Self-organization of disc-like molecules: chemical aspects
Sandeep
Kumar
Raman Research Institute, C.V. Raman Avenue, Sadashivanagar, Bangalore – 560 080, India. E-mail: skumar@rri.res.in; Fax: +91 80 23610492; Tel: +91 80 23610122
First published on 21st November 2005
Abstract
The hierarchical self-assembly of disc-shaped molecules leads to the formation of discotic liquid crystals. These materials are of fundamental importance not only as models for the study of energy and charge migration in self-organized systems but also as functional materials for device applications such as, one-dimensional conductors, photoconductors, light emitting diodes, photovoltaic solar cells, field-effect transistors and gas sensors. The negative birefringence films formed by polymerized nematic discotic liquid crystals have been commercialized as compensation foils to enlarge the viewing angle of commonly used twisted nematic liquid crystal displays. To date the number of discotic liquid crystals derived from more than 50 different cores comes to about 3000. This critical review describes, after an in-depth introduction, recent advances in basic design principles and synthetic approaches towards the preparation of most frequently encountered discotic liquid crystals.
 Sandeep Kumar | Sandeep Kumar was born at Chandausi, India on 1st December 1959 and obtained his PhD in Chemistry–Medicinal Chemistry from Banaras Hindu University, Varanasi in 1986 under Professor A. B. Ray. After postdoctoral research at the Hebrew University of Jerusalem, Israel with Professor E. Glotter, at Technion, Israel and at the Scripps Research Institute, La Jolla, USA with Professor E. Keinan and at the University of Mainz with Professor H. Ringsdorf, he joined the Centre for Liquid Crystal Research, Bangalore to start a new Chemistry lab in 1995. In 2002, he moved to the Raman Research Institute as a Senior Associate Professor. The Royal Society of Chemistry awarded him a journals grant for international authors in 2001. He has published 85 research papers in peer reviewed international journals. Several of his papers have been amongst the top ten accessed on the web. His current research interests include design, synthesis and applications of liquid crystals, conducting polymers and green chemistry. |
Introduction
Life on Earth begins with the self-organization of molecules. No life would be possible without the self-assembly of lipids into bilayers within the cell membrane. Numerous biological supramolecular structures, e.g., the spontaneous formation of the double helix of nucleic acids, collagen, microtubules, ribosomes, multisubunit enzymes, etc., result from self-assembly of organic molecules.1–6 In materials science, non-covalent interactions have been used to obtain well-defined self-assembled architectures in neat systems as well as in solvents.1–6 Liquid crystals (LCs) belong to one of such systems. Supramolecular interactions such as van der Waals forces, dipolar or quadrupolar interactions, charge-transfer interactions and hydrogen bonding play a crucial role in the formation of LCs and in the determination of their mesomorphic properties. Liquid crystalline state—the fascinating, intriguing, beautiful, mysterious, delicate fourth state of matter, is an intermediate state between the solid and the liquid. Liquid crystals are unique nanostructures with remarkable electronic and optoelectronic properties. Liquid crystal phases share some of the properties of both isotropic liquids and crystalline solids. While the molecules in these phases exhibit some positional and orientational order, they also behave as fluids. They combine both the fluidity of liquids and the anisotropy of crystals. These materials are extremely diverse since they range from DNA to high strength synthetic polymers like Kevlar and from small organic molecules like alkylcyanobiphenyls used in displays to self-assembling amphiphilic soap molecules. Their importance in life science has been well recognized and a number of biomesogens have been reported.7,8 Recently their biomedical applications such as in controlled drug delivery,9 protein binding,10 phospholipids labeling11 and in microbe detection12 have been demonstrated. On the other hand, in materials science, the numerous applications of LCs are ubiquitous in everyday life. Although LCs have diverse applications such as temperature-sensing (medical thermometers), solvents in chemical reactions, in chromatography, in spectroscopy, in holography, etc., they are primarily known for their extensive exploitation in electro-optical display devices such as watches, calculators, telephones, personal organizers, laptop computers, flat panel televisions, etc.13The serendipitous discovery of LCs in 1888 marks an important milestone in the history of scientific discoveries.14 When determining the melting point of cholesteryl benzoate, Friedrich Reinitzer, an Austrian botanist, noticed the unusual melting behaviour of this compound. It melts at 145.5 °C to form a cloudy liquid with unusual reflection colours. This opaque liquid then appears to melt again at 178.5 °C to a clear transparent liquid. This strange phenomenon of the existence of two melting points was explained by the German physicist Otto Lehmann and the term “fluid crystals” or “liquid crystals” was introduced. It should be noted that researchers as early as the 1850s actually dealt with liquid crystals but did not realize the uniqueness of the phenomena.15
Liquid crystalline materials are divided into two categories, the thermotropic and the lyotropic. When a substance passes between the solid, liquid crystal and liquid state and vice versa as a function of temperature and in the absence of solvent, the liquid crystalline phases are classified as thermotropic, while lyotropic phases form in the presence of a suitable solvent. Thus lyotropic mesophases, found abundantly in biological systems, are always mixtures, whereas thermotropic LCs could be single component or a mixture of many compounds.
Thermotropic LCs are generally further distinguished with respect to the molecular shape of the constituent molecules. The majority of pure compounds showing thermotropic mesophases have one distinctive feature in common: the rod-like shape of their molecules. The term “calamitic” has been used to describe LCs of rod-shaped molecules, in order to distinguish them from discotic liquid crystals (DLCs) composed of disc-like molecules. The so-called “banana” phases, formed by bent-core mesogens or bow-shaped molecules, have recently been added to the family of liquid crystalline materials.16 A few examples of board-like molecules forming “sanidic” phases have also been reported.17
The hierarchical self-assembly of disc-shaped molecules leads to the formation of DLCs.18 Vorlander in 1908 established his rule that liquid crystalline compounds must have a molecular shape as linear as possible. All the liquid crystalline materials prepared over about 90 years belonged to this family. However, in 1977 a group of Indian scientists reported that not only rod-like molecules, but also compounds with disc-like molecular shape are able to form mesophases.19 They prepared a number of benzene-hexa-n-alkanoates and from thermodynamic, optical and X-ray studies, it was established that these materials form a new class of LCs in which molecules are stacked one on top of the other in columns that assemble in a hexagonal arrangement.
It is interesting to mention that Vorlander in 1924 supposed the possibility of the existence of mesophases in leaf-shaped molecules but his attempts to realize any example with this behaviour had been unsuccessful probably because the molecules, he looked at, were devoid of flexible aliphatic chains. He mentioned in his article that leaf-shaped molecules do not form any liquid crystals at all. Of course, the same molecules surrounded by long aliphatic chains are now well-known for forming columnar mesophases.
In the early 1960s, anisotropic mesophases with nematic texture characteristics had been observed during the pyrolysis of graphitizable substances (carbonaceous mesophase).20 These mesophases were considered to be built up of flat polyaromatic molecules, however, and not by well-defined organic molecules. They were not stable and homogeneous in nature. Therefore, it was not possible to characterize these mesophases.
A majority of DLCs form columnar mesophases probably due to intense π–π interactions of polyaromatic cores. The core–core separation in a columnar mesophase is usually of the order of 3.5 Å so that there is considerable overlap of π-orbitals. As flexible long aliphatic chains surround the core, the intercolumnar distance is usually 20–40 Å, depending on the lateral chain length. Therefore, interactions between neighbouring molecules within the same column would be much stronger than interactions between neighbouring columns. Consequently, charge migration in these materials is expected to be quasi-one-dimensional. Conductivity along the columns in columnar mesophases has been reported to be several orders of magnitude greater than in the perpendicular direction.21,22 Thus the columns may be described as molecular wires (Fig. 1). Charge carrier mobility as high as 0.71 cm2V−1s−1 along the columns has recently been observed.23
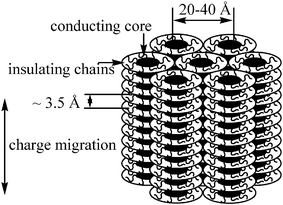 | ||
| Fig. 1 Schematic view of charge migration in columnar phase. | ||
The supramolecular assemblies of disc-shaped molecules have been extensively studied for the energy and charge migration in organized systems and their device applications such as, one-dimensional conductors, photoconductors, light emitting diodes, photovoltaic solar cells, field-effect transistors and gas sensors have been sought.24 The negative birefringence films formed by polymerized nematic DLCs have been commercialized as compensation foils to enlarge the viewing angle of commonly used twisted nematic liquid crystal displays.25 Currently the number of DLCs derived from more than 50 different cores comes to about 3000. The aim of this review is to discuss the chemistry of some of these materials.
Structures of discotic mesophases
Most of the discotics exhibit only one type of mesophase but a few examples with polymorphism are also known. Mesophases formed by disc-shaped molecules are primarily three types: (1) nematic, (2) columnar and (3) lamellar.The nematic phases of disc-shaped molecules can be sub-divided into three types: (a) nematic discotic (b) chiral nematic and (c) nematic columnar. In the nematic discotic phase, the molecules stay more or less parallel, having orientational order but no long-range positional order (Fig. 2a). The nematic phase of disc-shaped molecules is usually not miscible with the nematic phase of rod-shaped molecules, though they exhibit similar fluid Schlieren textures.26 However unlike the usual calamitic nematic, discotic nematic phase is optically negative, the director being the preferred axis of orientation of the disc-normals (or the short molecular axes). The symbol “ND” is used to denote discotic nematic mesophase. However, as the symmetry of a nematic phase formed by disc-shaped molecules is identical to that formed by rod-shaped molecules, it has recently been recommended27 to remove the subscript “D” from the symbol “ND”.
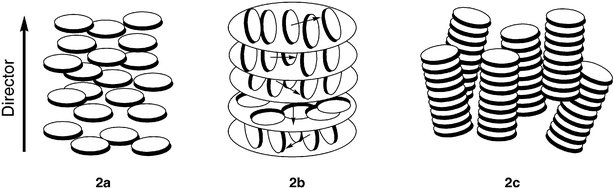 | ||
| Fig. 2 Schematic representation of (a) discotic nematic (b) helical structure of chiral nematic phase and (c) nematic columnar phase. | ||
Like chiral calamitic nematic or cholesteric phase, chiral discotic nematic mesophase, ND* also exists. The mesophase occurs in mixtures of discotic nematic and mesomorphic or non-mesomorphic chiral dopants, as well as in pure chiral discotic molecules.28,29 The helical structure of chiral discotic nematic phase is schematically shown in Fig. 2b.
The nematic columnar (NCol) phase is characterized by a columnar stacking of the molecules. However, these columns do not form two-dimensional lattice structures. They display a positional short-range order and an orientational long-range order (Fig. 2c).30
Columnar phases of discotic mesogens
In columnar mesophases, molecules assemble themselves one on top of the other in columns which are packed parallel on a two-dimensional lattice. The molecules may be arranged in a regular ordered manner or aperiodically (disordered). Depending on the order in the molecular stacking in the columns and the two-dimensional lattice symmetry of the column packing, the columnar mesophases may be classified in five classes: (a) columnar hexagonal phase, (b) columnar rectangular phase, (c) columnar oblique phase, (d) columnar plastic phase, and (e) columnar helical phase.Columnar hexagonal phase is characterized by a hexagonal packing of the molecular columns. Hexagonal mesophases are often denoted as Colho or Colhd where h stands for hexagonal and o and d for ordered or disordered stacking of the molecules. In both the cases, fluidity exists; only the correlation lengths are different and, therefore, it is recommended to discontinue o and d subscripts. The recommended abbreviation for columnar hexagonal phase is “Colh”. Fig. 3a illustrates the molecular packing in a Colh mesophase.
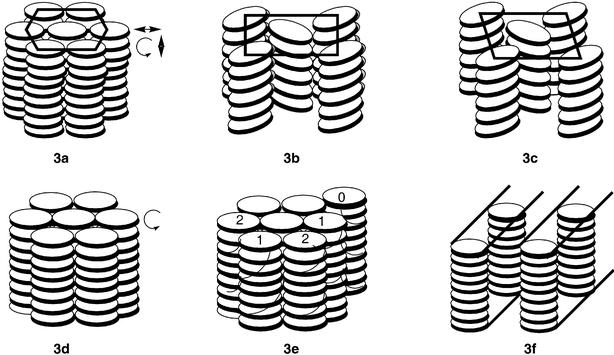 | ||
| Fig. 3 Schematic representation of (a) hexagonal columnar phase (b) rectangular columnar phase (c) columnar oblique phase (d) columnar plastic phase (e) helical phase and (f) columnar lamellar phase. | ||
The columnar rectangular mesophase consists of the stacking of the aromatic cores of the molecules in columns surrounded by the disordered aliphatic chains and packed in a rectangular fashion (Fig. 3b). Columnar rectangular phase is denoted by Colr. In the columnar oblique mesophase, the columns are arranged with an oblique unit cell (Fig. 3c).
Columnar plastic phase, denoted as Colp, has been identified recently in discotic liquid crystals.31 The phase is characterized by three-dimensional crystal-like order in a hexagonal lattice, while the discs within the columns are able to rotate about the column axis (Fig. 3d). In the Colh phase structural disorders such as non-parallel arrangement of the discs, longitudinal and lateral displacements and rotation around the columnar axis occur, while the motional freedom of discs in the Colp phase is restricted.
An exceptional mesophase structure with helical order has been demonstrated for a triphenylene derivative namely hexahexylthiotriphenylene (HHTT).32 In this so-called H phase helical columns develop which interdigitate in groups of three columnar stacks. The H phase found in HHTT is illustrated in Fig. 3e.
A layered structure is known to exist for mesophases of certain discotic compounds, e.g. bis(p-n-decylbenzoyl)methanato copper(II), some perylene derivatives, etc.33 Such a columnar lamellar mesophase which is denoted by the symbol ColL is shown in Fig. 3f. Another columnar mesophase with two-dimensional square lattice has recently been described in some metallomesogens.34
Chemistry of discotic liquid crystals
DLCs generally consist of flat, rigid aromatic cores surrounded by flexible chains. These materials often have two, three, four or six-fold rotational symmetry and three or more peripheral chains of three or more carbon atoms. However, there are many exceptions and materials with low symmetry, with a non-planar, non-aromatic core having shorter number of chains are also documented. Flexible chains are connected to the core via different linking groups such as ether, ester, benzoate, alkyne, etc. Fig. 4 represents a general molecular architecture for discotic mesogens.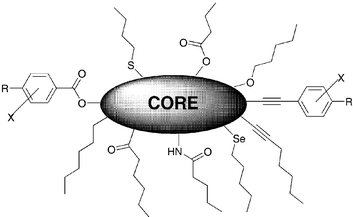 | ||
| Fig. 4 A general structural template for discotic mesogens. | ||
From the synthetic chemistry point of view, strategies to prepare discotic mesogens are fairly straightforward. Assembling an aromatic core followed by nucleophilic or electrophilic aromatic substitutions are most common in the synthesis of discotic liquid crystalline materials. While preparation of these materials is often but not always simple, their purification is usually difficult and tedious. This is primarily because of incomplete substitution of all the functional groups. Partial substitution of polyfunctional core gives a mixture of structurally similar products having almost identical Rf values on a chromatographic plate and their separation is generally very difficult. It is advisable to push the reaction towards completion by taking excess of the reagent and/or longer reaction time. However, in many cases, the excess of the reagent or longer reaction time cause side reactions and thus a complex mixture of the products. Finding optimum reaction conditions is the crucial part of the synthesis. The chemistry of some of the DLCs has been improved significantly in the last few years. Highly regioselective and high yielding methods are now available for the synthesis of some DLCs.
More than 50 different cores are known which on appropriate substitutions afford DLCs. These cores are mainly (a) aromatic hydrocarbons such as, benzene, naphthalene, anthraquinone, phenanthrene, pyrene, perylene, triphenylene, truxene, dibenzochrysene, decacyclene, dibenzopyrene, tetrabenzoanthracene, hexabenzocoronene; (b) macrocyclic cores such as, metacyclophane, tribenzocyclononatriene, tetrabenzocyclododecatetraene, phenylacetylene; (c) heterocyclic cores such as, benzpyrone, oxatruxene, thiatruxene, benzotrisfuran, tricycloquinazoline, bispyran, hexaazotriphenylene, pyrillium salt, coroneneimide, triazine, tetrathiofulvalene; (d) metallomesogens such as, β-diketone complexes, triketone complexes, dioxime complexes, tetraketone complexes, dithiolene complexes, benzalimine complexes, dibenzaldiimine complexes, pyrazolatogold complexes, porphyrin, tetraazoporphyrin, tetrapyrazinoporphyrazine, phthalocyanine; (e) saturated cores such as, cyclohexane, pyranose sugars, azamacrocycles and hexacyclans. Additionally, a number of discotic cores generated through non-covalent H-bonding are also known to display columnar mesophases.
Various options of synthesis pursued to prepare different DLCs are as follows. The simplest route is the attachment of chains to commercially available polyfunctional cores, e.g. esterification of naturally occurring scyllo-inositol. However, this option is limited as only a few polyfunctional cores suitable to prepare DLCs are commercially available. The direct electrophilic aromatic substitution of polycyclic aromatic hydrocarbons has been used in some cases to generate polyfunctionalized core or directly liquid crystalline materials but because of regioselectivity problem, such examples are also not common. The third option is the preparation of polyfunctional cores directly starting from unprotected functionalized precursor molecules, e.g., one-step preparation of hexahydroxyanthraquinone from gallic acid. However, this option is also limited as many functional groups are sensitive towards the drastic reaction conditions used to build the core. The most common method used to prepare DLCs involves construction of the core with protected functional groups, cleavage of the protecting groups and finally grafting pendant chains. Recently, particularly to prepare discotics with ether linkages, efforts have been made to avoid the protection and deprotection steps and thus, long aliphatic chains required to induce mesogenity were attached in the starting molecules and these then transformed directly to liquid crystalline materials using classical or standard synthetic methods of today. This methodology has also been applied to prepare a number of alkyl substituted (alkyl chains attached directly to the core) DLCs.
Although, several hundred DLCs have been synthesized, they can be sub-classified as esters (including benzoates and cyclohexanoates), ethers, thioethers, alkynes, ketones, amides and hydrocarbons. The paucity of the space does not allow discussing the chemistry of all discotics in detail and, therefore, the chemistry of most frequently encountered core systems and linking groups in the field of DLCs is presented here. In this article, emphasis has been given only to the chemistry of these materials and, therefore, unlike many other articles, thermal properties (mesomorphic behaviour) and structure–properties relationship have not been focused here.
Discotic monomers: esters and ethers
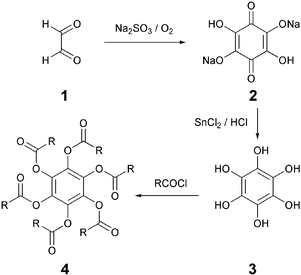
A large proportion of DLCs are polyphenylesters and polyphenyethers. Polyphenols (most commonly hexaphenols) of various cores were generally prepared following well established chemistry and then flexible aliphatic chains of variable length were grafted using classical chemistry. In recent times, efforts have been focused to prepare DLCs, particularly ethers, by direct cyclization of precursors with defined chains.Hexaalkanoyloxybenzenes 4 were the first DLCs prepared by Sadashiva in 1977.19 It is interesting to note that the first reports on the synthesis of these benzene hexaesters date back to as early as 1937, but their mesomorphic nature was not detected. The reaction of hexahydroxybenzene 3 with about 12 equivalents of an acid chloride at elevated temperature for about six hours yielded the desired liquid crystalline ester (Scheme 1). It should be noted that no solvent was used in this reaction. Commonly used esterification reaction conditions, i.e. refluxing the hexahydroxybenzene and acid chloride in dry pyridine, generally give complex mixture of products from which the desired product is very difficult to isolate. The hexahydroxybenzene was prepared from glyoxal. The intermediate compound, tetrahydroxy-1,4-benzoquinone 2 is now commercially available and, therefore, these discotics can be prepared in just two steps. A number of benzene hexaesters having normal alkyl chains, branched alkyl chains and heteroatoms in the alkyl chains have been prepared to understand the structure–mesomorphic properties relationship.35,36 Hexaethers of benzene were found to be non-liquid crystalline indicating that dipolar interactions of carbonyl group play pivotal role in the self-assembly of these molecules.
 | ||
| Scheme 1 Synthesis of hexaalkanoyloxybenzene derivatives. | ||
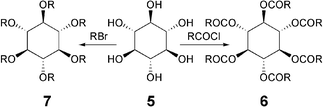
Structurally close to benzene hexaesters, mentioned above, are those of the saturated hexahydroxy cyclohexane. Commercially available scyllo-inositol 5 can be easily converted to its hexaesters 6 with an excess of acid chloride in trifluoroacetic acid or to hexaethers 7 by reacting with potassium hydroxide and alkyl halides (Scheme 2).37 These cyclohexanyl hexaesters and ethers display a rich mesomorphism. It is interesting to note that axially-substituted cyclohexane esters do not display any mesomorphism.
 | ||
| Scheme 2 Synthesis of cyclohexane-based DLCs. | ||
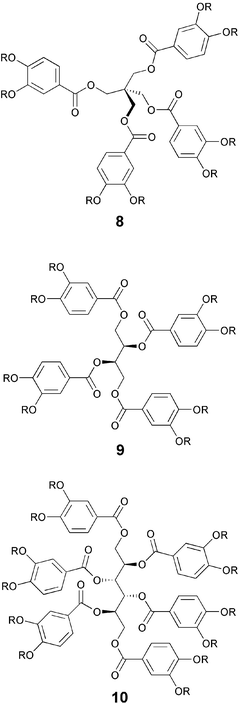
Similarly, benzoylation of commercially available alcohols such as pentaerythritol, D-threitol and D-mannitol with appropriately substituted benzoyl chlorides produces liquid crystalline peracylated pentaerythritol 8, D-threitol 9 and D-mannitol 10 derivatives.38,39 The tetrabenzoates of pentaerythritol 8 are interesting DLCs as they are derived from a tetrahedral dot-like molecule.38 However, it should be noted that the real core is not the central tetrahedral carbon atom but probably the part structure consisting of the carboxy groups, the aromatic rings and oxygen atoms of the ether linkage acts as the core. The columnar phase formation is essentially driven by the micro-segregation of their polar central cores from the non-polar flexible chains.
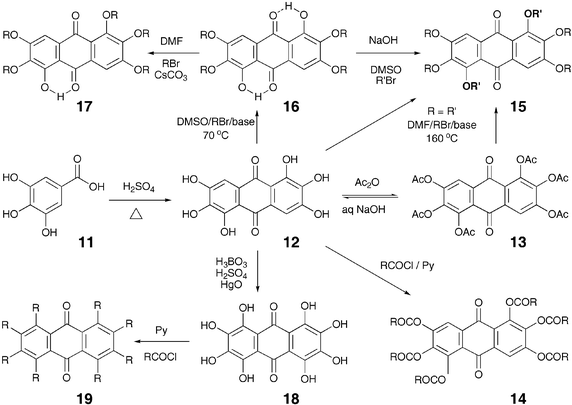
The 1,2,3,5,6,7-hexahydroxyanthraquinone 12 commonly known as rufigallol was obtained in low yield by acid-catalyzed self-condensation of gallic acid (Scheme 3). The crude yellow product is difficult to filter as it clogs badly. Purification of rufigallol can be achieved by centrifuging the crude product followed by acetylation of the dried product in acidic conditions. The hexaacetate 13 separates as yellow plates and can be recrystallized from acetic anhydride. Pure rufigallol, obtained by the hydrolysis of hexaacetate, on esterification with long-chain acid chlorides yields liquid crystalline hexaesters 14.40 Similarly, hexaethers 15 can be prepared by the etherification of the hexaphenol but they can be better made directly from the hexaacetate.41 Under milder etherification conditions, the hydrogen bonded 1 and 5 positions do not get alkylated and thus 1,5-dihydroxy-2,3,6,7-tetraalkoxy-9,10-anthraquinone 16 forms. This unequal reactivity leads to the preparation of several unsymmetrical rufigallol derivatives. Recently a few mono-hydroxy-pentaalkoxyanthraquinone derivatives 17 have also been prepared42 in low yields by partial alkylation of dihydroxy-tetraalkoxyanthraquinones, but the process is applicable only for a small scale and the purification of the product was tedious.
 | ||
| Scheme 3 Synthetic routes to anthraquinone-based DLCs. | ||
Oxidation of 1,2,3,5,6,7-hexahydroxyanthraquinone yields octahydroxy-anthraquinone 18 (Scheme 3) which on esterification with alkanoyl chlorides provides octa-substituted anthraquinone discotics 19.43 Billard and co-workers reported the first discotic liquid crystalline hexaesters of rufigallol in 1980 and since then about 100 different discotic liquid crystalline derivatives of this molecule have been prepared but no new efficient method to prepare rufigallol have been reported.
While dimerization of gallic acid in 98% H2SO4 at 100 °C gives rufigallol, its reaction with potassium persulfate and concentrated sulfuric acid at 160 °C produces flavellagic acid 20, which can be oxidized to ellagic acid 21 (Scheme 4). Both the compounds can be converted into DLCs 22–25 by esterification or etherification with appropriate aliphatic acid chlorides or alkyl halides.44 Of course, ellagic acid, though expensive, is now commercially available and can be used to prepare a variety of DLCs.
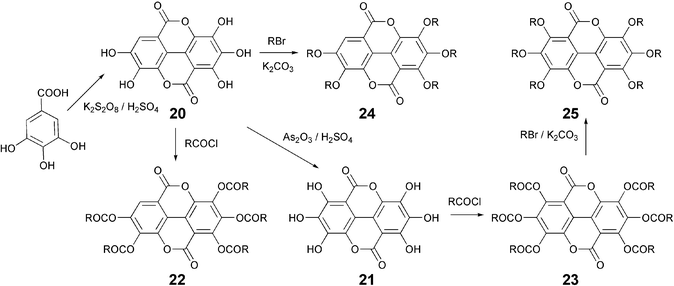 | ||
| Scheme 4 Synthesis of flavellagic and ellagic acid based DLCs. | ||
Metacyclophane is another example of polyphenolic discotic core which was prepared from unprotected phenol. Acid-catalyzed condensation of pyrogallol and 1,1-diethoxyethane furnished dodecahydroxy compound 26 in good yield.45 Esterification of 26 produced bowl-like DLCs 27 (Scheme 5).
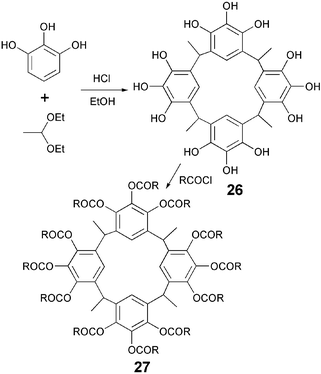 | ||
| Scheme 5 Synthesis of bowl-like metacyclophane DLCs. | ||
In all other polyphenolic discotic cores, free hydroxyl groups were created by the cleavage of protecting groups. Polymethoxyarenes have been commonly used as precursors for the synthesis of discotic esters and ethers as methoxy groups may be easily cleaved to furnish the corresponding phenols. For example, hexamethoxy derivatives of triphenylene, truxene, oxotruxene, thiatruxene, phenanthrene, tribenzocyclononatriene, tricycloquinazoline, have been employed as precursors in the synthesis of various DLCs based on these cores. These polyphenols are often sensitive to air oxidation and cannot be stored for a long time. The yields of final products largely depend upon the purity of these polyphenols and are often low. Moreover, partial oxidation and esterification or etherification generates a number of products and it is tedious to isolate the desired product from this complex mixture of products.
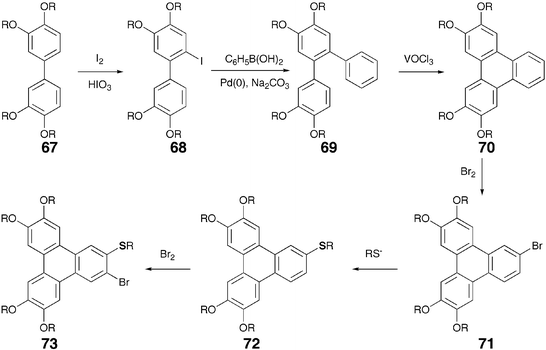
The best studied core system in the field of DLCs is triphenylene (TP). Though this symmetrical fused aromatic hydrocarbon has been known in the chemical literature for more than a century, as a core for DLCs it was introduced by Billard et al. in 1978.46 TP derivatives are thermally and chemically stable, their chemistry is fairly accessible, and they show a variety of mesophases having interesting electronic properties. This fostered numerous synthetic efforts to prepare a variety of TP based DLCs. At present more than 500 discotics based on a TP core are known in the literature. The chemistry and mesomorphic properties of TP-based DLCs have recently been covered in review articles47–49 and, therefore, only a brief account of the chemistry of these materials is presented here. Major developments in the chemistry of TP-based ethers and esters are as follows: (1) exploitation of the Musgrave's method to prepare a number of symmetrical TP hexaesters, ethers and benzoates. The synthesis involves oxidative trimerization of veratrol in H2SO4 using chloranil or iron(III) chloride as oxidant to get hexamethoxy-TP, complete demethylation of hexamethoxy-TP using BBr3, HBr or HI to obtain hexahydroxy-TP and finally esterification or etherification of this phenol with various acid chlorides or alkyl halides. The procedure is sluggish, low yielding and isolation of the pure product is tedious; (2) cyclic anodic trimerization of dialkoxybenzene followed by a chemical or electrochemical reductive work-up to produce hexalkoxy-TPs. This method is very attractive but applicable only on a small scale; (3) direct trimerization of long chain substituted dialkoxybenzenes to hexaalkoxy-TP discotics using only a catalytic amount of H2SO4 (0.3%) in dichloromethane followed by reductive work-up; (4) use of MoCl5 or VOCl3 for the above mentioned trimerization in the presence or absence of H2SO4. It should be noted that though a number of reagents are known for the oxidative coupling of phenols and phenolethers,50 only the above mentioned three reagents, FeCl3, MoCl5 or VOCl3 have so far been effectively utilized to prepare TP discotics; (5) development of several methods to prepare unsymmetrical TP discotics. The most successful is the biphenyl-phenyl oxidative coupling using the above mentioned reagents. The Suzuki and Nigishi type of reactions for the synthesis of biphenyl and terphenyl followed by oxidative cyclization to create TP nucleus with predefined substitutions have also been used; (6) synthesis of a number of alkoxy-hydroxy-TP (mono-, di- and trifunctionalized-TPs) using a variety of reagents such as, diphenylphosphide lithium, 9-Br-BBN or B-bromocatecholborane; (7) use of isopropoxy masking group in a biphenyl-phenyl oxidative coupling route for the preparation of mono- and dihydroxy substituted TPs; (8) isolation of the monofunctionalized-TP as a by-product of the oxidative trimerization reaction. However, it should be noted that separation of the monohydroxy-pentaalkoxy-TP from unreacted hexaalkoxy-TP is tedious and need highly efficient column chromatography. The job becomes easier if aluminium oxide is used as adsorbent instead of silica gel for column chromatography; (9) selective removal of hydroxyl group(s) from TP periphery followed by electrophilic substitutions; (10) substitution at the α-position and the synthesis of heptasubstituted TPs.
TP derivatives with side chains bearing terminal groups such as azobenzene,51 cyanobiphenyl,52 carbazole,53 ferrocene,54 fullerene,55 sugars,56 crown ethers,57 and mixed tails58–62 are some recent examples prepared using above mentioned methodologies. Common routes to prepare TP derivatives are out lined in Scheme 6.
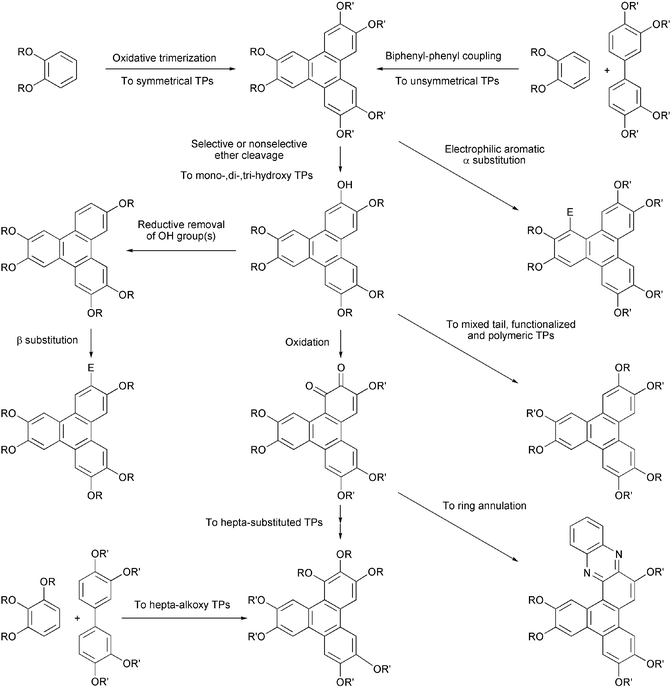 | ||
| Scheme 6 Common routes to triphenylene discotics. | ||
The charge and energy migration in triphenylene based discotic liquid crystals have been studied extensively and because of these properties, their potential applications in a number of devices have been envisaged. It has recently been demonstrated that the degree of order determines charge mobility in columnar liquid crystalline materials.63 It is expected that an increase in the orbital overlap area will lead to higher charge mobility. Dibenzo[fg,op]naphthacene (DBN), also named as dibenzopyrene (DBP), consists of 24 carbon atoms bound together in 6 adjoining C6-membered rings to form the planar disc-shaped aromatic molecule. The DBN core appeared in the Liquid Crystal field very recently but became the focus of attention because of the first ferroelectric switching of columnar mesophases exhibited by some of its derivatives.64 A number of octaalkoxy- and octaalkanoyloxy-DBP derivatives have been prepared and studied for various physical properties.
All the octasubstituted derivatives were synthesized following the method of Musgrave and Webster which involve the oxidation of 3,3′,4,4′-tetramethoxybiphenyl 28 by chloranil in 70% sulfuric acid to 2,5,6,9,12,13-hexamethoxydibenzo[fg,op]naphthacene-1,8-quinone 29 (Scheme 7). Cleavage of methoxy groups yields hexahydroxy-1,8-quinone 30. This can be reduced to octahydroxy-DBN 31. The octahydroxy-DBN can be converted to octaethers 32 or octaesters 33 by classical reactions. Reductive acetylation of the hexahydroxy-1,8-quinone followed by alkylation can also be used to prepare octasubstituted-DBN liquid crystals. Direct oxidation of tetrapentyloxybiphenyl by chloranil to hexapentyloxydibenzo[fg,op]naphthacene-1,8-quinone and its conversion to octapentyloxy-DBP by the above mentioned procedure has also been reported by the Ringsdorf group.65 Another reagent VOCl3, which has been used for the preparation of triphenylene discotics, can also be efficiently utilized under very mild reaction conditions for the preparation of these quinones.66
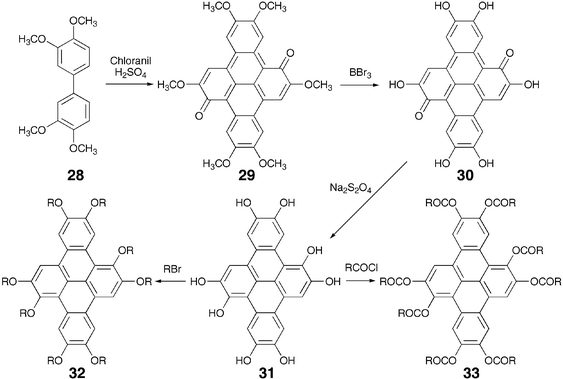 | ||
| Scheme 7 Synthesis of octa-substituted DBN derivatives. | ||
Being a larger core having more delocalized π electrons than triphenylene, a greater degree of π–π interaction and hence higher charge carrier mobility was expected in DBN derivatives. However, the charge carrier mobility in the octasubstituted DBN derivatives was found to be one order of magnitude lower than in the columnar phase of hexaalkoxytriphenylenes.67 This could be because of less ordered columnar packing due to the steric hindrance caused by the ‘way region’ alkoxy chains and this results in the lower charge carrier mobility. It was anticipated that the removal of these alkoxy chains from 1 and 8 positions would give a better core–core interaction and, therefore, high charge carrier mobility. This leads to the development of a novel, versatile and regiospecific synthesis of variable degree substituted dibenzo[fg,op]naphthacene derivatives.67 For the synthesis of various dibenzo[fg,op]naphthacene (DBN) derivatives 42 the strategy outlined in Scheme 8 was envisaged. The synthesis is based on the preparation of key intermediate tetraphenyl 41 starting from 3-nitrobromobenzene 34. The 2,2′-dibromo-4,4′dihydroxybiphenyl 36 was prepared by reductive dimerization of 34 followed by benzidine rearrangement. Alkylation of the diphenol 36 with appropriate 1-bromoalkane resulted in 2,2′-dibromo-4,4′dialkoxybiphenyls 37. The tetraphenyls 41 were prepared either by coupling 37 with dialkoxyboronic acids 39 or by first converting the dibromobiphenyls 37 to diboronic acids 38 and then reacting with dialkoxyiodobenzenes 39. Photocyclodehydrogenation of the tetraphenyls 41 in the presence of an excess of iodine furnished the desired 2,5,6,9,12,13-hexaalkoxy-DBN derivatives 42. The cyclization can also be achieved by oxidative coupling using FeCl3 as oxidizing agent. The versatility of the procedure has been demonstrated by preparing a number of hexa-substituted DBN derivatives.67 X-ray diffractometry results confirm that hexasubstituted dibenzonaphthacenes are more ordered than octasubstituted derivatives.
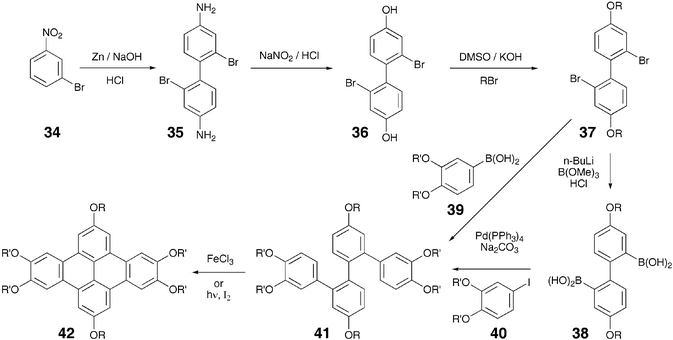 | ||
| Scheme 8 A rational synthesis of hexa-substituted DBN derivatives. | ||
The classical tetramerization of alkoxy-substituted pyrroles has been extensively used to prepare a number of porphyrin-based DLCs. Similarly, condensation of pyrrole with substituted benzaldehydes resulted in the synthesis of a number of liquid crystalline meso-tetrasubstituted porphyrins. Recently, expanded porphyrins were reported to display columnar mesophases.68 These hydrazinophyrins 43 were synthesized by the acid-catalyzed condensation of 3,4-dialkoxy-2,5-diformylpyrroles with one equivalent of hydrazine (Scheme 9). Although, the 2 + 2 condensation is also possible and has been realized in other cases, Sessler et al. obtained only the 4 + 4 product in 30–60% yield.68
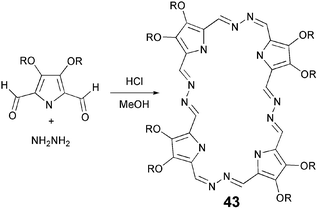 | ||
| Scheme 9 Synthesis of hydrazinophyrin. | ||
Like-wise phthalocyanine-based DLCs have been synthesized and studied extensively. Recently efforts have been directed to prepare large phthalocyanines such as 44. Following the traditional synthesis, these materials have also been prepared by the tetramerization of 1,2-dicyano-substituted precursors, obtained using recently developed approaches for the synthesis of unsymmetrical triphenylenes (Scheme 10).69
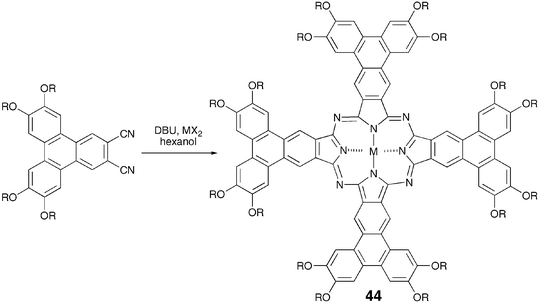 | ||
| Scheme 10 Synthesis of macrodiscotic triphenylenophthalocyanines. | ||
The coupling of a 1,2-diketone with a 1,2-diamine (Scheme 11) leads to the formation of a number of heterocyclic DLCs. The prominent recent examples are: diquinoxalino[2,3-a∶2,3-c]phenazine 45,70 dibenzoquinoxaline 46,71 bisphenazine 4772 and phenanthrophenazine 48.73 The 1,2-diamines were generally prepared by the reduction of dinitro compounds. The 1,2-diketones are often commercially available, e.g., hexaketocyclohexane, or can be prepared by the oxidation of tolanes or phenols. They can also be prepared by the Friedel Crafts acylation of substituted benzenes with oxalyl chloride.
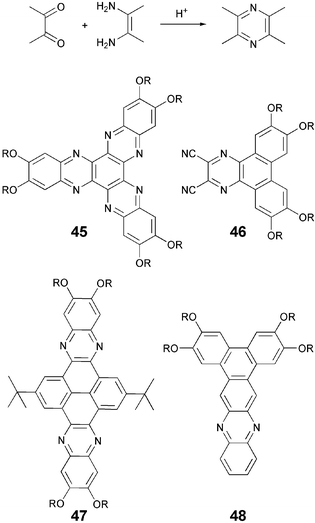 | ||
| Scheme 11 Coupling of a 1,2-diketone with a 1,2-diamine and some structures derived from this coupling. | ||
The dibenzo[g,p]chrysene 52 core was generated by the oxidative cyclization of diphenylphenanthrene 51 which in turn was prepared by a palladium-catalyzed annulation reaction of diphenylacetylene 50 with 2-iodobiaryl 49 (Scheme 12).74 The intermediate 51 can also be prepared by the photochemical cyclization of tetraphenylethene 54 which was prepared from benzophenone derivative 53via McMurry coupling.75
![Synthetic routes to dibenzo[g,p]chrysene derivatives.](/image/article/2006/CS/b506619k/b506619k-s12.gif) | ||
| Scheme 12 Synthetic routes to dibenzo[g,p]chrysene derivatives. | ||
Alkoxy derivatives of helicene and heterohelicene are very interesting as these are derived from a chiral core. Self-assembly of these helical cores attached with aliphatic chains produces hexagonal arrayed columns not only from the melt but also in solution.76 The modern synthetic methods applied for the synthesis of a helicene molecule is depicted in Scheme 13.
 | ||
| Scheme 13 Synthesis of helicene derivatives. | ||
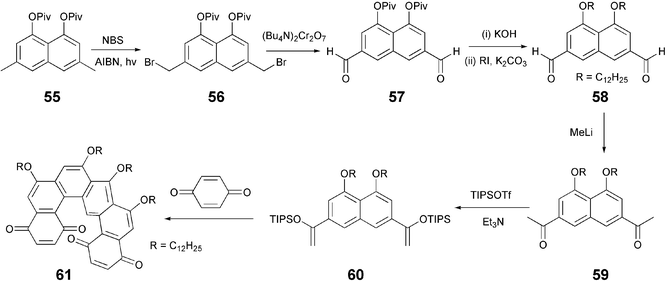
Connecting alkoxy-pyrazoles via metal bridges produces interesting DLCs like 6277 and 63.78 The trinuclear dendritic pyrazolate complexes of copper, silver and gold self-assemble through the metallophilic interactions to form photoluminescent superhelical fibres. The alkoxy-substituted Au(I) pyrazolate complex gives a light-emitting organogel, where the photoluminescence colour can be changed synchronously to the sol–gel transition.78 On the other hand, the Cu(I) analogue was reported to be useful for the preparation of thermally rewritable phosphorescent paper.79 These materials were synthesized simply by treating substituted pyrazole derivatives with metal salts in THF/Et3N at room temperature.
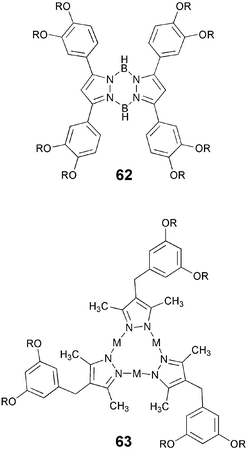
Discotic monomers: thioethers
The observation of very high one-dimensional charge carrier mobility in a TP thioether vitalized research activities in the field of discotic liquid crystals. The compound 2,3,6,7,10,11-hexahexylthio-TP forms a self-organized helical columnar phase at low temperatures with nearly crystalline order in addition to the normal columnar phase. The photoinduced charge carrier mobilities up to 0.1 cm2 V−1 s−1 were achieved in the H phase. With the exception of organic single crystals, these were the highest electronic mobility values reported till that time.63 Of course, now much higher mobilities have been achieved in some DLCs.23The regioselective bromination of TP hydrocarbon opened the route to prepare symmetrical 2,3,6,7,10,11-hexaalkylthio-TP derivatives. The synthesis of hexaalkylthio-TPs involves the thiolate anion substitution of 2,3,6,7,10,11-hexabromo-TP with excess of sodium alkylthiolate in a polar aprotic solvent DMEU at 100 °C for 1 to 2 hours in 40–55% yield. An improvement in this process was reported by generating the thiolates with sodium hydride in dry ether instead of sodium ethoxide in ethanol and subsequently heating with hexabromo-TP in DMEU at 70 °C for 30 min. This enhanced the yield to 78–84% and the product was less contaminated with side-products.80 These compounds can be obtained in high yields with very high purity by generating thiolates with potassium tert-butoxide in NMP and subsequently heating with hexabromo-TP at 70 °C for 25 minutes in the same solvent followed by quenching the reaction with an appropriate 1-bromoalkane (Scheme 14).81
 | ||
| Scheme 14 Synthesis of triphenylene thioethers. | ||
The mixed alkoxy-thioalkyl-TPs were designed in search of highly ordered helical phase. These materials were prepared by displacing β-halogen(s) by thioalkyl group(s). Corresponding alkoxy-bromo TPs were prepared using a combination of organometallic chemistry and oxidative coupling routes.82,83 An example is shown in Scheme 15. Thiolation was achieved under normal reaction conditions but in some cases a catalytic amount of tetrakistriphenylphosphine Pd(0) was used to improve the yield of the product. The mesophase behaviour of these compounds is dependent on the position and relative number of substituents. Most of the compounds in this mixed alkylthio-alkoxy series show only hexagonal columnar phase but one compound namely 3,6-bis(hexyloxy)-2,7,10,11-tetrakis(hexylthio)triphenylene is claimed to exhibit highly ordered helical phase.83
 | ||
| Scheme 15 Synthesis of mixed alkoxy-thioalkyltriphenylenes. | ||
Similarly, thioethers of many other cores such as tricycloquinazoline, hexaazatriisothianaphthene and hexaazotrinaphthalene were prepared by the nucleophilic displacement of halogen atoms. It is noteworthy that despite the poor solubility of haloarenes, the thiolation reaction proceeds smoothly in all the cases.
The molecules such as 74 in which six α-linked terthiophenes are connected to a central benzene core via a thioether linkage, display smectic and nematic phases. These molecules adopt an overall calamitic shape, rather than expected disc shape by folding terthiophene segments upward and downward. These molecules were prepared using the MacNicol reaction as shown in Scheme 16.84
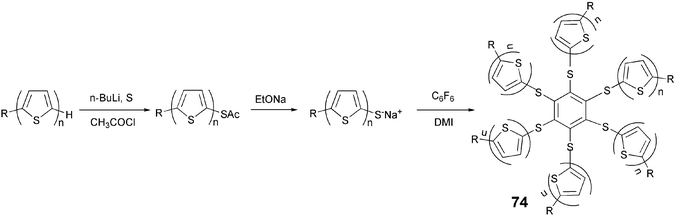 | ||
| Scheme 16 Synthesis of thiophene-linked star-shaped benzene molecules. | ||
The direct cyclization of preformed thioethers is now more common to prepare porphyrin, phthalocyanine (Pc) and subphthalocyanine (SubPc) based DLCs. Thus, the classical tetramerization of thioalkyl-substituted phthalonitrile afforded the octakis(alkylthio)-Pc while its trimerization in the presence of BCl3 furnished the hexakis(alkylthio)-SubPc (Scheme 17).85,86 SubPcs have found much interest recently due to the possibility of preparing unsymmetrically substituted phthalocyanines.
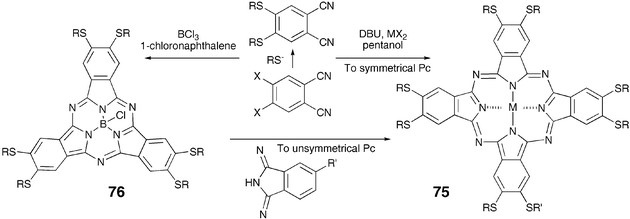 | ||
| Scheme 17 Synthesis of phthalocyanines and subphthalocyanines. | ||
Discotic monomers: alkynes
The radial and cyclic multiynes are extremely important in the field of DLCs as most of these materials show the less commonly observed discotic nematic phases. The Sonogashira coupling—a Pd-catalyzed, robust, reliable and general method for the preparation of unsymmetrical acetylene—was applied for their synthesis. For this cross-coupling reaction, the combination of Pd, phosphines and CuI in dry triethylamine is generally employed under inert conditions. Although, now that many ligand- and copper-free Pd-catalyzed Sonogashira cross-coupling protocols in various solvents and bases have been developed, these have not been much explored in the synthesis of alkyne-based DLCs.The radial multiynes 85 (Scheme 18) were introduced in the field of DLCs by Praefcke and coworkers in 1987.87 Subsequently, a large number of symmetrical, unsymmetrical and functionalized multiynes based on benzene, naphthalene, triphenylene, phthalocyanine, hexabenzocoronene cores were prepared and characterized. All these compounds were usually prepared by coupling bromoarenes with excess of acetylene using a mixture of (Ph3P)2PdCl2, CuI and Ph3P as catalyst in dry triethylamine (Scheme 18). The yield of final product depends on a number of factors such as the nature and amount of free acetylene, dry and inertness of the system, etc. Highly confusing yields ranging from 10% to 80% have been reported. The Glaser coupling—an oxidative coupling of terminal alkynes catalyzed by cuprous ion to give diynes, is the major side reaction. Therefore, a large excess of the alkyne is required because if the terminal alkyne is not available, a number of products due to partial alkynylation of haloarene are formed. The separation of pure product from this complex mixture is very difficult. Further, most of the terminal alkynes are not commercially available and, therefore, have to be prepared using multi-step synthesis. This is another constraint in the large scale synthesis of alkyne-based DLCs. Mainly two different methods, as depicted in Scheme 18, were practiced for the synthesis of aryl acetylenes. Traditionally, alkyl-substituted phenylacetylenes were prepared starting from benzene. Friedel–Crafts acylation of benzene with the long chain acid chlorides gave the alkanoylbenzenes (1-phenyl-1-alkanones) 77 in excellent yields. Wolff–Kishner or Clemmensen reduction of these ketones furnished alkylbenzenes 78. A second Friedel–Crafts acylation of these alkylbenzenes with acetyl chloride yielded alkyl substituted acetophenone (1-(4-alkylphenyl)ethanone)) 79 in moderate yields. Treatment of these acetophenones with PCl5 resulted 1-(4-alkylphenyl)-2-chloroethenes 80 which can be converted to terminal acetylenes (1-ethynyl-4-alkylbenzenes) 81 by dehydrhalogenation in moderate yields.
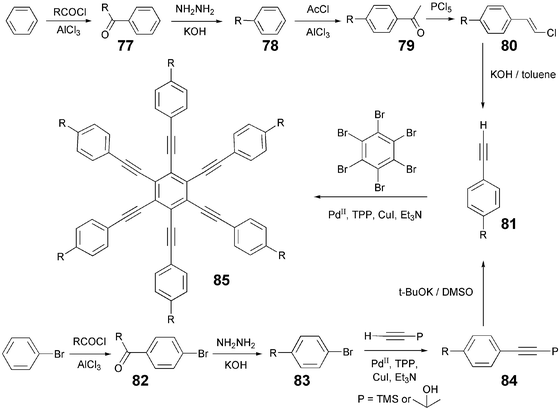 | ||
| Scheme 18 Synthetic routes to alkylphenylacetylene and its coupling with hexabromobenzene. | ||
Alternatively, these acetylenes were prepared using a combination of old and modern methods. Friedel–Crafts acylation of the bromobenzene with the acid chlorides furnished the 1-bromo-4-alkanoylbenzenes 82. Reduction of these ketones as mentioned above, gave 1-bromo-4-alkylbenzenes 83. Pd-catalyzed alkynylation of different alkyl-substituted bromobenzenes 83 with monoprotected acetylene like 2-methyl-3-butyn-2-ol or trimethylsilylacetylene afforded the protected phenylacetylenes 84 which were deprotected using KOH in refluxing toluene or in aqueous methanol to yield the phenylacetylenes 81. Both the methods give more or less same overall yield of free phenylacetylene 81. The synthesis of alkoxy-substituted phenylacetylenes is relatively simple and can be achieved starting from phenol or bromophenol following similar strategies. Some recent discotic liquid crystalline structures prepared using above protocols are shown in Fig. 5.88–91
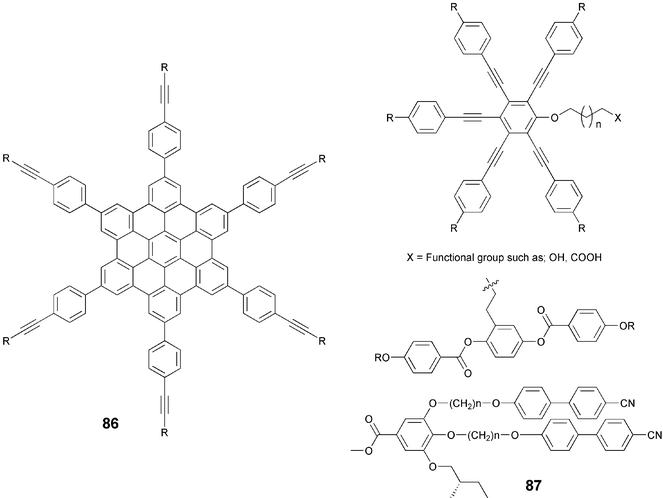 | ||
| Fig. 5 Structures of some recent discotic radial multiynes. | ||
Unlike the above mentioned alkynylbenzenes, Sonogashira coupling between hexa-iodobenzene and substituted 6-ethynylazulene did not yield the desired product 90. However, the inverse cross-coupling reaction of substituted 6-bromoazulene 89 with hexaethynylbenzene prepared in situ from hexakis(trimethylsilylethynyl)benzene 88 produced 90 in moderate yield (Scheme 19).92 A series of poly(6-azulenylethynyl)benzenes substituted with n-hexyloxycarbonyl chains at 1,3-positions in azulene rings i.e. hexakis-, 1,2,4,5-tetrakis- and 1,3,5-tris(6-azulenylethynyl)benzene derivatives were prepared using this methodology.92 The thermal behaviour of these materials was also very different than the hexalkynylbenzene derivatives. These materials displayed columnar phases instead of discotic nematic phase commonly observed in benzene based multiynes. This could be due to the strong interactions of dicarbonyl-substituted azulene moieties.
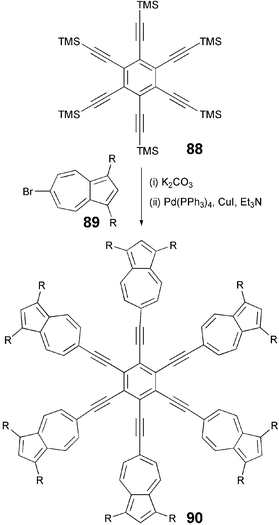 | ||
| Scheme 19 Synthesis of hexakis(6-azulenylethynyl)benzene. | ||
The direct coupling of pyrrole and an alkoxyarylpropynal under usual conditions of porphyrin synthesis produced the desired compound 93 in very poor yield. However, it can be prepared in better yield via a two-step process as shown in Scheme 20. Reaction of the aldehyde 91 with a large excess of pyrrole, in the presence of BF3.Et2O as catalyst at −25 °C produced the dipyrromethane 92 in good yield. Condensation of 92 with aldehyde 91 under similar reaction conditions, followed by oxidation of the resultant porphyrinogen with DDQ and insertion of metal, afforded the required mesogenic porphyrin 93 in 22–28% yield.93
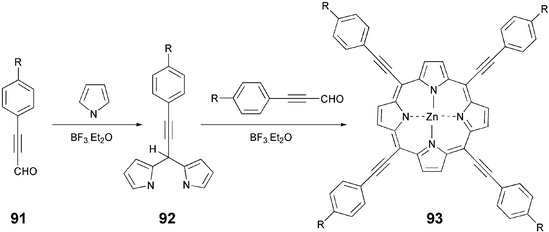 | ||
| Scheme 20 A two-step synthesis of tetraarylethynyl-substituted porphyrins. | ||
A number of cyclic acetylenes were reported to display nematic as well as columnar mesophases. These phenylacetylene macrocycles are interesting as their self-assembly may lead to aligned nanotubes. These materials were prepared using a well-defined stepwise, repetitive synthetic approach as shown in Scheme 21.94 It should be noted that a one-pot synthesis of such macrocycles was reported about 30 years ago. The copper salt of m-iodophenylacetylene on homocondensation gave a broad mixture of cyclic and noncyclic oligomers and polymers. The desired cyclic hexamer could be isolated in very low yield by extraction of all less rigid and, therefore, more soluble by products.95
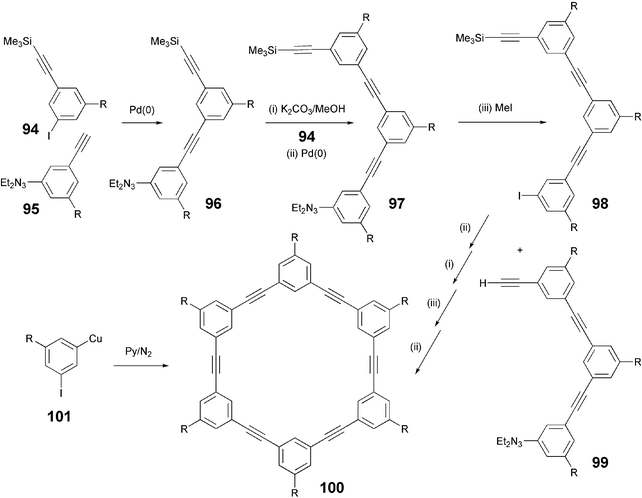 | ||
| Scheme 21 Synthesis of phenylacetylene macrocycles. | ||
Discotic liquid crystals like 103 with inverse topology (rigid periphery and flexible core) formed by cyclic acetylene-based macrocycles have recently been discovered.96 These macrocyclic discotics are unique as they carry less flexible side chains that point to the inside filling the internal cavity of the macrocycle. This architecture has recently been confirmed by single crystal X-ray analysis.97 A number of such nanosized DLCs were prepared by Hoger et al.via the alkylation of corresponding phenols using classical alkylation or under Mitsunobu reaction conditions. The diyne 103 is obtained by intermolecular Glaser coupling of the half disc (bis-acetylene) 102 (Scheme 22) which in turn can be prepared following methodologies described above.
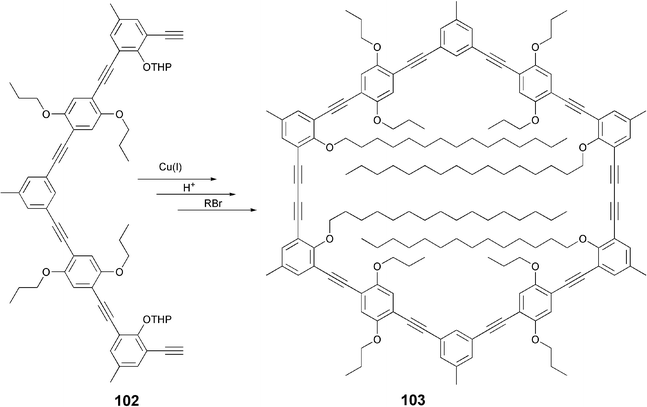 | ||
| Scheme 22 Synthesis of shape-persistent macrocycles. | ||
Alkyl-functionalized discotic monomers
Connecting flexible aliphatic chains to the core directly, instead of through a hetero atom, has a profound effect on the thermal behaviour of discotic mesogens. In the case of benzene-centred multiynes it was observed that when the peripheral alkyl chains are attached to the phenyl ring in the hexaalkynylbenzene via oxygen atom, the melting and clearing temperatures are higher compared to when the same alkyl chains are directly attached to the ring. This in conjunction with the observation that when the peripheral aliphatic side chains of various cores are branched, the mesophase is widened but the type of the mesophase formed is not affected by the introduction of branching in many cases, has been utilized to reduce the transition temperatures and enhance the phase widths of DLCs.98–100 In the case of triphenylene, direct attachment of the chains to the core completely destroys the mesomorphism. These non-liquid crystalline symmetrical hexaalkyltriphenylenes 105 were prepared using Pd-catalyzed cross-coupling between triphenylene hexatriflate 104 and arylzinc iodides (Scheme 23).101 The mixed alkyl-alkoxytriphenylenes 106 prepared from alkoxytriphenylene bromides and arylzinc iodides were also found to be non-liquid crystalline (Scheme 24).101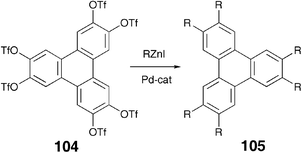 | ||
| Scheme 23 Synthesis of hexaalkyltriphenylenes. | ||
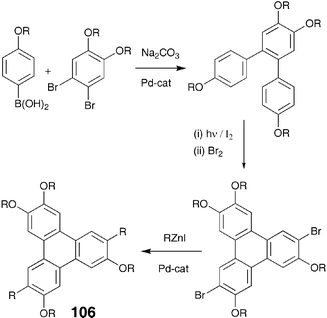 | ||
| Scheme 24 Synthesis of mixed alkoxy-alkyltriphenylenes. | ||
Similarly, trialkyldecacyclenes 108 prepared via the Wolff–Kishner reduction of liquid crystalline trialkanoyldecacyclenes 107 were reported to be non-mesomorphic.102 The unusual C3 symmetric decacyclene derivatives 107, in which aliphatic chains are connected to the core via keto group, were synthesized by Friedel–Crafts acylation of the commercially available decacyclene (Scheme 25). This reaction proceeds with high regioselectivity to give exclusively one isomer having C3 symmetry.102
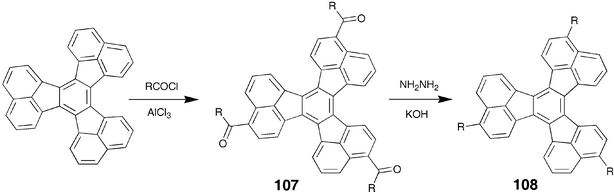 | ||
| Scheme 25 Synthesis of trisubstituted decacyclene derivatives. | ||
On the other hand, a number of alkyl-substituted cores showing columnar phases are known. The important examples of this group are porphyrin, phthalocyanine, tetrapyrazinoporphyrazine and hexabenzocoronene based DLCs, in addition to the radial multiynes described above. In most cases, alkyl-substituted precursors were cyclized to get the desired architectures. For example, Sugino et al. recently reported103 a dodecyl-substituted tetraphenylporphyrin metallomesogen 109. The compound was obtained via a typical porphyrin synthesis methodology i.e. condensing an alkyl-substituted benzaldehyde with pyrrole in propionic acid followed by metallation (Scheme 26). The compound exhibits a 3D plastic lamellar mesophase with a columnar structure. The axial hydrogen bond interaction in the stacked columnar structure played a vital role in enhancing the clearing point and stacking periodicity in the molecule.
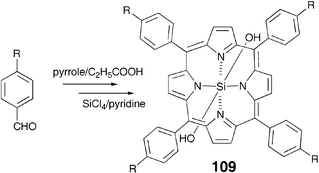 | ||
| Scheme 26 Synthesis of a tetraphenylporphyrin dihydroxysilicon(IV) complex. | ||
The rigid dendritic porphyrins with 8 or 16 long flexible alkyl chains on their periphery 110 and 111 were prepared using Suzuki coupling between the 5,10,15,20-tetrakis(3′,5′-dibromophenyl)porphyrin and corresponding alkylphenyboronic acids. Both dendrimers exhibited glass transition and very broad mesophase range.104
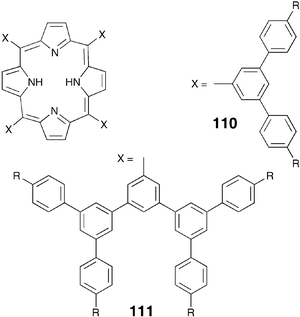
The tetraalkyl-substituted coronenediimides 115 can be prepared as shown in Scheme 27.105 The synthetic methodology is straight forward but a few precautions are necessary. While the Sonogashira–Hagihara coupling between 113 and substituted alkyne works well in triethylamine or sterically hindered piperidine derivatives, in the presence of piperidine the nucleophilic attack of the base takes place to give mono- and dipiperidine substituted products. The cyclization of 114 to 115 can be best carried out using a strong but non-nucleophilic base such as DBU. Nucleophilic bases such as KOH or potassium butoxide hydrolyzed the imide function. Sterically bulky acetylene hinders the cyclization to get the corresponding coronenediimides.
 | ||
| Scheme 27 Synthesis of tetraalkyl-substituted coronenediimides. | ||
Recently, the alkyl-substituted hexabenzocoronene-based DLCs gained much attention due to their high charge carrier mobilities and consequently these materials have been used to construct efficient photovoltaic solar cells and field-effect transistors.106 About a year ago, Hill et al. reported107 an example of an asymmetrically substituted amphiphilic hexabenzocoronene derivative that self-assembles into remarkably defect-free large nanotubular objects.
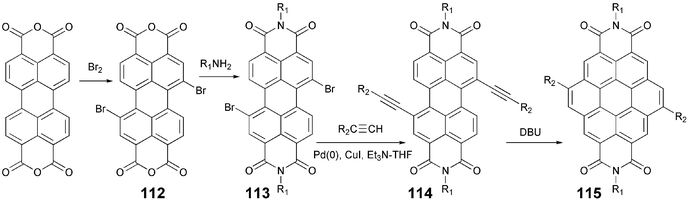
The symmetrical hexaalkyl-substituted HBCs 118 were most commonly prepared by cyclotrimerization of disubstituted diphenylacetylenes (tolanes) 116 under the catalytic action of Co2(CO)8 followed by the oxidative cyclodehydrogenation using anhydrous FeCl3 in high yields (Scheme 28). Alternatively, the sixfold bromination of hexaphenylbenzene 119 followed by the sixfold Suzuki coupling reaction can be used (Scheme 28) to prepare hexaphenyl HBC derivatives 121.108 The hexa(4-iodophenyl)-HBC prepared this way was used to synthesize a number of star-shaped HBC discotics.108 The synthesis of unsymmetrical and functionalized HBC derivatives 124 was achieved by [4 + 2]-cycloaddition of suitably substituted tolanes and 2,3,4,5-tetraarylcyclopenta-2,4-dien-1-ones 122 (Scheme 28).109 This versatile route offers the possibility of synthesizing a variety of novel unsymmetrical and functionalized HBC based DLCs.110
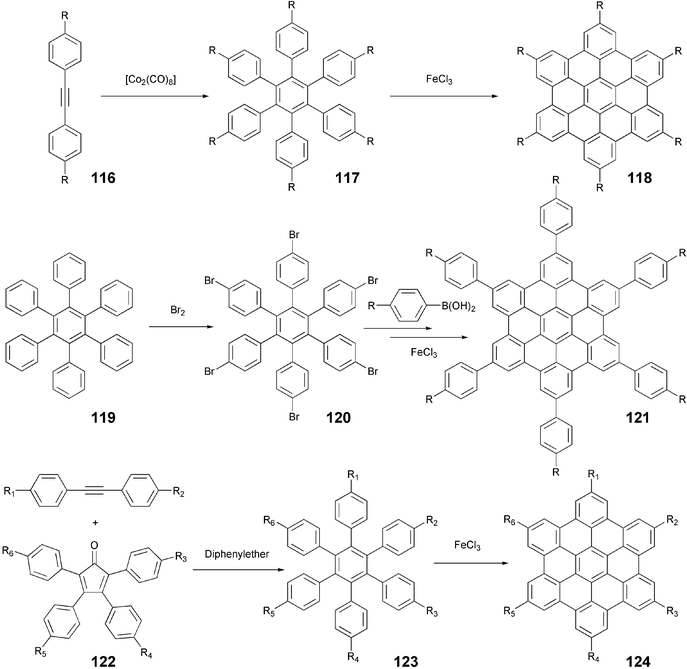 | ||
| Scheme 28 Synthetic routes to symmetrical and unsymmetrical HBC derivatives. | ||
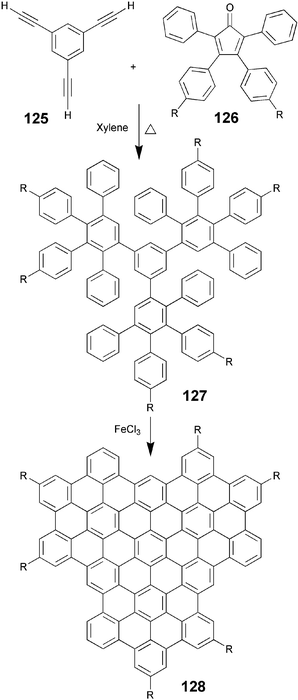
In continuation of the synthesis of large core DLCs, the Mullen group has recently reported the preparation of alkyl-substituted superphenalene-based DLCs.111 The compounds 128 with various normal and branched alkyl chains were prepared by a simple two-step synthesis. The Diels–Alder reaction of commercially available 1,3,5-triethynylbenzene 125 with excess cyclopentadienones 126 in o-xylene at elevated temperature yielded corresponding polyphenylene compounds 127. The oxidative cyclodehydrogenation of these polyphenylenes with FeCl3 in nitromethane afforded the desired superphenylenes (Scheme 29). It is remarkable that such a simple methodology can be utilized to generate a number highly complex-looking large-core molecules106 like 129.
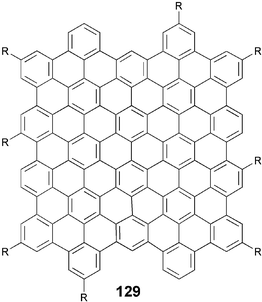
 | ||
| Scheme 29 Synthesis of superphenalene-based DLCs. | ||
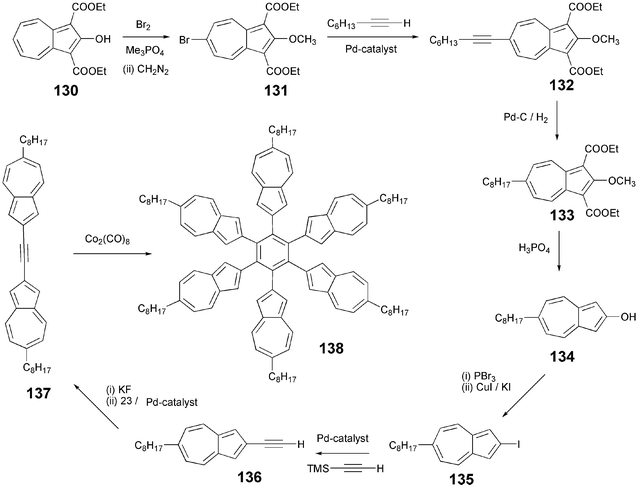
The hexakis(6-octyl-2-azulenyl)benzene 138 is another recent example of alkyl-substituted DLCs.112 Again, the cobalt-mediated cyclotrimerization of diazulene-substituted acetylene (6-octyl-2[(6-octyl-2-azulenyl)ethynyl]azulene) 137 produced the desired hexa(2-azulenyl)benzene derivative (Scheme 30). It is noteworthy that the cyclooligomerization of 137 using CpCo(CO)2 as catalyst did not yield the required product 138 but produced only a (η5-cyclopentadienyl)[tetra(2-azulenyl)cyclobutadiene]cobalt complex.
 | ||
| Scheme 30 Synthesis of hexakis(6-alkyl-2-azulenyl)benzene. | ||
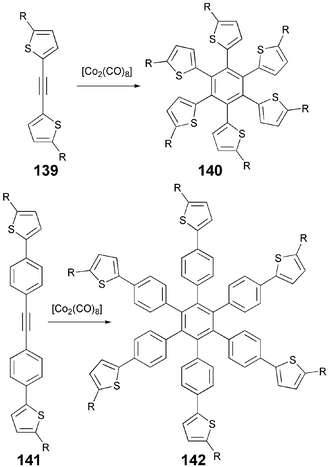
Similarly, Co2(CO)8-mediated cyclotrimerization of alkylthiophene-substituted tolanes 139 and 141 resulted in star-shaped benzene-based DLCs 140 and 142 (Scheme 31). The starting materials 139 and 141 can be easily prepared starting from alkyl-halo-thiophene using Sonogashira and Kumada-type organometallic reactions.113 The attachment of thiophene units only at 1,3,5-positions of a benzene molecule or at 2,4,6-positions of a triazine core does not yield liquid crystalline materials.114
 | ||
| Scheme 31 Alkylthiophene-substituted benzene-based star-shaped DLCs. | ||
Ionic discotic liquid crystals
Ionic DLCs have been known for a long time. The multipolar triphenylene amphiphile with six pyridinium head groups was reported by the Ringsdorf group to form lyotropic mesophases with water.115 Discotic ionic molecules containing 2,4,6-triarylpyrylium, 2,4,6-triarylpyridinium, crown ethers, 3,5-diaryl-1,2-dithiolium, and phthalocyanine moieties are also known to show mesomorphism. These materials have received renewed interest because of their potential applications as heat carriers in solar-thermal energy generators and as electrolytes for batteries and capacitors. Yoshio et al. reported one-dimensional ion transport in self-organized columnar ionic liquids.116 Despite the non-discotic shape of the trialkoxybenzene substituted imidazolium salt, it forms a columnar phase in which the ionic conductivities parallel to the columnar axis were found to be higher than those perpendicular to the axis.Recently, a number of columnar mesophase-forming triphenylene-tethered imidazolium ionic liquid crystals 143 were synthesized either by the quaternization of 1-methylimidazole with an ω-bromo-substituted triphenylene or by the quaternization of a triphenylene-substituted imidazole with methyl iodide.117,118 Similarly, a number of pyridinium salts tethered with hexaalkoxytriphenylene molecules were synthesized by the quaternization of pyridine with ω-bromo-substituted triphenylene derivatives.119
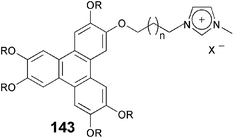
The tricycloquinazoline (TCQ) is an important core used to prepare a variety of DLCs.99 The hydrolysis of TCQ hexaacetate 144 generates the TCQ hexaanion 145 which on complexation with the double-tail surfactant didodecyldimethylammonium bromide 146 leads to highly organized nanostructures 147Scheme 32.120
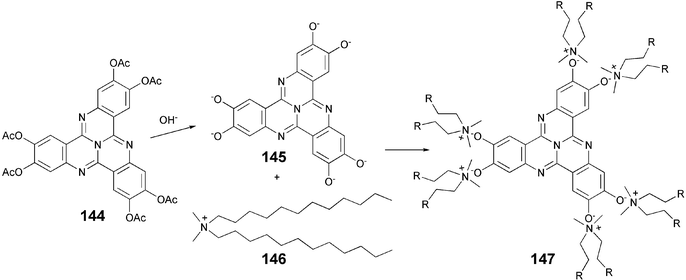 | ||
| Scheme 32 Preparation of ionic tricycloquinazoline derivatives. | ||
Miscellaneous discotic monomers
In principle, DLCs are composed of flexible aliphatic chains connected to central core where alkyl side chains act as a soft region and the core as a rigid region. The microphase segregation of polar aromatic part from non-polar aliphatic chains creates mesophases. However, recently it was demonstrated that molecules having an aromatic core as well as aromatic side chains can also display mesophases. The hexaazotriphenylene with six aromatic side chains such as compounds 148 and 149 were reported to be liquid crystalline.121 These molecules were prepared by condensation reaction of hexaaminobenzene with corresponding diarylketones which in turn were prepared by the Suzuki coupling between commercially available 4,4-dibromobenzil and corresponding arylboronic acids (Scheme 33)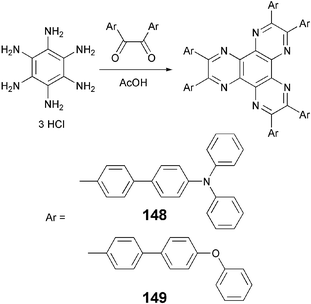 | ||
| Scheme 33 DLCs with aromatic core and aromatic side chains. | ||
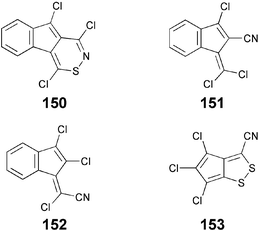
Excitingly a few examples of Colh phase forming DLCs such as 150–153 are also known which are completely devoid of side chains. The polarizable cyano groups or chlorine and sulfur atoms were considered as unusual soft parts in these molecules. These compounds were prepared by the treatment of various oximes with disulfur dichloride involving the initial abnormal Beckmann rearrangement of oximes to cyanides, followed by cyclization and/or exhaustive chlorination and dehydrochlorination.122
Alkaline condensation of 2,4,6-triazine with substituted benzaldehydes yields star-shaped stilbenoid-armed DLCs 154 (Scheme 34).123 These molecules display discotic nematic or columnar phases depending upon the substitution pattern around the core. Structurally similar benzene core based star-shaped compounds 155 could be obtained by Wittig–Horner reaction as shown in Scheme 34. The aldehydes used in the condensation reaction were prepared using a sequence of reactions starting from hydroquinone.
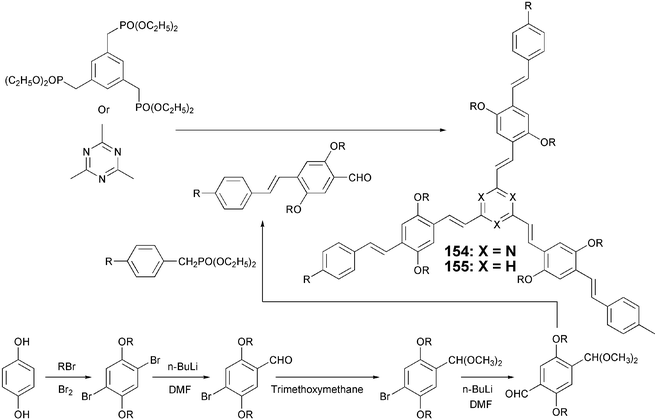 | ||
| Scheme 34 Synthesis of stilbenoid-armed star-shaped DLCs. | ||
Symmetrical porphyrin based DLCs are generally prepared from pyrroles. Recently a polysubstituted porphyrin 159 having olefinic and alkyl chains in the periphery was prepared from commercially available hemin-IX as shown in Scheme 35. Protoporphyrin IX dimethylester 156 can be oxidized to diformyl product 157 either directly or via bis-glycol. Reaction between Wittig reagent, methyl (triphenylphosphoranylidene)acetate, followed by saponification gives the tetra acid 158 which can be esterified with desired alkoxy-substituted phenol to yield asymmetrically β-polysubstituted porphyrin-based DLCs.124
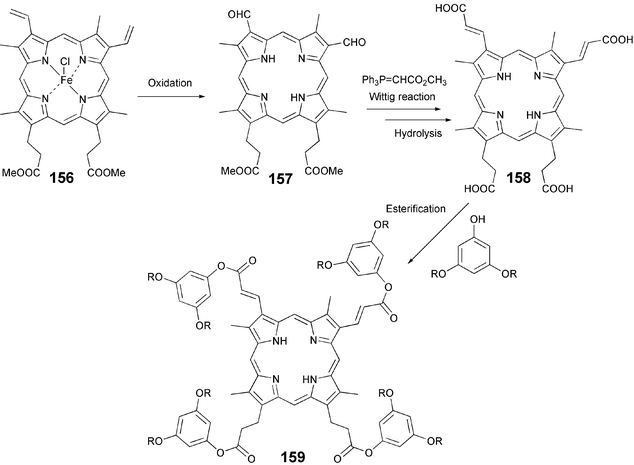 | ||
| Scheme 35 Synthesis of asymmetrically β-polysubstituted porphyrin-based DLCs. | ||
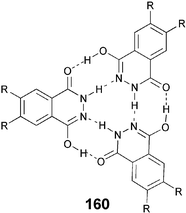
It is well accepted that columnar phases may occur via H-bonding between identical or different individual molecules. Suitably substituted 6,7-bis(alkoxy)2,3-dihydrophthalazine-1,4-diones 160 self-organize into columnar phases via triply H-bonded motif.125 Melamine-benzoic acid systems, trisamides, trisurea, hexamides and crowded arenas are some recent examples of this class of DLCs.126–129 It is normally a straightforward procedure to prepare individual molecules via classical organic reactions. The intermolecular H-bonded superstructure can be created by just mixing individual molecules in solvent followed by removal of solvent and annealing the residue in the isotropic state.
Discotic dimers, oligomers and polymers
A liquid crystal dimer, as its name implies, is composed of molecules containing two mesogenic groups either identically or differently linked via a flexible spacer or through a rigid spacer.130 Physical properties of liquid crystalline dimers are significantly different to that of conventional low molar mass liquid crystals because of restricted molecular motions. Dimers represent ideal model compounds for polymers or networks, due to their ease of purification and characterization and the possibility of freezing, in their mesophase, to a glassy state. Mono-functionalized discotic mesogens are the prerequisite to prepare dimers. The most commonly prepared dimers are the symmetrical twins in which two identical moieties are connected via a spacer. Often the spacer is a polymethylene O(CH2)nO chain but in some cases an ester or amide linkage in the middle of the spacer or at the terminal positions has also been used. These symmetrical dimers can be synthesized either in a single step by reacting the monohydroxy-discogen with 0.5 equivalents of the appropriate α,ω-dibromoalkane under classical etherification reaction conditions or in two steps. In the two-step procedure, the monohydroxy unit is first reacted with an excess of the appropriate α,ω-dibromoalkane to obtain the ω-brominated product that can be further reacted with monohydroxy-discogen to get the desired dimer. This two-step process is particularly important to prepare non-symmetrical dimers. The symmetrical discotic dimers with rigid diacetylene spacer were prepared by the dimerization of free monoacetylene-substituted monomers using Glaser coupling. Linking two discotic molecules via a short rigid spacer has a dramatic effect on the mesomorphic behaviour of these materials.131Discotic oligomers (trimers, tetramers, pentamers, hexamers and star-shaped molecules) are essentially two types, linear, in which three or more discotic units are connected in head to tail fashion with flexible spacers and star-shaped in which discotic fragments are tethered to a central polyfunctional core. Mono- and difunctionalized precursors are required to prepare linear molecules. On the other hand, star-shaped molecules can be easily prepared by attaching monofunctionalized discotics to a central polyfunctional core using classical reactions such as etherification, esterification and amidation.
While monofunctionalized discogens with polymerizable end groups such as acrylate, methaacrylate, olefin, on polymerization or connecting to a preformed polymer provide side-chain polymers, difunctionalized discotic molecules with polymerizable end groups generate main-chain polymers and a combination of the two furnishes discotic elastomers. A range of synthetic methods including polymer homologous reactions, free radical polymerizations, optical polymerizations, ring-opening metathesis polymerizations and condensation polymerizations have been used to prepare these polymers. Because of scarcity of space, structures of these materials have not been presented here. Interested readers may see a recent review article on triphenylene-based discotic liquid crystal dimers, oligomers and polymers.49
Applications of discotic liquid crystals
The negative birefringence films produced by polymerized nematic discotic LCs are the first successful commercial application of DLCs. These films are utilized to improve the viewing angle of classical twisted nematic liquid crystal displays (TNLCD).25 Recently it has been demonstrated that monomeric discotic nematic liquid crystals can be used instead of calamitic nematic liquid crystals to overcome the viewing angle problem of TNLCD. The LCD prepared using a hexalkynylbenzene based discotic nematic liquid crystal100 showed a wide and symmetrical viewing angle and no reversal of contrast ratio in any direction.132 However, because of the very high viscosity of the material, the device was extremely slow.The columnar phases of metallomesogens have the electrical properties of molecular semiconductors. On the other hand, the columnar phases of pure polynuclear aromatic mesogens like the TP compounds are insulators, but they can be made to conduct by doping. When an aligned sample of hexa-n-hexyloxy-TP or hexa-hexylthio-TP was doped with about 1 mol% of the electron acceptor AlCl3, the conductivity parallel to the columnar axis (σ∥) increases by several orders of magnitude.21,22 Further, σ∥ is more than 1000 times greater than σ⊥, the conductivity measured in the perpendicular direction. This is a striking proof of the quasi-one-dimensional nature of these conductors (Fig. 1).
Discotic liquid crystals based on various aromatic cores such as triphenylene, phthalocyanine, tricycloquinazoline, etc., have recently emerged as a new class of fast photoconducting materials. Photoinduced charge carrier mobility (hole mobility) of the order of 10−3 cm2 V−1 s−1 has been reported in the Colh phase of hexapentyloxytriphenylene. Charge carrier mobility up to an order of magnitude higher than Colh phase is observed in the Colp phase. In the three-dimensionally ordered helical phase of hexahexylthiotriphenylene, charge carrier mobility 10−1 cm2 V−1 s−1 has been reported.63 While HBC-based discotics exhibit carrier mobility 0.46 cm2 V−1 s−1, remarkably high charge carrier mobility up to 0.71 cm2 V−1 s−1, which is comparable to the mobility of amorphous silicon (1 cm2 V−1 s−1) has been observed in phthalocyanine-based DLCs.23
The generation of light by applying an electric field (electroluminescence) has been reported to be highly efficient for the conjugated organic polymers such as polyphenylenevinylene (PPV). High mobilities are an advantage for application in light emitting diodes (LEDs) in order to obtain high current densities and hence high light intensities. As mentioned above, the charge carrier mobilities in discotic liquid crystals are high and, therefore, they can be used in electroluminescence devices. Discotic liquid crystals have recently been used both as hole-transporting and electron transporting materials to construct organic LEDs.133 However, their luminescence efficiency was very low and more research efforts, particularly to find efficient electron transporting discotics are required.
The light-induced electron-hole generation, separation and migration is an important process for the conversion of light to electric or chemical energy (solar cells; photosynthesis), the latter being the basis of life on earth. Efficient solar cells based on single crystalline organic materials and doped organic polymers have been reported. However, single crystals are difficult and costly to process, a disadvantage which can be overcome with the design and synthesis of low-molar mass and polymeric discotic liquid crystals because their columnar structure resembles the aromatic stacking in single crystalline conductors. Gregg et al. studied photovoltaic effects in symmetrical cells filled with discotic liquid crystalline porphyrin complexes.134 Photovoltaic effect comparable with that of some of the better organic solar cells was found. However, the authors did not study the charge mobility in the mesophase itself but utilized the liquid crystalline properties to fill the cell and to promote macroscopic order, which, on cooling, provided polycrystalline films. Very recently, Schmidt-Mende et al. utilized discotic liquid crystalline hexabenzocoronene as the hole-transporting layer and a perylene dye to construct a p/n-type photovoltaic solar cell.135 The device made by Schmidt-Mende et al. exhibits a peak quantum efficiency of 34% and power efficiency of up to ∼2%.
Summary and future
The field of DLCs is, relatively speaking, a young field. Even so, many significant advances have been achieved in a short span of time. More than 3000 DLCs, from monomers to polymers, have been synthesized and extensively studied for various physical properties. The field is experiencing an exponential growth and commanding its own dedicated international conferences. In spite of their scarcity, discotic nematic liquid crystals have actually found commercial application in the multibillion dollar liquid crystal display industry. Discotic molecules are of interest for their intriguing supramolecular architectures. Their strong π–π interactions within a column lead to high electronic mobilities, a property that is essential in the development of eco-friendly organic photovoltaic solar cells or transistors.In this review, by means of representative examples, we have analyzed different classes of DLCs. For want of space, many discotic monomers, oligomers, polymers, discotic dendrimers, hydrogen-bonded DLCs and discotic metallomesogens could not be covered. However, in most cases, the chemistry follows more or less same guidelines described in above sections. A number of different classical chemical reactions may be used to synthesize innumerable DLCs. These include a variety of condensation, cyclization, esterification, etherification, amidation, aromatic substitution, and others. With the advancement of synthetic organic chemistry, it is often possible to tailor-make a variety of DLCs with desired physical properties. It is now possible to link various subunits of the molecule together in controlled fashion to produce core systems with almost any substitution pattern. Moreover, the possibility to tune the properties by simple mixing with other liquid crystalline or non-liquid crystalline materials and metal nanoparticles seems very attractive.136–138
There are several significant challenges remaining for the commercial use of DLCs. One of the most important issues is their large-scale synthesis. Most of the discotics prepared so far are only in milligram or gram scale. Translating synthetic methodologies on a large scale is often not straightforward and needs significant development work. Impurities are a big problem in the LC field as they render a large effect on the mesomorphic properties and on the device performance. Therefore, liquid crystalline materials must be purified with utmost care. Distillation under reduced pressure, which is an efficient industrial method to purify low molar mass materials, can not be used to purify discotics due to their high melting and boiling points. Chromatographic methods are generally not commercially viable and therefore, other methods such as zone refining, fractional crystallization, etc., should be explored. Probably more discotic materials with lower melting and isotropic temperatures and preferably forming stable glassy state must be designed.
Although the amount of LCs in general and DLCs in particular, produced commercially is negligible compared to the huge amount of other industrial chemicals polluting our environment yet liquid crystal chemists should take responsibility to find “greener” methods for their production. Synthesis of liquid crystalline materials in ionic liquids or using microwave dielectric heating could be a good beginning. Chemists, physicists, materials scientists and engineers must work together to understand the many facets of the research problems that are being uncovered as the field grows. Therefore, if the question is: what is the future of these intriguing functional supramolecular systems? The simple answer is: “very bright”.
Acknowledgements
I would like to thank Professor B. K. Sadashiva and Professor N. V. Madhusudana for helpful discussions.References
- J.-M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995 Search PubMed.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed. Engl., 1988, 27, 113 CrossRef.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS.
- P. G. de Gennes and J. Prost, The Physics of Liquid Crystals, Clarendon Press, Oxford, 1994 Search PubMed.
- T. Kato, Science, 2002, 295, 2414 CrossRef CAS.
- J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13, 2661 RSC.
- G. H. Brown and J. J. Wolken, Liquid Crystals and Biological Structures, Academic Press, New York, 1979 Search PubMed.
- G. T. Stewart, Liq. Cryst., 2003, 30, 541 CrossRef CAS.
- S. J. Watson, H. F. Gleeson, A. D'Emanuele, S. Serak and V. Grozhik, Mol. Cryst. Liq. Cryst., 1999, 331, 2235 CAS.
- V. K. Gupta, J. J. Skaife, T. B. Dubrovsky and N. L. Abbott, Science, 1998, 279, 2077 CrossRef CAS.
- J. M. Brake, M. K. Daschner, Y. Y. Luk and N. L. Abbott, Science, 2003, 302, 2094 CrossRef CAS.
- S. V. Shiyanovskii, T. Schneider, I. I. Smalyukh, T. Ishikawa, G. D. Niehaus, K. Doane, C. J. Woolverton and O. D. Lavrentovich, Phys. Rev. Lett. E, 2005, 71, 020702 CAS.
- B. Bahadur (ed.), Liquid Crystals-Application and Uses, Vol I–III, World Scientific, Singapore, 1990 Search PubMed.
- F. Reinitzer, Monatsh. Chem., 1888, 9, 421; for English translation see Liq. Cryst., 1989, 5, 7 Search PubMed.
- W. Heintz, J. Prakt. Chem., 1855, 66, 1 Search PubMed.
- T. Niori, T. Sekine, J. Watanabe, T. Furukawa and H. Takezoe, J. Mater. Chem., 1996, 6, 1231 RSC.
- O. H. Schonherr, J. H. Wendorf, H. Ringsdorf and P. Tschirner, Makromol. Chem., Rapid Commun., 1986, 7, 791 CrossRef.
- For recent reviews on DLCs see, e.g., B. Donnio, D. Guillon, R. Deschenaux and D. W. Bruce, Compr. Coord. Chem., 2003, 7, 357 Search PubMed; R. J. Bushby and O. R. Lozman, Curr. Opin. Colloid Interface Sci., 2002, 7, 343 Search PubMed; C. Tschierske, Annu. Rep. Prog. Chem., Sect. C, 2001, 97, 191 Search PubMed; D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill (ed.), Handbook of Liquid Crystals, Wiley-VCH, Weinheim, 1998, Vol. 2B RSC; S. Chandrasekhar and S. Kumar, Science Spectra, 1997, 66 RSC.
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Pramana, 1977, 9, 471 Search PubMed.
- J. D. Brooks and G. H. Taylor, Nature, 1965, 206, 697 CAS.
- V. S. K. Balagurusamy, S. K. Prasad, S. Chandrasekhar, S. Kumar, M. Manickam and C. V. Yelamaggad, Pramana, 1999, 53, 3 Search PubMed.
- N. Boden, R. J. Bushby, A. N. Cammidge, J. Clements and R. Luo, Mol. Cryst. Liq. Cryst., 1995, 261, 251 CrossRef CAS.
- K. Ohta, K. Hatsusaka, M. Sugibayashi, M. Ariyoshi, K. Ban, F. Maeda, R. Naito, K. Nishizawa, A. M. van de Craats and J. M. Warman, Mol. Cryst. Liq. Cryst., 2003, 397, 25.
- N. Boden and B. Movaghar, in Handbook of Liquid Crystals, ed. D. Demus, J. Goodby, G.W. Gray, H.-W. Spiess and V. Vill, Wiley-VCH, Weinheim, 1998, Vol. 2B, Chapter IX; See also Reference 45 Search PubMed.
- K. Kawata, Chem. Rec., 2002, 2, 59 Search PubMed.
- R. Pratibha and N. V. Madhusudana, Mol. Cryst. Liq. Cryst. Lett., 1985, 1, 111 Search PubMed.
- M. Baron, Pure Appl. Chem., 2001, 73, 845 CrossRef CAS.
- C. V. Yelamaggad, V. Prasad, M. Manickam and S. Kumar, Mol. Cryst. Liq. Cryst., 1998, 325, 33 CrossRef CAS.
- G. Heppke, D. Kruerke, C. Lohning, D. Lotzsch, D. Moro, M. Muller and H. Sawade, J. Mater. Chem., 2000, 10, 2657 RSC.
- K. Praefcke, D. Singer, B. Kohne, M. Ebert, A. Liebmann and J. H. Wendorff, Liq. Cryst., 1991, 10, 147 CrossRef CAS.
- B. Glusen, W. Heitz, A. Kettner and J. H. Wendorff, Liq. Cryst., 1996, 20, 627 CrossRef.
- E. Fontes, P. A. Heiney and W. H. De Jeu, Phys. Rev. Lett., 1988, 61, 1202 CrossRef CAS.
- A. M. Giroud-Godquin and J. Billard, Mol. Cryst. Liq. Cryst., 1981, 66, 147 CrossRef.
- K. Hatsusaka, K. Ohta, I. Yamamoto and H. Shirai, J. Mater. Chem., 2001, 11, 423 RSC.
- I. Tabushi, K. Yamamura and Y. Okada, J. Org. Chem., 1987, 52, 2502 CrossRef CAS.
- D. M. Collard and C. P. Lillya, J. Am. Chem. Soc., 1989, 111, 1829 CrossRef CAS.
- B. Kohne and K. Praefcke, Angew. Chem., Int. Ed. Engl., 1984, 23, 82 CrossRef.
- A. Pegenau, T. Hegmann, C. Tschierske and S. Diele, Chem. Eur. J., 1999, 5, 1643 CrossRef CAS.
- A. Pegenau, P. Goring, S. Diele and C. Tschierske, Liquid Crystals: Chemistry and Structure, ed. M. Tykarska, R. Dabroski and J. Zielinski, Proc. SPIE-Int. Soc. Opt. Eng., 1998, vol. 3319, p. 70 Search PubMed.
- J. Billard, J. C. Dubois, C. Vaucher and A. M. Levelut, Mol. Cryst. Liq. Cryst., 1981, 66, 115 CrossRef.
- C. Carfugna, P. Iannelli, A. Roviello and A. Sirigu, Liq. Cryst., 1987, 2, 611 CrossRef CAS.
- S. Kumar, J. J. Naidu and S. K. Varshney, Liq. Cryst., 2003, 30, 319 CrossRef CAS.
- J. Billard, Z. Luz, R. Poupko and H. Zimmermann, Liq. Cryst., 1994, 16, 333 CrossRef CAS.
- H. Zimmermann, J. Billard, H. Gutman, E. J. Wachtel, R. Poupko and Z. Luz, Liq. Cryst., 1992, 12, 245 CrossRef CAS.
- E. Dalcanale, A. Du. Vosel, A. M. Levelut and J. Malthete, Liq. Cryst., 1991, 10, 185 CrossRef CAS.
- J. Billard, J. C. Dubois, N. H. Tinh and A. Zann, Nouv. J. Chim., 1978, 2, 535 Search PubMed.
- A. N. Cammidge and R. J. Bushby, Handbook of Liquid Crystals, Vol. 2B, ed. D. Demus, J. Goodby, G.W. Gray, H.-W. Spiess and V. Vill, Wiley-VCH, Weinheim, Chap. VII, 1998 Search PubMed.
- S. Kumar, Liq. Cryst., 2004, 31, 1037 CrossRef CAS.
- S. Kumar, Liq. Cryst., 2005, 32, 1089 CAS.
- G. Lessene and K. S. Feldman, in Modern Arene Chemistry, ed. D. Astruc, Wiley-VCH, Weinheim, 2002 Search PubMed.
- M. L. Rahman, C. Tschierske, M. Yusoff and S. Silong, Tetrahedron Lett., 2005, 46, 2303 CrossRef CAS; Y. Shimizu, A. Kurobe, H. Monobe, N. Terasawa, K. Kiyohara and K. Uchida, Chem. Commun., 2003, 1676 RSC.
- P. H. J. Kouwer, J. Pourzand and G. H. Mehl, Chem. Commun., 2004, 66 RSC.
- M. Manickam, G. Cooke, S. Kumar, P. R. Ashton, J. A. Preece and N. Spencer, Mol. Cryst. Liq. Cryst., 2003, 397, 99; M. Manickam, M. Belloni, S. Kumar, S. K. Varshney, D. S. Shankar Rao, P. Ashton, J. A. Preece and N. Spencer, J. Mater. Chem., 2001, 11, 2790 RSC; M. Manickam, S. Kumar, J. A. Preece and N. Spencer, Liq. Cryst., 2000, 27, 703 CrossRef CAS.
- G. Cooke, A. Radhi, N. Boden, R. J. Bushby, Z. Lu, S. Brown and S. L. Heath, Tetrahedron, 2000, 56, 3385 CrossRef CAS.
- M. Manickam, A. Smith, M. Belloni, E. J. Shelley, P. Ashton, N. Spencer and J. A. Preece, Liq. Cryst., 2002, 29, 497 CrossRef CAS.
- J. Barbera, A. C. Garces, N. Jayaraman, A. Omenat, J. L. Serrano and J. F. Stoddart, Adv. Mater., 2002, 13, 175.
- R. J. Bushby, Q. Liu, O. R. Lozman, Z. Lu and S. R. McLaren, Mol. Cryst. Liq. Cryst., 2004, 411, 293 CrossRef.
- R. J. Bushby, N. Boden, G. A. Kilner, O. R. Lozman, Z. Lu, Q. Liu and M. A. Thornton-Pett, J. Mater. Chem., 2003, 13, 470 RSC.
- A. N. Cammidge and H. Gopee, Mol. Cryst. Liq. Cryst., 2003, 397, 117.
- M. T. Allen, S. Diele, K. D. M. Harris, T. Hegmann, B. M. Kariuki, D. Lose, J. A. Preece and C. Tschierske, J. Mater. Chem., 2001, 11, 302 RSC; M. T. Allen, K. D. M. Harris, B. M. Kariuki, N. Kumari, J. A. Preece, S. Diele, D. Lose, T. Hegmann and C. Tschierske, Liq. Cryst., 2000, 27, 689 CrossRef CAS.
- S. Kumar, M. Manickam, S. K. Varshney, D. S. S. Rao and S. K. Prasad, J. Mater. Chem., 2000, 10, 2483 RSC.
- S. J. Cross, J. W. Goodby, A. W. Hall, M. Hird, S. M. Kelly, K. J. Toyne and C. Wu, Liq. Cryst., 1998, 25, 1 CrossRef CAS.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Häussling, K. Siemensmeyer, K. H. Etzbach, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141 CrossRef CAS; A. M. van de Craats, J. M. Warman, M. P. deHaas, D. Adam, J. Simmerer, D. Haarer and P. Schuhmacher, Adv. Mater., 1996, 8, 823 CrossRef CAS.
- H. Bock and W. Helfrich, Liq. Cryst., 1992, 12, 697 CrossRef CAS.
- P. Henderson, H. Ringsdorf and P. Schuhmacher, Liq. Cryst., 1995, 18, 191 CrossRef CAS.
- S. Kumar and S. K. Varshney, Synthesis, 2001, 305 CrossRef CAS.
- S. Kumar, J. J. Naidu and D. S. S. Rao, J. Mater. Chem., 2002, 12, 1335 RSC.
- J. L. Sessler, W. Callaway, S. P. Dudek, R. W. Date, V. Lynch and D. W. Bruce, Chem. Commun., 2003, 2422 RSC.
- A. N. Cammidge and H. Gopee, Chem. Commun., 2002, 966 RSC.
- C. W. Ong, S. Liao, T. H. Chang and H. Hsu, Tetrahedron Lett., 2003, 44, 1477 CrossRef CAS; M. Lehmann, G. Kestemont, R. G. Aspe, C. Buess-Herman, M. H. J. Koch, M. G. Debije, J. Piris, M. P. de Haas, J. M. Warman, M. D. Watson, V. Lemaur, J. Cornil, Y. Geerts, R. Gearba and D. A. Ivanov, Chem. Eur. J., 2005, 11, 3349 CrossRef CAS.
- E. J. Foster, J. Babuin, N. Nguyen and V. E. Williams, Chem. Commun., 2004, 2052 RSC.
- J. Hu, D. Zhang, S. Jin, S. Z. D. Cheng and F. W. Harris, Chem. Mater., 2004, 16, 4912 CrossRef CAS.
- S. Kumar and M. Manickam, Mol. Cryst. Liq. Cryst., 2000, 338, 175 CrossRef CAS.
- S. Kumar and S. K. Varshney, Mol. Cryst. Liq. Cryst., 2002, 378, 59 CrossRef CAS.
- A. Schultz, S. Laschat, S. Diele and M. Nimtz, Eur. J. Org. Chem., 2003, 2829 CrossRef CAS.
- L. Vyklicky, S. H. Eichhorn and T. J. Katz, Chem. Mater., 2003, 15, 3594 CrossRef CAS; K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899 CrossRef CAS.
- J. Barbera, R. Gimenez and J. L. Serrano, Chem. Mater., 2000, 12, 481 CrossRef CAS.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179 CrossRef CAS.
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS.
- W. K. Lee, P. A. Heiney, J. P. McCauley and A. B. Smith, III, Mol. Cryst. Liq. Cryst., 1991, 198, 273 CrossRef CAS.
- S. Marguet, D. Markovitsi, P. Millie, H. Sigal and S. Kumar, J. Phys. Chem. B, 1998, 102, 4697 CrossRef CAS.
- S. Kumar and J. J. Naidu, Liq. Cryst., 2002, 29, 899 CrossRef; J. A. Rego, S. Kumar, I. J. Dmochowski and H. Ringsdorf, Chem. Commun., 1996, 1031 RSC.
- A. N. Cammidge and H. Gopee, J. Mater. Chem., 2001, 11, 2773 RSC.
- S. Inoue, S. Nishiguchi, S. Murakami, Y. Aso, T. Otsubo, V. Vill, A. Mori and S. Ujiie, J. Chem. Res., 1999, 596 RSC.
- B. A. Minch, W. Xia, C. L. Donley, R. M. Hernandez, C. Carter, M. D. Carducci, A. Dawson, D. F. O'Brien and N. R. Armstrong, Chem. Mater., 2005, 17, 1618 CrossRef CAS.
- S. H. Kang, Y.-S. Kang, W.-C. Zin, G. Olbrechts, K. Wostyn, K. Clays, A. Persoons and K. Kim, Chem. Commun., 1999, 1661 RSC.
- B. Kohne and K. Praefcke, Chimia, 1987, 41, 196 CAS.
- J. Wu, M. D. Watson, L. Zhang, Z. Wang and K. Mullen, J. Am. Chem. Soc., 2004, 126, 177 CrossRef CAS.
- P. H. J. Kouwer, G. H. Mehl and S. J. Picken, Mol. Cryst. Liq. Cryst., 2004, 411, 387 CrossRef.
- P. H. J. Kouwer, W. F. Jager, W. J. Mijs and S. J. Picken, J. Mater. Chem., 2003, 13, 458 RSC.
- R. W. Date and D. W. Bruce, J. Am. Chem. Soc., 2003, 125, 9012 CrossRef CAS.
- S. Ito, H. Inabe, N. Morita, K. Ohta, T. Kitamura and K. Imafuku, J. Am. Chem. Soc., 2003, 125, 1669 CrossRef CAS.
- L. R. Milgrom, G. Yahioglu, D. W. Bruce, S. Morrone, F. Z. Henari and W. J. Blau, Adv. Mater., 1997, 9, 313 CrossRef CAS.
- J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 2655 CrossRef CAS.
- H. A. Staab and K. Neunhoeffer, Synthesis, 1974, 424 CrossRef CAS.
- S. Hoger, V. Enkelmann, K. Bonrad and C. Tschierske, Angew. Chem., Int. Ed., 2000, 39, 2268 CAS.
- S. Hoger, X. H. Cheng, A. Ramminger, V. Enkelmann, A. Rapp, M. Mondeshki and I. Schnell, Angew. Chem., Int. Ed., 2005, 44, 2801 CrossRef.
- D. M. Collard and C. P. Lillya, J. Am. Chem. Soc., 1991, 113, 8577 CrossRef CAS.
- S. Kumar, D. S. Shankar Rao and S. Krishna Prasad, J. Mater. Chem., 1999, 9, 2751 RSC.
- S. Kumar and S. K. Varshney, Angew. Chem., Int. Ed., 2000, 39, 3140 CrossRef CAS; S. Kumar, S. K. Varshney and D. Chauhan, Mol. Cryst. Liq. Cryst., 2003, 396, 241 CrossRef CAS.
- A. N. Cammidge and H. Gopee, Mol. Cryst. Liq. Cryst., 2003, 397, 177.
- E. Keinan, S. Kumar, R. Moshenberg, R. Ghirlando and E. J. Wachtel, Adv. Mater., 1991, 3, 251 CAS.
- T. Sugino, J. Santiago, Y. Shimizu, B. Heinrich and D. Guillon, Liq. Cryst., 2004, 31, 101 CAS.
- M. Kimura, Y. Saito, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, J. Am. Chem. Soc., 2002, 124, 5274 CrossRef CAS.
- U. Rohr, C. Kohl, K. Mullen, A. M. van de Craats and J. Warman, J. Mater. Chem., 2001, 11, 1789 RSC.
- C. D. Simpson, J. Wu, M. D. Watson and K. Mullen, J. Mater. Chem., 2004, 14, 494 RSC.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. ishii and T. Aida, Science, 2004, 304, 1481 CrossRef CAS.
- J. Wu, M. D. Watson and K. Mullen, Angew. Chem., Int. Ed., 2003, 42, 5329 CrossRef CAS.
- A. Fechtenkotter, N. Tchebotareva, M. Watson and K. Mullen, Tetrahedron, 2001, 57, 3769 CrossRef CAS.
- S. Ito, M. Wehmeier, J. D. Brand, C. Kubel, R. Epsch, J. P. Rabe and K. Mullen, Chem.–Eur. J., 2000, 6, 4327 CrossRef CAS.
- Z. Tomovic, M. D. Watson and K. Mullen, Angew. Chem., Int. Ed., 2004, 43, 755 CrossRef CAS.
- S. Ito, M. Ando, A. Nomura, N. Morita, C. Kabuto, H. Mukai, K. Ohta, J. Kawakami, A. Yoshizawa and A. Tajiri, J. Org. Chem., 2005, 70, 3939 CrossRef CAS.
- Y. Geng, A. Fechtenkotter and K. Mullen, J. Mater. Chem., 2001, 11, 1634 RSC.
- F. Cherioux, L. Gaurent and P. Audebert, Chem. Commun., 1998, 2225 RSC.
- R. Keller-Griffith, H. Ringsdorf and A. Vierengel, Colloid Polym. Sci., 1986, 264, 924 CAS.
- M. Yoshio, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2004, 126, 994 CrossRef CAS.
- S. Kumar and S. K. Pal, Tetrahedron Lett., 2005, 46, 2607 CrossRef CAS.
- J. Motoyanagi, T. Fukushima and T. Aida, Chem. Commun., 2005, 101 RSC.
- S. Kumar and S. K. Pal, Tetrahedron Lett., 2005, 46, 4127 CrossRef CAS.
- J. Kadam, C. F. J. Faul and U. Scherf, Chem. Mater., 2004, 16, 3867 CrossRef CAS.
- T. Ishi-i, T. Hirayama, K. Murakami, H. Tashiro, T. Thiemann, K. Kubo, A. Mori, S. Yamasaki, T. Akao, A. Tsuboyama, T. Mukaide, K. Ueno and S. Mataka, Langmuir, 2005, 21, 1261 CrossRef CAS.
- J. Barbera, O. A. Rakitin, M. Blanca Ros and T. Torroba, Angew. Chem., Int. Ed., 1998, 37, 296 CrossRef CAS.
- H. Meier, M. Lehmann, H. C. Holst and D. Schwoppe, Tetrahedron, 2004, 60, 6881 CrossRef CAS.
- M. Castella, F. Lopez-Calahorra, D. Velasco and H. Finkelmann, Chem. Commun., 2002, 2348 RSC.
- M. Suarez, J.-M. Lehn, S. C. Zimmerman, A. Skoulios and B. Heinrich, J. Am. Chem. Soc., 1998, 120, 9526 CrossRef CAS.
- J. Barbera, L. Puig, J. L. Serrano and T. Sierra, Chem. Mater., 2004, 16, 3308 CrossRef CAS.
- M. L. Bushey, T.-Q. Nguyen and C. Nuckolls, J. Am. Chem. Soc., 2003, 125, 8264 CrossRef.
- R. I. Gearba, M. Lehmann, J. Levin, D. A. Ivanov, M. H. J. Koch, J. Barbera, M. G. Debije, J. Piris and Y. H. Geerts, Adv. Mater., 2003, 15, 1614 CrossRef CAS.
- J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759 CrossRef CAS.
- C. T. Imrie and P. A. Henderson, Curr. Opin. Colloid Interface Sci., 2002, 7, 298 Search PubMed.
- S. Kumar and S. K. Varshney, Org. Lett., 2002, 4, 157 CrossRef CAS.
- G. G. Nair, D. S. S. Rao, S. K. Prasad, S. Chandrasekhar and S. Kumar, Mol. Cryst. Liq. Cryst., 2003, 397, 245.
- I. Seguy, P. Destruel and H. Bock, Synth. Met., 2000, 111–112, 15 CrossRef CAS and references therein.
- B. A. Gregg, M. A. Fox and A. J. Bard, J. Phys. Chem., 1990, 94, 1586 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkotter, K. Mullen, E. Moons, R. H. Friend and J. D. Mackenzie, Science, 2001, 293, 1119 CrossRef CAS.
- N. Boden, R. J. Bushby, G. Cooke, O. R. Lozman and Z. Lu, J. Am. Chem. Soc., 2001, 123, 7915 CrossRef CAS.
- L. Y. Park, D. G. Hamilton, E. A. McGehee and K. A. McMenimen, J. Am. Chem. Soc., 2003, 125, 10586 CrossRef CAS.
- S. Kumar and V. Lakshminarayanan, Chem. Commun., 2004, 1600 RSC.
| This journal is © The Royal Society of Chemistry 2006 |