiently at room temperature to produce amines and phosphine oxides (Scheme 36A). Both phosphines and azides are nearly inert to biological molecules. Therefore, the classical Staudinger reaction is a good candidate for the chemical modification of bio-molecules.
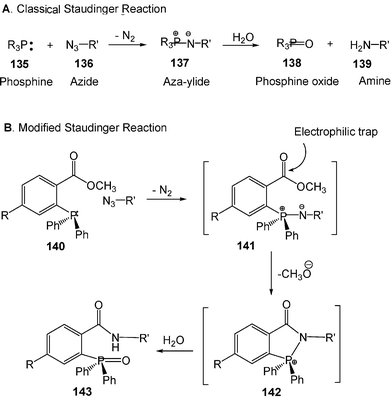 | ||
| Scheme 36 Classical and modified Staudinger reactions. (Reprinted with permission from ref. 40. Copyright 2000, American Association for the Advancement of Science.) | ||
However, the aza-ylides are not stable in water which limits their application for constructing a new and useful covalent bond under physiological conditions. Using an electrophilic trap, Bertozzi and co-workers developed an aqueous Staudinger reaction that would ultimately produce a stable amide bond (Scheme 36B). With this modified Staudinger reaction, the water-soluble biotinylated phosphine 145 couples with azides installed within cell surfaces to produce stable cell-surface adducts 146 (Scheme 37).40
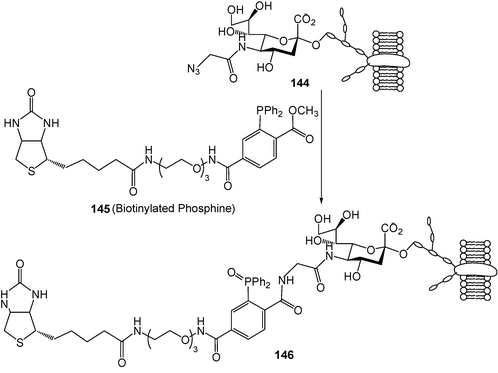 | ||
| Scheme 37 Reaction of biotinylated phosphine with cell surface azido sialic acid. (Reprinted with permission from ref. 40. Copyright 2000, American Association for the Advancement of Science.) | ||
9. Conclusion and outlook
Extraordinary attention has been paid to organic reactions in water in the past decade and research and development in this area is still increasing exponentially.41 This review provides an introductory overview of this field. The types of organic reactions in water are now as diverse as those in organic solvents. It is worthy of note that completely new reactivities of reactions have been discovered in water. The wide application of aqueous organic reactions also provides a driving force for the future development of this area.Acknowledgements
We are grateful to NSERC, NSF and NSF-EPA Joint Program for a Sustainable Environment for support of our research. L.C. thanks a Cancer Research Fellowship by the Tulane University Cancer Center. C.J.L. is a Canada Research Chair (Tier I) at McGill University.References
- R. Breslow, Acc. Chem. Res., 2004, 37, 471 CrossRef CAS.
- L. Chen and C.-J. Li, Org. Lett., 2004, 6, 3151 CrossRef CAS; C. J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. Barry Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS.
- K. C. Nicolaou, H. Xu and M. Wartmann, Angew. Chem., Int. Ed., 2005, 44, 756 CrossRef CAS.
- R. N. Butler, W. J. Cunningham, A. G. Coyne and L. A. Burke, J. Am. Chem. Soc., 2004, 126, 11923 CrossRef CAS.
- S. Otto, G. Boccaletti and J. B. F. N. Engberts, J. Am. Chem. Soc., 1999, 121, 6798 CrossRef CAS.
- S. Kong and J. D. Evanseck, J. Am. Chem. Soc., 2000, 122, 10418 CrossRef CAS.
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229 CrossRef.
- M. Sakai, M. Ueda and N. Miyaura, Angew. Chem., Int. Ed., 1998, 37, 3274.
- T. Hayashi, Synlett, 2001, 879 CrossRef CAS.
- C.-J. Li and Y. Meng, J. Am. Chem. Soc., 2000, 122, 9538 CrossRef CAS.
- T. S. Huang, Y. Meng, S. Venkatraman, D. Wang and C.-J. Li, J. Am. Chem. Soc., 2001, 123, 7451 CrossRef CAS.
- C. C. K. Keh, C. M. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 4062 CrossRef CAS.
- K.-T. Tan, S.-S. Chng, H.-S. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2003, 125, 2958 CrossRef CAS.
- S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209 CrossRef CAS.
- Y. Mori, K. Manabe and S. Kobayashi, Angew. Chem., Int. Ed., 2001, 40, 2816 CrossRef CAS.
- K. Manabe, S. Iimura, X.-M. Sun and S. Kobayashi, J. Am. Chem. Soc., 2002, 124, 11971 CrossRef CAS.
- S. Kobayashi, T. Hamada and K. Manabe, J. Am. Chem. Soc., 2002, 124, 5640 CrossRef CAS.
- H. Yorimitsu, T. Nakamura, H. Shinokubo, K. Oshima, K. Omoto and H. Fujimoto, J. Am. Chem. Soc., 2000, 122, 11041 CrossRef CAS.
- H. Yorimitsu, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 2001, 74, 225 CrossRef.
- S. Mecking, A. Held and F. M. Bauers, Angew. Chem., Int. Ed., 2002, 41, 544 CrossRef CAS.
- D. M. Lynn, B. Mohr, R. H. Grubbs, L. M. Henling and M. W. Day, J. Am. Chem. Soc., 2000, 122, 6601 CrossRef CAS.
- R. H. Crabtree, The Organometallic Chemistry of Transition Metals, 2nd Edn, Wiley Interscience; New York, 1988 Search PubMed.
- B. Cornils, W. A. Herrmann, R. Schlogl, Eds., Catalysis from A to Z: A Concise Encyclopedia, John Wiley & Sons, 2000 Search PubMed.
- C.-J. Li, Acc. Chem. Res., 2002, 35, 533 CrossRef CAS.
- K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., Int. Ed., 2004, 43, 1576 CrossRef CAS.
- M. Lautens, A. Roy, K. Fukuoka, K. Fagnou and B. Martin-Matute, J. Am. Chem. Soc., 2001, 123, 5358 CrossRef CAS.
- X. H. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653 CrossRef CAS.
- C. M. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 9584 CrossRef CAS.
- C. M. Wei and C.-J. Li, J. Am. Chem. Soc., 2002, 124, 5638 CrossRef CAS.
- M. Matsushita, K. Kamata, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 6632 CrossRef CAS.
- Y. Uozumi and R. Nakao, Angew. Chem., Int. Ed., 2003, 42, 194 CrossRef CAS.
- K. Saito, T. Tago, T. Masuyama and H. Nishide, Angew. Chem., Int. Ed., 2004, 43, 730 CrossRef CAS.
- D. Sloboda-Rozner, P. L. Alsters and R. Neumann, J. Am. Chem. Soc., 2003, 125, 5280 CrossRef.
- M. R. Biscoe and R. Breslow, J. Am. Chem. Soc., 2003, 125, 12718 CrossRef CAS.
- A. Schlatter, M. K.Kundu and W.-D. Woggon, Angew. Chem., Int. Ed., 2004, 43, 6731 CrossRef CAS.
- F. Rodrigues, Y. Canac and A. Lubineau, Chem. Commun., 2000, 2049 RSC and references therein.
- U. M. Lindstrom, R. Ding and O. Hidestal, Chem. Commun., 2005, 1773 RSC.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS.
- For recent leading reviews, please see: C.-J. Li, Chem. Rev., 2005, 105, 3095 Search PubMed; U. M. Lindstrom, Chem. Rev., 2002, 102, 2751 CrossRef CAS.
- C.-J. Li, Tetrahedron, 1996, 52, 5643 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |