Imaging the dynamics of gas phase reactions
Michael N. R.
Ashfold
*a,
N. Hendrik
Nahler
a,
Andrew J.
Orr-Ewing
a,
Olivier P. J.
Vieuxmaire
a,
Rachel L.
Toomes
b,
Theofanis N.
Kitsopoulos
b,
Ivan Anton
Garcia
c,
Dmitri A.
Chestakov
c,
Shiou-Min
Wu
c and
David H.
Parker
c
aSchool of Chemistry, University of Bristol, Bristol, UK BS8 1TS. E-mail: mike.ashfold@bris.ac.uk; Fax: +44 117 9250612; Tel: +44 117 9288312/3
bDepartment of Chemistry, University of Crete and Institute of Electronic Structure and Laser, Foundation of Research and Technology-Hellas, 711 10, Heraklion, Crete, Greece
cUniversity of Nijmegen, Department of Molecular and Laser Physics, Toernooiveld 1, 6525 ED, Nijmegen, The Netherlands
First published on 18th October 2005
Abstract
Ion imaging methods are making ever greater impact on studies of gas phase molecular reaction dynamics. This article traces the evolution of the technique, highlights some of the more important breakthroughs with regards to improving image resolution and in image processing and analysis methods, and then proceeds to illustrate some of the many applications to which the technique is now being applied—most notably in studies of molecular photodissociation and of bimolecular reaction dynamics.
1. Introduction
The huge technological progress seen in the past quarter century has enabled spectacular advances in our ability to probe the detailed dynamics of gas phase reactions. Fifty years ago, flash photolysis was still in its infancy.1 Norrish and co-workers were able to record the spectra of vibrationally excited O2 products from the reactions of ground state oxygen atoms with NO2 and ClO2.2 Shortly thereafter, Cashion and Polanyi were reporting first observations of infrared (IR) chemiluminescence from vibrationally excited products of bimolecular reactions.3 Studies of the dynamics of molecular photodissociation processes started to become feasible. Pulsed broadband ultraviolet (UV) excitation of a static sample of, for example, ClNO was shown to yield highly vibrationally excited NO products.4 By the late 1970’s, tuneable dye lasers had found their way into the laboratories of many of the leading players in the reaction dynamics community, and the implementation of techniques like laser induced fluorescence (LIF),5,6 coherent anti-Stokes Raman spectroscopy (CARS)7 and resonance enhanced multiphoton ionisation (REMPI)8 was beginning to allow detailed investigation of the internal (e.g. rotation, vibration, spin–orbit, lambda doublet, etc.) quantum state population distributions within a wide range of products from both unimolecular (i.e. photofragmentation) and bimolecular reactions.First measurements of product velocity distributions also appeared around this time, following the introduction of photofragment translational spectroscopy (PTS) methods.9,10 These experiments involved pulsed photolysis of a chosen target molecule, seeded in a molecular beam, and measurement of the times-of-flight (TOFs), and hence the velocity distributions, of the resulting photofragments. Product selectivity was achieved by employing electron impact ionisation and mass spectrometric detection—a method that is both sensitive and universal, though not quantum state specific. Fragments arising in a molecular photodissociation may be formed with an anisotropic distribution of recoil velocities. PTS experiments allow measurement of product angular distributions also, through use of polarized photolysis laser radiation and/or rotating sources or detectors.11 These concepts have been extended to bimolecular collision processes, using two crossed molecular beams (at least one of which is ‘chopped’ to give the zero of time necessary for any TOF experiment).
Such experiments have yielded a wealth of dynamical information. For example, such studies can reveal whether a given reaction yields translationally or internally excited products, the nature of the internal vibrational and rotational excitation (if present), and whether or not the product scattering is anisotropic. Since the form of the product energy disposal reflects the details of the forces acting between the embryonic fragments at their moment of creation, such measurements provide insight into the nature of the electronic states—and the potential energy surfaces (PESs)—controlling the fragmentation (or collision) process. Consider the case of photodissociations, for example. Products formed with fast, anisotropic recoil velocity distributions are generally viewed as arising in one of two ways. In the first of these, the molecules of interest are excited to, and dissociate on, a repulsive PES. Alternatively, it may be that the photo-excited molecules have to evolve, non-radiatively, from the initially prepared state to another excited PES, upon which fragmentation then occurs—either directly, or via passage over an exit channel barrier. The radiationless transition (predissociation) involved in the latter mechanism may involve a time delay comparable to (or longer than) the rotational period of the photo-excited parent molecule. Such rotation of the molecular frame in the period between excitation and fragmentation is one mechanism for degrading fragment recoil anisotropy. Conversely, there are many other photofragmentations that yield essentially isotropic product recoil velocity distributions, peaking at velocities close to zero. Energy and angular momentum conservation dictates that most of the excess energy (i.e. the energy supplied by the photon over and above that required to break the bond) is partitioned into internal (vibrational) excitation of the one or more molecular products. Such ‘statistical’ energy disposal tends to arise in cases where the photo-excited molecule evolves via a radiationless transition (normally internal conversion (IC)) to the ground electronic state, redistribution of energy among the various vibrational modes in the highly internally excited ground state molecule that results, and subsequent unimolecular decay.12–14
An obvious next challenge was to measure velocity distributions of products formed in specified internal quantum states. Optical spectroscopy offers the clearest route to product state selection; provided the product of interest, P, has a well resolved and well characterised LIF or REMPI excitation spectrum, population in any chosen quantum state (q) of P can be monitored simply by tuning to the appropriate probe laser wavelength. Let us again focus attention on the (simpler) case of a pulsed photolysis process. The resulting P(q) fragments are created in a localised volume, at a well-defined instant in time, and begin to expand in a series of concentric spheres the radii of which (at any particular instant in time) will be determined by the recoil velocities. These are known as Newton spheres. Each pair of product quantum states will expand with its own associated Newton sphere. Several methods have been developed to allow determination of the (one-dimensional (1-D)) speed distribution of quantum state selected products. For example, the P(q) products can be tagged by REMPI, and the resulting ions extracted into a TOF mass spectrometer (MS). In an appropriately designed experiment, the temporal form of the P+ peak in the TOF spectrum will be sensitive to (i) the extraction voltages employed, (ii) any time delay between creation (photolysis) and REMPI tagging and/or switching on the extraction voltages, and (iii) the original speed distribution of the P(q) fragments along the TOF axis. Simulation allows determination of (iii)—one of the main objectives of such experiments.15 Similar arguments apply in the case of photo-initiated bimolecular reactions, wherein a photolysis laser is used to create fragments (e.g. Cl atoms). These serve as one of the reactants in a subsequent bimolecular reaction, one or more of the products of which (e.g. HCl) are then probed, state-specifically, by REMPI–TOF methods.16 Rydberg tagging methods offer higher TOF resolution, but sample only that small fraction of P products that happen to be formed with recoil velocities falling within the solid angle subtended by the detector; such methods have thus far been limited to H17–21 and O22,23 atom products.
Doppler spectroscopy offers an alternative route to determining 1-D speed distributions of state selected reaction products. This technique relies on the fact that the precise frequency at which a P(q) product of interest will absorb (in an LIF or REMPI detection scheme) will depend on the projection of its recoil velocity along the probe laser propagation axis. The lineshape of the probe transition will thus be sensitive to this 1-D speed distribution (i.e. through the distribution of Doppler shifts). How well this distribution can be determined depends on a number of factors, including:
(i) The fragment speed: fast moving fragments can be investigated with greater confidence since the Doppler shift scales linearly with the lab. frame velocity, v. Self-evidently, the intrinsic kinematics of a system plays a crucial rôle. Given that the Doppler shift is proportional to v, and that this is the sum of the centre of mass velocity vCM and the velocity w of the detected fragment in the centre of mass frame, a simple vector diagram suffices to confirm that the ‘best’ conditions (i.e. those for which the range of lab. frame velocities will be maximal) is obtained when vCM and w are equal. Conversely, if w is small compared with vCM, then the product velocity in the lab. frame will be very similar to vCM irrespective of the scattering angle, and the spread of detected Doppler shifts will be small;
(ii) The velocity spread of the parent molecules (in the case of a photolysis experiment) or in the collision encounters (in the case of a bimolecular reaction dynamics experiment); and
(iii) The probe laser bandwidth—which, ideally, should be as small as possible relative to the Doppler contribution to the overall spectral linewidth.
Interpreting Doppler lineshapes requires some care and caution, since quite different 3-D velocity distributions can give rise to rather similar 1-D Doppler projections. Velocity aligned Doppler spectroscopy, wherein Doppler lineshapes of the selected P(q) fragments are probed with progressively longer time delays between the photolysis and probe laser pulses, offered one route to removing potential ambiguities.24 Many other studies have demonstrated how comparisons of the Doppler lineshapes of a given P(q) product recorded with different interaction geometries (e.g. counter-propagating versus orthogonal photolysis and probe laser propagation axes), pump and probe laser polarisations, and/or different probe transitions (e.g. ΔJ = 0 versus ΔJ = ±1 transitions) can reveal the extent of correlations between various vectorial properties of the P(q) products: informative examples include product orientation and alignment (i.e. the distribution of the product angular momentum (J) relative to μ, the parent transition dipole moment, and thus to εphot, the electric vector of the photolysis radiation) and correlations between J and the product recoil velocity, v.25,26 The concept of Doppler lineshape analysis has been extended and applied, very profitably, to studies of products (e.g. OH) arising in photo-initiated bimolecular collision processes.27
Imaging techniques, the focus of this Invited Article, give two-dimensional (2-D) velocity projections of a given P(q) product, and enable detailed study of any alignment and/or orientation of the products, and of any correlations between the product v and J vectors. The first illustration of the use of ion imaging methods to study a molecular photodissociation process appeared in 1986.28 This pioneering investigation involved pulsed laser photolysis of CH3I molecules, at a wavelength of 266 nm. C–I bond fission resulted in formation of CH3 radicals and I atoms. Energetically, at least, it is possible that these I atom products could be formed in their ground (2P3/2) state or in the spin–orbit excited (2P1/2) state—which lies 7603 cm−1 higher in energy. By suitable choice of probe wavelength, 2 + 1 REMPI allowed state-selective sampling of just CH3(v″ = 0) fragments. The resulting cloud of CH3+ ions expanded with a velocity distribution dictated by the energetics and dynamics of the dissociation process, but was simultaneously accelerated through a TOF-MS onto a time and position sensitive detector. Analysis of the 2-D image of these CH3+ ion impacts showed that the I atoms partnering the CH3(v″ = 0) fragments were predominantly in their spin–orbit excited state (this conclusion followed from measurement of the image radius, which scales with the fragment recoil velocity) and—from the shape of the image—that the fragmentation of the parent molecule occurred promptly following an excitation the transition moment for which was aligned parallel to the C3 symmetry axis.
Imaging studies have come a very long way since Chandler and Houston’s ground-breaking demonstration work.29–33 Image resolution has improved greatly.34 Image processing and analysis methods have developed substantially.35 Most striking of all, however, has been the range and the diversity of the scientific problems to which imaging techniques have now been usefully applied. This Invited Article will chart many of the more important technical developments, highlight the impact of imaging methods in studies of molecular photodissociations and of bimolecular collision processes, and conclude with a very brief summary of many other imaging applications that remain outside the scope of this review.
2. Experimental methods
Imaging methods for characterizing the products of molecular dynamics processes have advanced rapidly in the past 15 years, both in quality of data and in range of application. Imaging now sets the standard for studies of photodissociation, is becoming well established for inelastic scattering, and has recently provided beautifully resolved data of crossed-beam chemical reaction products. In this section we describe the current experimental state-of-the-art of imaging,34 its advantages and limitations, and prospects for the future. We limit our discussion to ion and velocity map imaging, which are single-product detection methods. Coincidence imaging methods36 including the elegant electron–ion correlated imaging method37 for photodissociation/ionization studies are not included here.The main limitation to imaging resolution in all early ion imaging experiments was the birthplace of the detected species. Imaging experiments generally employ a 2-D position sensitive detector (PSD) to measure velocity at a fixed flight time from its position on the detector. For most experiments, especially those designed to capture the entire Newton sphere, the uncertainty in determining distance arises from the uncertainty in where the product particle of interest was formed. Even when using a focused laser beam, e.g., for photodissociation, the starting volume is as long as the confocal parameter of the laser beam, typically a few mm, compared to a typical PSD radius of 20–40 mm. Imaging took a great leap forward in 1997 with the realisation that it was possible to remove the starting volume from the uncertainty in determining velocity, using velocity mapping38 of ionic Newton spheres. Velocity mapping has a three-fold advantage: the Newton spheres are superimposed, open lenses without grids are used for the electrodes, and the removal of any sensitivity to the precise birthplace of the ion also means that a large starting volume can be used. This allows lower ion densities and thus reduced blurring from effects of space charge, i.e., the mutual repulsion of ions of like-charge. Grids lower the lens transmission and perturb the ion trajectories. Space charge, the bane of ion measurements, changes the nascent velocity of the investigated species. Velocity map imaging (VMI) works directly for ionic Newton spheres formed in processes such as photoionization,38,39 photodissociation of state-selected cations,40 electron impact ionisation,41 photo-detachment,42–44 Penning or associative ionisation,45 and ion-molecule reactions.46 For other experiments such as photodissociation,47 inelastic,48 or reactive scattering,49 and deflection of molecular beams by a laser-formed lens,50 the initially formed neutral Newton spheres are converted to ions and then velocity-mapped.
2.1 Apparatus design
Ion imaging, Fig. 1, involves the following experimental steps. (a) creation of Newton spheres (in this example by photodissociation); (b) conversion of the photofragment spheres to ion spheres by laser ionization; (c) projection of the ion Newton spheres onto a 2-D detector; and (d) recovery of the desired information from the 2-D image, either directly from the middle slice of the ion packet (experimental slicing), or via a mathematical transformation. The result of (d) should be equivalent to taking a thin slice through the middle of the Newton sphere formed in step (a).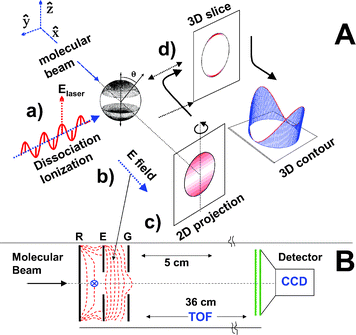 | ||
| Fig. 1 A: The imaging approach for measuring Newton spheres from photodissociation. (a) Photodissociation of molecules in a molecular beam using a linearly polarized laser with the polarization direction parallel to the detector face. (b) Conversion of the photofragment molecules making up the Newton spheres into ions by laser ionization. (c) Projection of the ion spheres onto a two-dimensional (2-D) detector. (d) Mathematical transformation of the 2-D image back to the three-dimensional data of step (a). This slice through the middle of the Newton sphere is displayed in (d) as either a false colour 2-D diagram or 3-D contour diagram. With experimental slicing techniques it is possible to avoid step (d). B: Apparatus layout showing the electrostatic lens used for velocity map imaging of photodissociation. A skimmed molecular beam passes through a small hole in the repeller (R) plate. Laser beam(s) cross the molecular beam between the repeller and extractor (E) plates. Field lines are shown for the electrostatic immersion lens, which is created by open, flat, annular electrodes. Ions formed are accelerated past the ground (G) electrode and fly through the time-of-flight tube to the imaging detector, which is monitored by a CCD camera. The dimensions of a typical apparatus are shown, with electrostatic lens spacing of 15 mm and flight tube length 360 mm. | ||
2.2 Ultimate resolution
An optimized dual-chevron mounted multi-channel plate detector assembly with event centroiding software can estimate the arrival position of an ion to within ∼20 μm over the 20 mm detector radius. This implies for the apparatus shown in Fig. 1 a velocity resolution of 0.1%, thus requiring a CCD camera resolution of 1000 × 1000 (1 M) pixels. A top-grade detector of 60 mm radius and 10 μm spatial resolution would yield a 0.02% velocity resolution and require a 5000 × 5000 (25 M pixel) camera. Velocity resolution ≤0.1% is comparable to the highest resolution competitive methods such as Rydberg tagging.13,18Ion-imaging and VMI actually map the square root of the kinetic energy, i.e.E1/2—all particles with the same E1/2 arrive at the same radius on the detector regardless of their mass/charge ratio. For a typical imaging apparatus shown in Fig. 1 a maximum repeller voltage of +5000 V limits the maximum E to ∼4 eV, which is ∼7000 m s−1 for an O atom product. The ultimate velocity resolution of 0.1% corresponds to 7 m s−1 in this case.
An image recorded with a 1 M pixel camera must usually register ∼2 M ions, corresponding to 20 000 laser pulses at 100 ion pulse−1, or 33 minutes recording time using a 10 Hz laser. This is around the maximum time that all other experimental conditions such as laser pulse energy, molecular beam concentration, nanosecond timing circuits, etc., can be kept constant. 100 ion pulse−1, however, can result in significant distortion of the image due to space charge.51 A safer 1 ion pulse−1 would require a kilohertz laser system, and very fast CCD system to avoid down-time during image acquisition, neither of which are readily available at present. This ultimate resolution, however, has not yet been achieved due to a variety of effects listed in the next section.
2.3 Practical aspects of imaging techniques
Improvements, limitations, and new applications of imaging are discussed here, organized following the creation–conversion–projection–recovery scheme of the imaging experiment.The transverse velocity spread, which is mapped onto the image, is small for most molecular beams, typically corresponding to a temperature of ∼1–2 K. For example, Softley and coworkers53 have measured a transverse velocity corresponding to a temperature of ∼1 K in a spatial imaging study of electric field deflection of high Rydberg states of H2. Gericke and coworkers54 have reported a detailed analysis of parent beam velocity spreads on imaging resolution for (1 + 1) ionization of NO and estimated a contribution of just 0.04 m s−1 (∼2 μK) for an experimental geometry designed to minimize this effect. For bimolecular imaging studies the velocity spread in the parent beams is the dominant limiting factor in image resolution. A discussion of various different approaches for lowering the effects of velocity spreads in crossed-beam studies appears in section 4.
The rotational temperature of the parent molecule can play an important role in the appearance of the image. Wrede et al.55 evaluated the effects of the rotational distribution in a cooled supersonic beam on product images. Near the threshold for photodissociation, the 15 K rotational distribution of their IBr parent molecule beam added a 25 m s−1 FWHM spread to the 60 m s−1 Br atom product velocity. Each internal state of the parent molecule yields a separate but non-resolved set of Newton spheres, leading to an apparent broadening of the image. Furthermore, near the threshold for photodissociation, parent rotation can compete with direct dissociation, i.e., the axial approximation becomes invalid. This tends to reduce any angular anisotropy in the distribution of product recoil velocities—to an extent that increases with increasing rotation.56 For many molecular photodissociation studies, the distribution of populated rotational states in the parent molecule limits the ultimate resolution of the photofragment image.
Vibrational cooling in a supersonic beam is usually much less efficient compared to rotational cooling. A resident vibrational population when rotationally cooled usually leads to well separated rings in the images. Vibrational population in several different modes of CH3I resulted in resolved rings in the CH3 and I fragment images.57
Saturation due to the use of excessive dissociation (or detection) laser flux has the effect of lowering the anisotropy of the surface pattern of the image, by exciting molecules with increasingly less favourable orientations with respect to the laser polarization. High laser pulse energies also increase the probability of multiphoton absorption, which can introduce new and overlapping product channels. Chestakov et al.58 have shown that focusing the dissociation laser with a cylindrical lens not only lowers saturation effects, but is also favourable for ‘sheet-dot’ optical slicing as discussed later.
As mentioned previously, space charge due to too many ions in the ionization volume can significantly lower the image resolution. Wolf and Helm51 have shown that as few as two ions in the ionization volume can measurably perturb the ion trajectories. This density limitation includes all ions formed—not just the selectively-detected ions. Another reason for keeping the ion count as low as possible is to avoid under-counting when using event counting software (as described later).
Ionization can induce a small but significant velocity-changing ionrecoil in the parent ion due to the excess energy above the ionization threshold (i.e. 3hν ≫ IP for a 2 + 1 REMPI process). Detailed studies of ion-recoil have been reported for Rb,51 and NO.54 Recoil effects are most pronounced for H atoms—as has been demonstrated in some detail in studies of hydrogen halide photolyses59,60—but can play a resolution-limiting role for any product. Ionization induced recoil takes place after the photolysis (or collision) process and thus affects all nascent product velocities in the same way, i.e., the ion-recoil image is projected on every point of the velocity map. Recoil effects thus increase quadratically when converting velocity to kinetic energy. Table 1 lists the ion-recoil velocities of typical product species using the most common REMPI detection methods. Detection of O(3P) oxygen atoms at 226 nm, for example, adds an ion-recoil image of diameter 40 m s−1, to all velocities. This greatly exceeds the previously estimated ultimate velocity resolution of 7 m s−1. Recoil is significant even for such heavy (and slow) species as Br and I. Avoidance of ion-recoil effects will require the development of more complicated REMPI detection schemes.
| Product | REMPI detection scheme and excitation wavelength | Recoil radius/m s−1 |
|---|---|---|
| H | 2 + 1; 243 nm | 425 |
| O | 2 + 1; 226 nm | 20 |
| NO | 1 + 1; 226 nm | 14 |
| Br | 2 + 1; 260 nm | 6.5 |
| I | 2 + 1; 271 nm | 5 |
State-selective ionization requires a resonant bound–bound transition, which implies that each fragment will exhibit a velocity-dependent Doppler-shift. The ionization laser wavelength must thus be scanned back and forth equally over the full Doppler profile, which can be a time-consuming and error-prone procedure. Doppler-free two-photon techniques have been proposed as one way of overcoming this problem.61 This approach involves retro-reflecting or splitting the ionization laser into two counter-propagating time-overlapped beams tuned to the centre of the Doppler profile. Each beam independently creates a partial (sub-Doppler) image but the entire image appears when one photon from each beam is absorbed. The ratio of the Doppler-free to single-beam images is enhanced when the laser bandwidth is a small fraction of the Doppler width. For this reason, images of energetic H atoms with their wide Doppler profiles are the most favourable targets for Doppler-free ionization when using a normal (>0.1 cm−1) bandwidth tuneable dye laser for ionization. Unfortunately, as Table 1 showed, H atoms are also most susceptible to recoil problems with 2 + 1 REMPI. This problem has been relieved by use of a 3 + 1 REMPI scheme involving 364.6 nm photons.60 The energy sum of four such photons corresponds to threshold ionization, which solves the recoil problem also. Under the experimental conditions demonstrated thus far, the homogeneous linewidth of the final one photon transition is found to exceed the Doppler width (presumably because of power broadening of the ionization step in the case of delayed pulsed field extraction,60 or because of lowering of the ionisation threshold by the electric field present in the source region in the case of more traditional ion imaging62), thus obviating the need for Doppler-scanning.
Field-free initial conditions. Electrostatic fields can re-orient state-selected molecules, ionize atoms or molecules excited to Rydberg states, and deflect charged species from the interaction zone. For all these types of species, it is crucial that field-free conditions are maintained until after the photo-excitation or collision of interest has taken place. Bergmann and coworkers63 have described an imaging apparatus that includes a permanently field-free ionization zone and velocity mapping conditions for projecting the ions. They report photodissociation studies of electronically state-selected alkali molecules. Weisshaar and coworkers46 used a different approach in their VMI studies of ion-molecule reactions. They maintain the repeller and extractor lenses at 0 V until the reaction has taken place, and pulse the fields on later for VMI of TOF-selected reaction products. Harland et al.41 have described the field-switching conditions needed to combine electron-molecule scattering and VMI. Chestakov et al.58 have described conditions where optimal velocity mapping for photodissociation occurs under field-free conditions followed by pulsing on the electrostatic lens fields.
Electrostatic lens designs. Once the neutral Newton spheres are converted to ion spheres the electrostatic lens assembly accelerates them into a time-of-flight region and onto the PSD. The original VMI lens design used flat annular electrodes spaced 15 mm apart with 20 mm open inner diameters for the accelerator and ground electrodes. Vrakking et al.64 have described how addition of an Einzel lens after the velocity mapping lenses can enable significant (>20) magnification of low velocity electron images. Wrede et al.55 carried out an extensive ion trajectory analysis that illustrated improvements in resolution that can be obtained by using more complex electrode shaping. Vrakking65 showed that use of a genetic algorithm for the electrostatic lens design (such as described in ref. 66), but optimized for velocity mapping, should yield even better resolution. Townsend et al.67 and Lin et al.68 have both reported lens designs with ‘zooming’ capability that provide velocity mapping without ‘pan-caking’ the image. These are discussed later, in the section on Slicing.
Steering. Even with the best construction methods, the image rarely arrives exactly at the centre of the detector without a little help. Active steering methods can be implemented by tilting the electrostatic lens38 or with deflection electrodes (such as a 12-pole design demonstrated by Suzuki and colleagues52) that are placed in the TOF region, after the electrostatic lens. This steering lens design has been used to displace a velocity mapped image of Na+ product atoms to an off-axis detector so as to avoid detector contamination by the primary metal atom beam.63 Neumark and coworkers43 use steering electrodes to deflect their anion beam away from an on-axis photoelectron detector. In a similar type of pulsed, mass-selected, anion photodissociation experiment, Bordas and coworkers69 used an off-axis beam geometry and static imaging fields chosen so as to project the photoelectron image without substantially deflecting the anion beam.
Detector gating. Mass selection is achieved by selectively gating the gain of the PSD at the TOF arrival time of the species of interest. For conventional VMI the detector gain should remain constant during the full arrival time of the ion packet which, for high velocity Newton spheres, can be as long as 50–60 ns.58 Velocity mapping conditions do not provide as high mass-resolution as, for example, Wiley-McLaren space focusing conditions. Using an overly long detector gate can lead to overlap with a neighbouring mass species, while a too-short detector gate results in slicing of the image, as described in the next section. When detecting photoelectrons, nanosecond gating of the PSD synchronized with the excitation laser can be used to generate time-resolved images of competing electron emission processes. Bordas and coworkers69 have described such an experiment for electrons emitted from photoexcited xenon atoms and small carbon cluster anions.
Event counting. By 1998 the benefits of using the event counting/centroiding mode of modern CCD cameras for imaging studies were starting to appear.70,71 Centroiding improves the resolution when using detectors that produce large single-event spot sizes, especially with image intensified CCD cameras. For all detectors, the event counting or thresholding mode alleviates inhomogeneous position-dependent gain problems associated with the PSD. Event counting can lead to undercounting artefacts, however, when two events occur close together on the detector. Furthermore, not all inversion programs can handle the highly pixelated nature of an event-counted image.
A variation of the forward convolution approach, involves simulation and fitting of a set of ‘‘basis images’’ to the experimental images in order to extract the parameters of interest. The most popular of these methods at present is BASEX,73 which expands the projection in a basis set of functions that are analytical projections of well-behaved Gaussian-like functions. The original 3-D image is then reconstructed as a linear combination of these well-behaved functions. Another example of the iterative approach exploits the similarities that exist for cylindrically symmetric systems between the form of the angular and radial distributions of the 3-D distribution and of its 2-D projection.74 While, like the Abel inversion and BASEX, the method is limited to distributions with an axis of symmetry, the returned 3-D distributions are virtually noise-free. The above-mentioned approaches, as well as the back-projection ‘onion-peeling’ method,75 have been compared for a variety of experimental conditions in ref. 35. While no method excels for all practical conditions, the BASEX method was found to be, in general, a very efficient and easy-to-use method.
Several new approaches have appeared since the evaluation in ref. 35. Bass et al.76 presented a Fourier moment image fitting method based on Dixon’s bipolar moment formalism.25 This method allows extraction of speed and angular distributions, as well as angular momentum alignment and orientation information, from raw images, without the necessity of cylindrical symmetry. The method also allows an efficient angular smoothing of noisy images. In order to use the Fourier moment analysis scheme, however, it is necessary to collect a set of images on two different REMPI transitions and using a series of experimental geometries, defined by the polarizations and relative propagation directions of the pump and probe laser beams. Processing of images with a large number of rings is also slow in this approach. Manzhos and Loock77 introduced another approach, based on pattern recognition, which does require cylindrical symmetry. As in the Fourier moment method, image parameters are extracted without inversion, and the exact image centre and any distortion from circularity are automatically identified. This method compares well with the ‘onion-peeling’ approach from the same authors and is most effective for images with well-spaced rings.
Slicing. Since the inception of ion imaging the advantages of recording only the central portion of the Newton sphere instead of crushing the full 3-D sphere were obvious. An approximation to the final recovered thin slice is measured directly, without the need for an inversion treatment. In some cases this procedure is invalid due to product alignment effects72 or to an inhomogeneous flux-to-density transformation,78 or it is just too noisy; alternative experimental methods have thus been introduced which ‘slice’ out the desired information directly from the 3-D distribution. Kitsopoulos and coworkers79 were the first to show that experimental slicing can be combined with the high resolution advantages of velocity mapping. Slicing has grown in importance and several different approaches have emerged which are shown in Fig. 2 and discussed in more detail in this section.
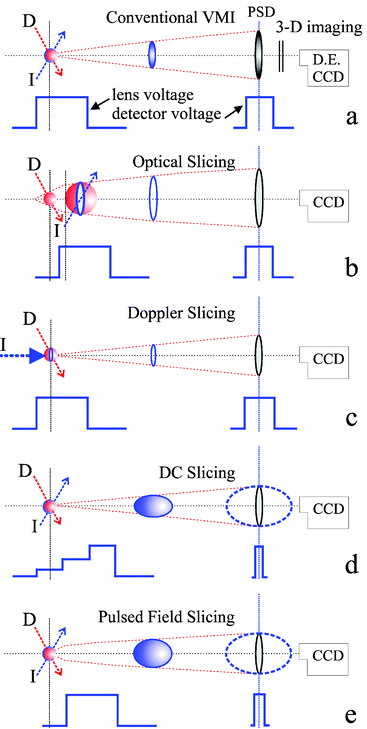 | ||
| Fig. 2 Experimental approaches for obtaining a slice through the central portion of the Newton sphere. (a) Conventional velocity map imaging (VMI) set-up, combined with Double Exposure CCD camera, coupled with a fast position sensitive detector. Position and arrival time are recorded for each ion. Static electric fields are applied to the repeller and extractor lens and a normal detector gate (>100 ns) is used. (b) Optical slicing, using a spatial and temporal delay between dissociation and ionization. Only the centre slice of the neutral sphere is ionized. The electrostatic lens field is turned on before the ionization laser pulse. (c) Doppler slicing, where a narrow-bandwidth ionization laser propagates along the time-of-flight axis and is tuned to ionize the subset of molecules with vz = 0. (d) DC slicing where velocity mapping of the full ion sphere without ‘pancaking’ takes place, using a stepped series of electrode voltages. A narrow (<20 ns) detector gate selectively amplifies the centre slice of the ion sphere. (e) Pulsed-field or field-free slicing, where the ionic Newton spheres are formed field-free and projected under velocity mapping conditions after a time delay onto the detector. The centre slice of the ion packet is selected using a narrow detector gate as in (d). | ||
a. (x,y,t) Camera system. Conventional VMI can be combined with a Double Exposure CCD camera coupled with a fast position sensitive detector in the configuration sketched in Fig. 2a. A ∼1 ns time resolution is possible, as Dinu et al. showed in their Coulomb explosion study of I2+ ions induced using 200 fs laser pulses.80 With this system the position and arrival time of each ion is recorded, and the slice can be selected directly from the full image data set. Static electric fields are applied to the repeller and extractor lens and a normal (>100 ns) detector gate is used. Under normal VMI conditions the Newton sphere is distorted along the TOF (z) direction on arrival at the detector. Conditions leading to velocity mapping without extensive ‘pancaking’ will yield a more useful slice, as described in (d) below.
b. Optical slicing. By using a spatial and temporal delay between dissociation and ionization as shown in Fig. 2b, it is possible to ionize only the central slice of the Newton sphere. The electrostatic lens field is turned on before the ionization laser pulse. Fig. 3 illustrates three different approaches to optical slicing, using either a ‘sheet’ laser beam cross section, formed by focusing with a cylindrical lens, or a ‘dot’ beam cross section from a normal spherical lens. The first, most intuitive, approach employs a sheet–sheet geometry (Fig. 3a) for dissociation and ionization, as demonstrated by Suzuki and coworkers81 in an ion-imaging apparatus. Using a dot–dot configuration (Fig. 3b), Suits and coworkers67 demonstrated excellent quality of image slicing, but the ionization laser must be rastered up and down to fill in the image. The counter-intuitive sheet-dot configuration (Fig. 3c), described by Chestakov et al.,58 takes full advantage of velocity mapping, since none of the detected ions are created in the same place.
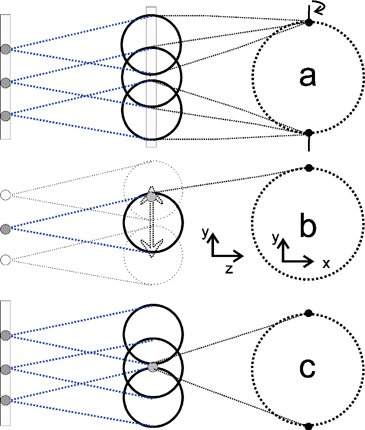 | ||
| Fig. 3 Optical slicing methods. (a) sheet–sheet slicing, (b) dot–dot slicing, where it is necessary to raster the ionising laser beam up and down, (c) sheet-dot slicing with fixed laser beams. | ||
The advantages of optical slicing are two-fold: space charge and saturation effects are reduced, and the kinetic energy resolution is improved. The time delay introduced in optical slicing results in a larger starting volume of ions than in the other slicing methods, which reduces space charge effects. The large origin volume is not a problem when using VMI. The photon flux is also lower when focussing the dissociation laser with a cylindrical lens, reducing saturation effects. Furthermore, all ions of interest are collected at the detector, which is efficient and allows mass selection using a standard (∼100 ns) high voltage detector switch. Another important advantage of optical slicing arises from the narrow slice width. By paying attention to beam shaping it is possible to reduce the width of the thin sheet to ∼50 μm. The ratio of slice width to sphere width for a 2000 μm neutral Newton sphere diameter is <3% in this case, which is often smaller than that achieved when slicing by delayed extraction methods—described in the next section. One drawback of optical slicing methods is the homogeneity required of the ‘sheet’ laser radiation.
c. Doppler slicing. With Doppler slicing (Fig. 2c), a narrow-bandwidth ionization laser propagates along the TOF axis and is tuned to ionize the subset of molecules with vz = 0. While this approach is difficult to implement for high-resolution photofragment imaging—where the pulsed valve and detector are in the way—it is quite easy for reactive scattering. Arikawa demonstrated Doppler slicing for photodissociation ion-imaging,82 using a slightly off-axis ionization laser. Chandler and coworkers48 have used this approach for crossed-beam scattering (where the molecular beams are perpendicular to the imaging detector axis). In this case, a small prism positioned just before the detector was used to deflect the ionization laser beam. With the optical method and Doppler slicing methods the ion packet is already sliced, thus a narrow detector gate is not necessary. This approach offers the same space-charge advantages as optical slicing (b) and, furthermore, Doppler-scanning is unnecessary. Its main limitation is the need for a narrow bandwidth laser.
d. DC slicing. The goal in DC (Fig. 2d) and pulsed field (Fig. 2e) slicing is to adjust the apparatus conditions so as to achieve a longer ion packet arrival time, Δtarrival, while at the same time maintaining VMI conditions. When Δtarrival is long enough, a short (<20 ns) voltage pulse at the detector can be used to record, selectively, the center of the ion packet. The arrival time spread is a consequence of the ion turn-around time, tturn-around, that fragments travelling in the –Z direction need to reverse their direction. For a homogeneous field, tturn-around is proportional to the ion mass, m. Light ions therefore ‘crush’ more easily than heavy ions. Ion trajectory analyses67,79 show that the spread of ion arrival times (Δt) is independent of the flight tube length, and roughly follows the relationship Δt ∝ E1/2/VR, where VR is the repeller voltage. For high VR, Δtarrival values <50 ns are typical. With the DC slicing method67,68 (Fig. 2d), Δtarrival is lengthened by using extra electrodes to extend the region along the ion flight path where acceleration by the electrostatic lens takes place. This approach is easy to implement, and the DC status ensures a homogeneous lens effect. Furthermore, the image size becomes less dependent on the position of ionization along the Z-axis, and a limited amount of ‘zooming’ is possible. However, this method cannot achieve the same degree of slicing (i.e. large values of Δtarrival) as pulsed-field slicing.
e. Delayed pulsed-field extraction slicing. In this case the ionic Newton spheres are formed in field-free conditions and projected onto the detector under velocity mapping conditions by pulsing on the repeller and extractor fields after a time delay. The centre slice of the ion packet is selected using a narrow detector gate as in DC slicing. Delayed extraction by electric field switching is a standard method in photofragment TOF velocity analysis methods.83 With the imaging technique, the photodissociation and fragment ionization occurs field-free and the expanding ion cloud is pushed towards the MCP detector by high voltage applied 200–800 ns after the probe laser pulse. The challenge is to find conditions where true zero-fields are obtained and distortions due to the open-lens electrodes used in VMI are avoided. Kitsopoulos and coworkers60 circumvented the open-lens problem by introducing an extra grid-covered electrode that was used for the field pulsing. With an ultra-fine (40 line mm−1) mesh, this grid electrode does not distort the image measurably, and allows slicing of light ions such as H+, which is not yet possible with DC slicing. Chestakov et al.58 have since demonstrated pulsed-field slicing without the use of a grid. A further advantage of pulsed field slicing is its applicability where field-free conditions are needed—e.g. in crossed-beam studies of ion-ion or ion-neutral reactions.46 This advantage was also demonstrated in photodissociation studies of I2+ ions, up to 2 μs after their formation.58 Chestakov et al.58 have also offered a comparison of the relative merits of pulsed-field, DC and optical slicing. Use of a pulsed-field allowed slicing of images where DC slicing was not successful. In cases where both were successful, DC slicing was found to yield less distorted images. Optical slicing was found to work best, however, as long as the laser beam profiles were sufficiently homogeneous. In most cases, the sliced image was an improvement over an inverted crushed image, mainly because slicing offers a substantial improvement in signal to background noise ratio.
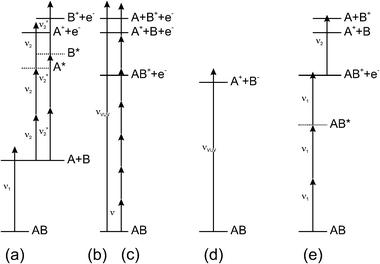 | ||
| Fig. 4 Possible routes to formation of fragment ions: (a) the ‘traditional’ ion imaging experiment, in which the selected neutral photofragment of interest is ionised, quantum state selectively, by (here 2 + 1) REMPI; dissociative ionisation, following single VUV (b) or intense multiphoton (c) photo-excitation; (d) ion-pair formation—normally, as shown here, by absorption of a single VUV photon; and (e) photolysis of parent molecular ions which, here, are shown being formed state-selectively by an appropriate REMPI scheme. | ||
| Probed species | Precursor |
|---|---|
| Ar | ArKr205 |
| Br | HBr,206,207 BrCl,71,208 Br2,56,209–213 IBr,55,213–215 CH3Br,216,217 CHBr3218 CF3Br,219 CF2ClBr,220 CCl3Br,221 C2H3Br,222,223 C2F3Br,223 C3H3Br,224 CH3COBr225 |
| C(1D) | CO226,227 |
| Cl | Cl2,67,213,228–236 HCl,206,237,238 BrCl,71,208,239,240 ICl,241 HOCl,242 Cl2O,242 ClNO,76,243,244 ClN3,245 Cl2CO,246 Cl2CS,247 Cl2SO,244 S2Cl2,248 CH3Cl,249 CH2BrCl,250 CCl4,244 CCl3Br,221 CF2ClBr,220 CH3COCl,251 C2H3Cl,252 C3H3Cl,224 (CH3)3COCl,244 C3H5Cl,253 2-chlorobutane,254 CH2Cl255,256 |
| H, D | H2,257,258 HCl,121 HBr,121 HI,62,259,260 H2S,261 (D2O)2,n,262 H2CO,263 HNCO,264 C2H2,265 CH4,266,267 CH3SH,261 C2H4,268 C2H6,268 C2H5SH,269 C3H8,268 C3H7SH,269 pyrrole,270–272 OD,273 CH2Cl,255 C2H3274 |
| I | HI,275 CH2I2,276,277 CH3I,278–280 (CH3I)n,281 CF3I,282,283 cyclopropyl iodide,284 C6H5I285 |
| Kr | ArKr,205 Kr2205 |
| N(2D) | NO,286 N2O47,287,292 |
| N(2P) | N2O292 |
| O(3P) | NO,286,288,289 N2O,290–292 NO2,293–296 O2,259,297–302 O3,303–305 SO2,303,306,307 OClO308,309 |
| O(1D) | O2,302,310 O3,58,201,303,311,312 N2O45,47,290,292,313–317 |
| O(1S) | N2O292 |
| S(3P) | CS2,318,319 OCS,320 SO2321 |
| S(1D) | CS2,318,319 OCS,75,320–326 SO2,321 ethylene sulfide327 |
| S(1S) | OCS328 |
| Fe | FeO329 |
| H2 | CH4266 |
| CO | OCS,330–334 H2CO,335 HNCO,19,264,336–339 CH2CO,340 CH3COCl,251 dicyclopropyl ketone341 |
| N2 | N2O,342 ClN3343 |
| NO | NO2,48,49,51,228,243,293,294,296,344–347 ArNO,348 (NO)2,349–355 2-chloro-2-nitrosopropane,356 nitrosobenzene357 |
| O2(a1Δg) | O3358 |
| SO | SO2321 |
| 3CH2 | CH2Cl255 |
| CH3,CD3 | CH3Br,217 CH3I,28,33,34,278,279,359–363 (CH3I)n,281 (CH3)2S,364,365 CH3S2CH3,366 CH3COCl,251 CH3CHO367 |
| HCO | CH3CHO367 |
Even if we restrict the discussion to small molecule photodissociations, and ion (rather than electron) detection, however, the literature still contains many variations on this ‘traditional’ theme. Suzuki and coworkers have performed similar imaging experiments using femtosecond laser pulses. Analysis of images recorded with different time delays between the photolysis (pump) and REMPI (probe) pulses has provided information about the timescales of fragmentation processes occurring in molecules like acetyl chloride84 and acetone.84,85 Several alternative routes to ion formation are also illustrated in Fig. 4. For example, using vacuum ultraviolet (VUV) light rather than REMPI allows ionisation, and thus imaging, of all photoproducts—albeit with a loss of the product quantum state specificity.86,87 Ionic fragments arising as a result of dissociative ionisation induced by VUV radiation (Fig. 4(b)),88–90 or by non-resonant multiphoton excitation with intense ultrafast laser pulses80,91–100 (Fig. 4(c)) have been imaged also. Given that dissociative ionisation must result in (at least) three particles, imaging just one charged product is often of only limited value, and coincidence methods must be the preferred route to unravelling the detailed dynamics of such processes.101 Ion-pair products, (Fig. 4(d)), most notably the R+ and/or X− products arising in the photolysis of various of the alkyl halides (RX), are also amenable to study by ion imaging methods.102–105 In most such cases studied to date the ion pairs have been formed following monochromatic excitation, and the anion has been a closed shell atomic species like Cl−. Energy conservation then requires that the image of the cation (or the anion) will display a ring for each R+(p) product state populated in the dissociation process—thereby offering a route to spectroscopic term values for the various R+(p) quantum states if the image resolution is sufficient. Imaging methods also offer new routes to studying the photolysis of van der Waals complexes,106–110 and of molecular ions. Cation photofragmentations studied by imaging methods include CHBr3+111 and ClN3+112 and, at higher levels of state selectivity, Br2+,40,113 BrCl+,114,115 OCS+,116 C2H4+117 and CF3I+.118,119 Such ion fragmentation studies involve a reversal of the roles of the lasers employed in the traditional imaging experiment. Laser 1 is now used to prepare the parent molecular ion of interest, via a REMPI excitation scheme chosen so as to provide the highest parent to fragment ion ratio and maximal parent state specificity. Photolysis of the resulting parent ions, with a second pulsed laser, yields a fragment ion and a neutral product. The former can be imaged directly and, as with the ion pair studies, image analysis can yield new and detailed spectroscopic, thermochemical and/or photophysical data for both the parent and fragment ions.
The remainder of this Section focuses on illustrations of the insights into molecular photofragmentation dynamics that can be gained from high resolution ion imaging studies.
3.1 BrCl photolysis
As Table 2 illustrates, the technique has been applied extensively to studies of the visible and near UV photochemistry of halogen molecules. Though apparently simple, these molecules have proven to be an excellent test-bed both for technique refinement and for testing approximations inherent in the Born–Oppenheimer approximation. Consider BrCl as an illustrative example. Both halogen atoms have 2P3/2 ground states; their respective 2P1/2 spin–orbit excited states (henceforth labelled X*) lie at Eso(Br) = 3685.24 cm−1 and Eso(Cl) = 882.35 cm−1. Fig. 5(a) shows that no fewer than 23 separate electronic states (including the ground state) correlate with the four possible asymptotes associated with X and/or X* products. Not all of these excited states can be populated via an electric dipole allowed transition from the ground X1Σ+(Ω = 0+) state, however. Ion imaging methods have provided what, to date, is the most detailed interpretation of the number and nature of the excited electronic states contributing to the visible and near UV absorption spectra of BrCl,208 and of other halogens and interhalogens like Cl2230 and IBr.55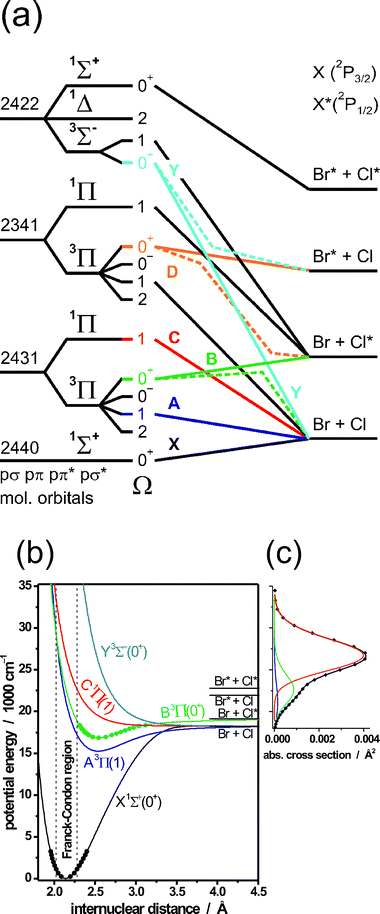 | ||
Fig. 5 (a) Diabatic/adiabatic correlation diagram for BrCl. The left hand columns show the occupancy of the valence molecular orbitals (pσpπ pπ*pσ*) for the various electronic configurations, the term symbol of the electronic state and its projection quantum number Ω. Diabatic and adiabatic correlations are shown as solid (—) and dashed (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) lines, respectively. (b) Diabatic potential energy curves for the X, A, B, C and Y states of BrCl, using the same colour scheme as for the diabatic correlations in (a). (c) Room temperature absorption spectrum of BrCl, illustrating its deconvolution into partial cross-sections associated with A–X, B–X and C–X excitations, colour coded so as to match the relevant excited state potentials in (b). (Adapted from ref. 55 and 208). ) lines, respectively. (b) Diabatic potential energy curves for the X, A, B, C and Y states of BrCl, using the same colour scheme as for the diabatic correlations in (a). (c) Room temperature absorption spectrum of BrCl, illustrating its deconvolution into partial cross-sections associated with A–X, B–X and C–X excitations, colour coded so as to match the relevant excited state potentials in (b). (Adapted from ref. 55 and 208). | ||
Fig. 6 shows representative 2-D slices through reconstructed 3-D recoil distributions of fragments arising in BrCl photolysis, together with the accompanying velocity distributions. Image (A) was obtained by monitoring 35Cl atoms (via the 35Cl+ ion) following excitation of BrCl at a wavelength (defined in air) of 561.65 nm. The corresponding vacuum wavenumber (ṽphot = 17800 cm−1) is some 226 cm−1belowD0(79Br−35Cl).120 The various rings are thus due to dissociation of vibrationally excited BrCl molecules, with v″ = 1, 2 and 3. (Analysis of images such as these shows that the BrCl sample used in this experiment was rotationally ‘cold’ (Trot ∼ 30 K) but had a vibrational state population distribution characterised by a temperature close to 298 K. This enabled investigation of the excitation wavelength dependent product yields and recoil anisotropies as a function of parent vibrational state.) Strikingly, the Br + Cl products arising from dissociation of BrCl molecules in their v″ = 1 and v″ = 2 states at this wavelength are seen to have different recoil anisotropies; the former recoil preferentially perpendicular to the electric vector of the photolysis laser radiation (εphot), whereas those formed from parent molecules with v″ = 2 display a parallel recoil anisotropy.
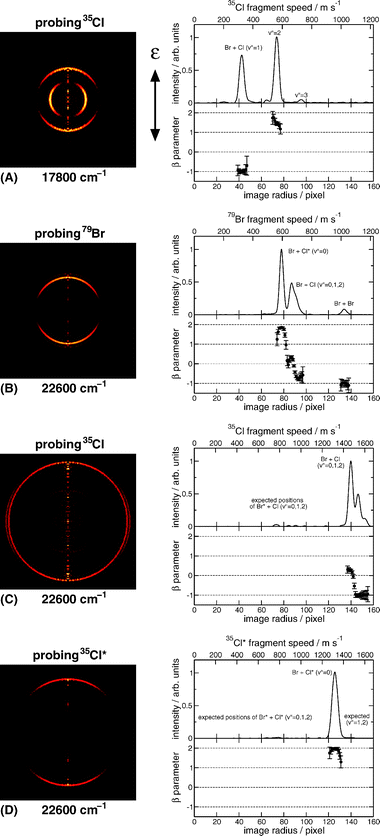 | ||
| Fig. 6 2-D slices through a selection of reconstructed 3-D recoil distributions of fragments arising in BrCl photolysis (with εphot vertical in the plane of the displayed image), together with their accompanying velocity distributions and the associated recoil anisotropies: (A) ṽphot = 17 800 cm−1, monitoring 35Cl fragments; (B)–(D) ṽphot = 22 600 cm−1 monitoring, respectively, 79Br, 35Cl and 35Cl* fragments. (Adapted from ref. 208). | ||
More quantitatively, the angular variation of the product yield is characterized by the beta (β) parameter. Photo-excitation preferentially selects those molecules whose transition moments (μ) are aligned parallel to εphot. Direct dissociation then occurs on a time-scale that is much shorter than a classical rotational period. The resulting fragments will recoil along what was the bond axis of the photo-excited molecule, i.e. they will display a spatial anisotropy that can be traced back to the original μ·ε interaction. The resulting photofragment angular distribution is given by
| I(θ) = (1 + βP2(cos θ))/4π, | (1) |
Fig. 5(b) provides the explanation for the very different recoil anisotropies measured for the Cl fragments arising in the near threshold dissociation of BrCl(v″ = 1 and 2) molecules. Photoexcitation at energies just above the first dissociation threshold can result in population of both the A3Π(Ω = 1) and B3Π(Ω = 0+) states of BrCl. The former state remains poorly characterised, but analogy with other halogens and interhalogens indicates that it will have a potential minimum and correlate diabatically with ground state products. The B state also displays a potential minimum but, in contrast, correlates diabatically with the spin–orbit excited products Br + Cl*. As we discuss later, the B state is subject to an avoided crossing (with the so-called Y state, of 0+ symmetry) at extended bond lengths, with the result that B state molecules prepared by photon absorption from the ground state can follow an adiabatic pathway to ground state Br + Cl products. This adiabatic pathway involves passage over a (small) energy barrier, the magnitude of which is sufficient to block dissociation of BrCl(B) molecules prepared by 17800 cm−1 excitation of BrCl(v″ = 1) molecules. A ← X excitation is thus the only fragmentation route available for these molecules at energies just above the first dissociation threshold. A ← X excitation involves a perpendicular (ΔΩ = +1) transition. The angular distribution of the ground state Br + Cl products that result should thus peak at θ = 90°, as observed for dissociation of the BrCl(v″ = 1) molecules. BrCl(v″ = 2) molecules possess ∼440 cm−1 more internal (vibrational) energy. Photo-excitation of these molecules at 17800 cm−1 provides enough energy for dissociation to Br + Cl products on both the diabatic A state and the adiabatic B state potentials. The observed angular distribution of the Br + Cl products derived from excitation of BrCl(v″ = 2) molecules peaks at θ = 0° and shows very little intensity at θ = 90°, implying that these products arise (almost exclusively) from a parallel (ΔΩ = 0) excitation process and that the B ← Xv″=2 absorption cross-section at this wavenumber is therefore much greater than that for the A ← Xv″=2 transition.
Images (B)−(D) in Fig. 6, obtained at λphot = 442.35 nm (ṽphot = 22 600 cm−1), serve to illustrate the increasing complexity of images recorded at shorter excitation wavelengths. The image radius is proportional to the fragment recoil velocity, i.e. to E1/2, so peaks arising from different parent v″ levels dissociating to a common limit become progressively less well-separated. All four asymptotes associated with the various X and X* product combinations are energetically possible at this photolysis energy. Cl* products are observed but, fortunately, Br* products only contribute at much shorter excitation wavelengths. In principle, therefore, an image of the ground state Br products should contain rings associated with both Br + Cl and Br + Cl* products, the analysis of which should yield the Cl*/(Cl + Cl*) product branching ratio, Γ, and the angular anisotropies of the respective product channels. In practice, analysis is complicated by the possible formation of Br atoms from dissociation of Br2 (that is present in equilibrium with BrCl and Cl2) and, in the present experiments, by the accidental degeneracy (to within the experimental resolution) between Br + Cl products arising from dissociation of BrCl(v″ = 0) molecules and Br + Cl* products from dissociation of BrCl(v″ = 2) molecules. Nonetheless, these complications can all be circumvented by careful imaging measurements of the Br, Cl and Cl* products, as a function of excitation wavelength, thereby yielding Γ and the angular anisotropies of the two fragmentation yields—as a function of v″ and of ṽphot.
As Fig. 7(a) shows, Br + Cl* products constitute about one third of the total yield from dissociation of BrCl(v″ = 0) molecules excited at threshold, but Γ rises to ∼0.7 at ṽphot ∼ 22 000 cm−1 and declines thereafter. Γ values derived following excitation from v″ > 0 levels show greater sensitivity to ṽphot, reflecting the nodal structure in the initial state vibrational wavefunctions. All observed Br + Cl* products display parallel (β = +2) recoil anisotropy, consistent with their being formed, exclusively, by photoexcitation to the B(0+) state and subsequent dissociation via the diabatic pathway. Measurements of the angular anisotropy of the Br + Cl products are more revealing since, as Fig. 7(b) shows, β is sensitive to both v″ and ṽphot. From Fig. 5(b), we see that ground state products can arise as a result of diabatic dissociation following excitation to the A(1) state, adiabatic dissociation following excitation to the B(0+) state, and diabatic dissociation following excitation to the C(1) state. Given knowledge of the total absorption spectrum of BrCl,120 the availability of such experimental data has allowed derivation of ṽphot dependent partial cross-sections for parallel and perpendicular absorption, and thus deconvolution of the parent absorption spectrum into contributions associated with excitation to the A3Π(1), B3Π(0+) and C1Π(1) excited states—as shown in Fig. 5(c). Such a deconvolution of the continuum absorption spectrum of BrCl gives information on the repulsive walls of these three excited state potentials, in the vertical Franck–Condon region. Taken together with existing spectroscopic data for the ground electronic state and for the bound levels supported by the B state potential, this has allowed determination of key regions of the potential energy curves for, and transition moments to, these three excited states. The diabatic potentials so derived were shown in Fig. 5(b). The solid curves in Fig. 7(a) and (b) show Γ(v″) and β(v″) values obtained by wavepacket calculations assuming the potentials shown in Fig. 5(b), parameters for (and transition moments from the X state to) the various excited states as listed in ref. 208, and a coupling matrix element between the bound (B) and dissociative (Y) diabatic states in the region of their crossing that has a value of VB/Y = 80 cm−1. The excellent agreement between experiment and theory serves to highlight the value of careful, high resolution ion imaging methods in ‘continuum state spectroscopy’ studies of this kind.
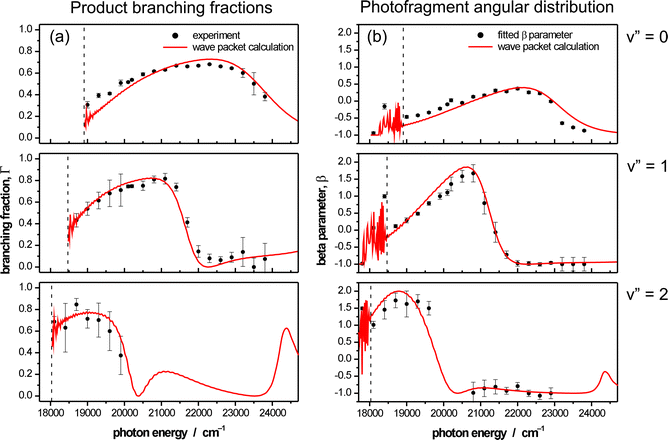 | ||
| Fig. 7 (a) Product branching fractions, Γ, and (b) angular anisotropy parameters, β, for the Br + Cl products resulting from photolysis of jet-cooled BrCl molecules with v″ ≤ 2, plotted as a function of excitation wavenumber. Error bars in each case represent the ±1σ statistical error band on the individual measurements. The curves in (a) and (b) depict, respectively, the calculated Γ(v″) and β(v″) values obtained by wavepacket calculations assuming the X, A, B, C and Y state potentials shown in Fig. 5(b), parameters for (and transition moments from the X state to) the various excited states as listed in ref. 208 and a coupling matrix element between the diabatic B and Y states in the region of their crossing VB/Y = 80 cm−1. The dashed vertical lines in each plot indicate the respective threshold dissociation energies. The rapid oscillatory structure at lower wavenumber in each calculated β(v″) versusṽphot plot is associated with predissociation of bound B state levels by coupling to the Y state. (Adapted from ref. 208). | ||
3.2 CH3I photolysis
Our second example in this section centres on the much-studied photodissociation of CH3I molecules following excitation at wavelengths within the so-called A-band.123 The A-band of CH3I is a region of continuous absorption spanning the wavelength range 220–350 nm. We have already alluded to the historical significance of CH3I photolysis at 266 nm: it provided the first illustration of the ion imaging method.28 Magnetic circular dichroism measurements, and a range of experimental photofragmentation studies and high quality ab initio quantum calculations have led to the conclusion that three optically allowed transitions, each deriving from an σ* ← n electron promotion, contribute to the A-band absorption, and that all three excited states dissociate rapidly, yielding CH3 fragments together with ground (I) and spin–orbit excited (I*) iodine atoms.123 The spin–orbit splitting between the I and I* states is Eso = 7603 cm−1. Fig. 8 shows approximate cuts along the C–I dissociation coordinate of interest. Absorption from the X1A1 ground state is dominated by a parallel transition to the 3Q0 (2A1) state that correlates adiabatically to CH3 + I* products. This absorption is straddled by two much weaker perpendicular bands, to the 3Q1 (2E) and 1Q1(3E) states, both of which correlate with ground state products. As with most previous discussions of alkyl halide photochemistry, we will find it convenient to label excited states using both the Mulliken notation124 (e.g.3Q0) and the appropriate C3v symmetry labels. Interpretations to date all assume conservation (or near-conservation) of C3v symmetry during C–I bond fission, and recognise the presence of a conical intersection at extended C–I bond lengths that allows flux excited to the 3Q0 PES to undergo a non-adiabatic transition to the 1Q1 PES. The photon energies corresponding to A-band absorption are considerably greater than the C–I bond strength [D0(H3C–I) ∼ 19270 cm−1]. Irrespective of whether the partner fragment is I or I*, the available energy following A-band excitation is sufficient to allow formation of the CH3 fragments in a range of vibrational (vi) and rotational (N,K) states. The equilibrium geometry of the CH3 moiety changes from pyramidal (sp3 hybridised) to planar (sp2 hybridisation) as the photo-excited CH3I molecule evolves to products and, as a result, the eventual CH3 fragments show significant excitation in ν2, the umbrella bending vibrational mode.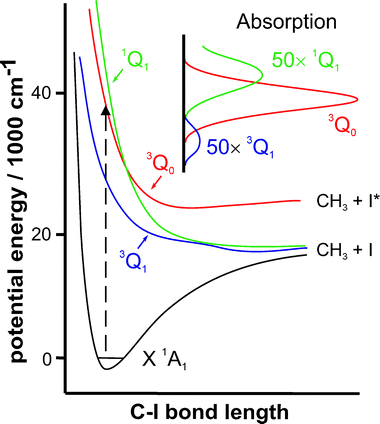 | ||
| Fig. 8 Sections through the potential energy surfaces of the ground (X1A1) and first three excited states of CH3I, plotted as a function of C–I bond extension. The inset shows a deconvolution of the A band absorption spectrum into partial cross-sections associated with the 3Q1 ← X, 3Q0 ← X and 1Q1 ← X transitions. (Adapted from ref. 279). | ||
This population spread among many quantum states of the CH3 product makes a ‘complete’ continuum state spectroscopy study of an alkyl halide like CH3I much more difficult than was the case for a diatomic interhalogen. I and I* fragments can be detected, quantum state selectively, by 2 + 1 REMPI, and imaged. The analysis of such images gives a measure of the internal energy distributions in the CH3 fragments that accompany I (or I*) formation at the λphot of interest and of the angular anisotropies of the respective products. Fig. 9(a) shows wavelength dependent β-parameters for both I and I* fragments determined in this way. However, to determine the quantum yield of either product (i.e. the corresponding I*/(I + I*) branching ratio, Γ) we also need the REMPI linestrength factors for the chosen I and I* probe transitions (i.e. the respective I and I* detection sensitivities). These are normally calculable in the case of atoms, though it is usually reassuring if they can be checked, independently—for example, by comparison with branching ratios determined by an alternative route (e.g. by non-state selective ionisation using VUV photoionisation or electron impact methods). Fig. 9(b) shows the best estimate λphot dependent Γ values determined from high resolution ion imaging studies of CH3I photolysis. MPI at wavelengths ∼333 nm, resonance enhanced at the two photon energy by the 2A2″(3pz) Rydberg state, provides a convenient, quantum state-selective detection method for CH3 (and CD3) fragments. Analysis of CH3+ ion images obtained following CH3I photolysis at any given λphot allows determination of the relative yields of I* and I products formed in conjunction with the particular CH3(vi,N,K) state (or states) probed at the chosen REMPI probe wavelength. By way of illustration, Fig. 9(b) also shows λphot dependent Γ values determined by analysis of imaging CH3(v = 0) fragments only. The determination of the overall quantum yield of (say) I* products in this way, irrespective of the internal state of the partner CH3 fragment, would involve much more work, however. It would first be necessary to repeat such measurements at many different REMPI wavelengths, chosen so as to probe the many different populated CH3(vi, N, K) states. Then, once again, to determine the overall I* quantum yield, we would need to determine the appropriate linestrength (calibration) factors for converting each of the measured CH3 REMPI signal intensities into the corresponding quantum state populations.
![Plots showing the deduced λphot dependence of: (a)
β(I*) and β(I) derived from analysis of I*
(●) and I (□) images and (b)
Γ, the [I*]/([I]
+
[I*]) branching fraction. Γ(all v)
(Δ) is derived from measured I and I* REMPI line intensities together with the appropriate linestrength factors, and constitutes the best estimate of the branching fraction averaged over all partner states of the CH3 co-fragment. Γ(v
= 0)
(■) is derived from analysis of CH3(v
= 0) fragments, and represents the branching ratio for forming I* and I fragments specifically in association with CH3(v
= 0) co-fragments. The solid and dashed/dotted lines in (a) and (b) illustrate different possible interpolations through the input β(I) and Γ(all v) data sets, respectively. The derived λphot dependence of σpar, the fraction of the total absorption probability associated with 3Q0
← X1A1 excitation and of the 3Q0
∼ > 1Q1 curve crossing probability Pcc are shown in (c) and (d), respectively. (Adapted from ref. 279).](/image/article/2006/CP/b509304j/b509304j-f9.gif) | ||
| Fig. 9 Plots showing the deduced λphot dependence of: (a) β(I*) and β(I) derived from analysis of I* (●) and I (□) images and (b) Γ, the [I*]/([I] + [I*]) branching fraction. Γ(all v) (Δ) is derived from measured I and I* REMPI line intensities together with the appropriate linestrength factors, and constitutes the best estimate of the branching fraction averaged over all partner states of the CH3 co-fragment. Γ(v = 0) (■) is derived from analysis of CH3(v = 0) fragments, and represents the branching ratio for forming I* and I fragments specifically in association with CH3(v = 0) co-fragments. The solid and dashed/dotted lines in (a) and (b) illustrate different possible interpolations through the input β(I) and Γ(all v) data sets, respectively. The derived λphot dependence of σpar, the fraction of the total absorption probability associated with 3Q0 ← X1A1 excitation and of the 3Q0 ∼ > 1Q1 curve crossing probability Pcc are shown in (c) and (d), respectively. (Adapted from ref. 279). | ||
Further subtleties reveal themselves when such an analysis is actually attempted. The difference between Γ(all v) and Γ(v = 0) in Fig. 9(b) indicates that the I*/I branching ratio must vary with the vi state of the accompanying CH3 partner. This is believable—in principle at least: there must be certain CH3(vi) product states that, simply on energy conservation grounds, could be formed in conjunction with I products, but not with I* products. In reality, dynamics rather than simple energetics appears to control the branching between I* and I products. Γ values determined by analysing images of CH3 fragments formed in different v2 states are found to depend sensitively on the precise REMPI wavelength used to probe the CH3(v2) product125—implying that the relative probabilities of I* and I product formation are even sensitive to the particular rotational (N,K) state of the CH3(vi) partner. Careful measurements of I* and I REMPI signal intensities, combined with best-estimate I and I* linestrength factors, are thus considered to provide the most robust branching ratio data yet available. As in the case of BrCl, spin–orbit excited halogen atoms (here I*) are found to arise exclusively as a result of a parallel excitation (i.e. to the 3Q0 state); subsequent direct dissociation yields products with a (near limiting) recoil anisotropy and β(I*) ∼ +2. Ground state I products formed at λphot ≤ 280 nm also show large positive β values. At first glance this is a most surprising result since, as Fig. 8 showed, the 1Q1 and 3Q1 states (both of which correlate adiabatically with ground state I atoms) are both reached by perpendicular excitations from the X state. The observation of β(I) > 0 highlights the importance of non-adiabatic coupling between the 3Q0 and 1Q1 states at extended C–I bond lengths. Full analysis of the data available from high resolution imaging studies allows estimation of the wavelength dependence of (i) σpar, the fraction of the total absorption probability associated with 3Q0 ← X1A1 excitation (Fig. 9(c)), and (ii) the 3Q0 ∼ > 1Q1 curve crossing probability Pcc (Fig. 9(d)). σpar is seen to drop at λphot >280 nm, reflecting the growing contribution from 3Q1 ← X1A1 absorption. β(I) remains positive, however, indicating that σpar has still not dropped to zero and that Pcc increases as λphot increases and the C–I separation velocity declines (Fig. 9(d)). Panels 9(a) and 9(b) show alternative interpolations between the input data points for, respectively, β(I) and Γ(all v) (one shown by a solid line, the other by, respectively, dashed and dotted lines); these alternative ‘fits’ result in minor differences in the deduced wavelength dependence of σpar and Pcc as shown in panels 9(c) and 9(d). The most plausible decomposition of the A band absorption derived from such analyses279 is shown in the inset to Fig. 8.
The CH3 and CD3 fragments resulting from photolysis of CH3I (and CD3I) at wavelengths within the A band illustrate another subtlety associated with the REMPI detection process. Different rotational lines in the REMPI spectrum appear with different relative intensities depending on the polarisation axis of the probe laser (εprobe). In general, a molecule with rotational quantum number J (or an atom with total electronic angular momentum quantum number J) has 2J + 1 sub-levels with mJ = J, J−1, …, −J, where mJ is the projection of J onto a space fixed axis (e.g. on the direction of εprobe). If levels with the same J but different |mJ| have different populations (i.e. the ensemble has a quadrupole moment), the sample is said to be aligned. An oriented distribution also involves a non-statistical mJ state population distribution but, in this case, the distribution will be dipolar, with states of the same |mJ| but opposite sign having different populations. A number of groups have worked through the necessary angular momentum algebra for determining the population (i.e. the rank k = 0 state multipole), orientation (k = 1) and alignment (k = 2) moments of a distribution (in which J is assumed to have cylindrical symmetry about the axis of the recoil velocity, v) that is, as here, probed with light of defined polarisation via a coherent multiphoton excitation process.126 Analysis of the CH3 (or CD3) case is further complicated by the open shell character of the radical, and by the fact that most ‘lines’ in the spectrum are actually blends of transitions involving a common N but a range of (molecule fixed) projection quantum numbers K. Detailed analysis of the REMPI spectra of CH3 fragments resulting from photolysis of jet-cooled CH3I molecules has revealed a strong propensity for any initial rotation about the parent C3 symmetry axis (with associated quantum number K) to be conserved in the CH3 fragment, whereas parent rotation about axes perpendicular to the principal rotation axis is coupled into product translational motion. The conservation of K results in a marked (εphot,v,N) vector correlation (where v is the fragment recoil velocity and N the rotational angular momentum of the CH3 radical), and thus a strong alignment of the methyl radicals. Impulsive energy release during the C–I bond fission can result in an extra ‘kick’ of a few rotational quanta of rotational angular momentum into tumbling motion perpendicular to the C3 axis, leading to N > K. Not surprisingly, given the deduced contribution of parent rotation to the ultimate CH3 product state population distribution and angular momentum alignment, the detailed form of these distributions is found to depend quite sensitively upon the molecular beam expansion conditions employed. Yet more detailed insights into propensities for K conservation have been derived from photolysis studies of CH3I molecules in specifically prepared (by use of a hexapole field) J, K, m states.362
3.3 Photofragment alignment and orientation
Imaging methods are eminently suited to investigating possible alignment (and orientation) of both atomic and molecular photofragments. From the foregoing it is evident that it is possible to have an oriented ensemble of any fragments with J ≥ 1/2, and an aligned distribution of any fragments with J ≥ 1. Modulation of the measured photofragment angular distribution, additional to that expected simply on the basis of the recoil anisotropy term (eqn (1)), can be one clear indicator of non-zero orientation or alignment contributions. Some early photofragment images293,313 hinted at the presence of higher order terms in the angular distribution, which were seen to deviate from that predicted by eqn (1). Subsequent imaging studies succeeded in determining |mJ| population distributions in, for example, S(1D2) and O(1D2) atoms from photolysis of, respectively, OCS323 and N2O.315 A yet more detailed picture of the photodissociation dynamics, including coherence effects, could be obtained by determining the full angular momentum density matrix for such atomic fragments. This can be derived, by fitting measured differences between images recorded with appropriate combinations of pump and probe laser polarisation and/or propagation direction.126We return to the example of BrCl to illustrate some of the types of insights that can be obtained. The left hand and centre panels in Fig. 10 are all images of the ground state Cl(2P3/2) atoms resulting from photolysis of jet-cooled BrCl molecules at λphot
= 467.16 nm (ṽphot
= 21 400 cm−1). From the foregoing, we know that the co-fragment is a ground state Br atom. As in most imaging experiments, the photolysis and probe laser beams used to record these images counter-propagated along an axis (X) orthogonal to the extraction/detection axis (Z). Images (a), (b), (d) and (e) were all recorded with εphot aligned parallel to Y, i.e. vertically in the imaging plane, but each involves a different probe laser polarisation. The four images were obtained by ramping through four different probe laser polarisations on successive laser shots, while simultaneously scanning repeatedly back and forth across the Doppler profile of the
| Cl(…3p5; 2P3/2) →→ Cl(…3p4(3P)4p1; 2P1/2) → Cl+ + e− |
 | ||
| Fig. 10 Images of Cl(2P3/2) atoms from photolysis of BrCl at 467.16 nm: (a) and (b) were obtained with εphot aligned parallel to Y (i.e. perpendicular to the TOF axis (Z), and vertical in the plane of the displayed image) and εprobe linearly polarised along, respectively, Y (the MY image) and Z (MZ). (c) displays the alignment anisotropy image {MZ − MY}. (d) and (e) show the corresponding MXR and MXL images, obtained with εphot aligned parallel to Y and εprobe respectively right and left circularly polarised, while (f) shows the alignment-free combination image {MZ + MY − 1/3 (MXR + MXL)}. Images (g) and (h) were recorded with left circularly polarised photolysis radiation and, respectively, left and right circularly polarised probe laser radiation; {M′XR − M′XL}, the difference between these two images, is shown in (i). The difference images (c) and (i) are both plotted using the convention blue = positive, white = zero and red = negative, though each has been normalised independently to allow use of the full colour range. | ||
Here we begin to see the real value of recording images like those shown in Fig. 10(a) and 10(b) under otherwise equivalent conditions. By focussing on the alignment image {MZ − MY}, we are able to cancel out the recoil anisotropy contribution that dominates the individual MZ and MY images. Conversely, alignment effects can be negated by focussing on the combination image {MZ + MY − 1/3 (MXR + MXL)}, (Fig. 10(f)), which is invertible, and from which the recoil anisotropy can simply be derived by fitting to eqn (1). Clearly, β ∼ 0 in the particular case of BrCl photolysis at 467.16 nm, consistent with the comparable parallel and perpendicular partial absorption cross-sections deduced at this wavelength from the earlier deconvolution of the BrCl absorption spectrum (Fig. 5(c)). The {MZ − MY} image is a non-invertible, squashed 2-D projection. Its detailed form depends sensitively upon λphot. Alignment images such as this can be analysed however, by fitting in terms of a suitably weighted set of four alignment basis functions which are themselves calculated from limiting combinations of the alignment anisotropy parameters, labelled s2, α2, η2, γ2 and γ′2.126,240 In a similar way, insights into the product orientation can be obtained from analysis of {MXR − MXL} difference images recorded with εphot aligned along the 45° bisector of Y and Z, or with circularly polarised photolysis laser radiation. The bottom row of Fig. 10 shows illustrative images recorded with left circularly polarised 467.16 nm photolysis radiation and, respectively, left (g) and right (h) circularly polarised probe laser radiation. These images, which we label M′XL and M′XR, are clearly different—implying some orientation of the Cl fragments. Analysis of the {M′XR − M′XL} difference image (i) yields information about the odd (k = 1 (and 3)) moments of the angular momentum polarisation.
Equations for converting such laboratory frame parameters into a complementary set of polarisation (a(k)q(p)) parameters expressed in the molecular frame72,127 are available in the literature.126 Each of the parameters can be ascribed a physical origin. For example, a(2)0(∥) and a(2)0(⊥) describe contributions to photofragment alignment as a result of incoherent excitation via parallel and perpendicular transitions, respectively. The values of these parameters can be used to obtain relative populations of the different mJ states of the photofragments (where mJ is the projection of J on v). In the case of Cl fragments from BrCl, we find that the value of a0(2)(//) is consistently positive at photolysis wavelengths from 420–480 nm, with a mean value of ∼ +0.5. This indicates that the Cl(2P3/2) atoms are formed preferentially in |mJ| = 3/2 rather than |mJ| = 1/2 states,16 following absorption via a parallel transition to a state with Ω = 0+ and dissociation on potentials of this symmetry. The reverse is true for mJ state populations that result from excitation to, and dissociation via, one or more states with Ω = 1, accessed by a perpendicular transition, for which a0(2)(⊥) = −0.5 across the range of wavelengths.
Preferential population of certain mJ states can be understood from the long-range correlations of the adiabatic molecular potential energy curves to particular atomic states of the Cl and Br products described by quantum numbers J and mJ. However, recent analysis128 demonstrates that adiabatic dissociation of BrCl on either the first adiabatic Ω = 0+ state (denoted as B(0+)) or one of the two low-lying Ω = 1 states (the A(1) and C(1) states), all of which correlate asymptotically to the lowest Br + Cl limit, cannot give rise to the observed alignment (and orientation) effects. Indeed, the value of a0(2)(//) depends upon the probability, pBX, of non-adiabatic transfer of dissociating flux from the B(0+) potential to the ground electronic state, X(0+) and subsequent interference between adiabatic and non-adiabatic dissociation pathways because of a phase shift of the nuclear wavefunctions on the two potentials (denoted by ΔΦBX). The alignment parameter must vanish for a purely adiabatic dissociation (i.e.pBX = 0), so the non-zero value is a clear indicator of the occurrence of non-adiabatic processes—probably at long range. Similarly, non-zero alignment following excitation to, and dissociation on, the Ω = 1 potentials is indicative of non-adiabatic dynamics between the A(1) and C(1) states, as well as a third state with Ω = 1 that also correlates to Br + Cl products. Again, the magnitude of the alignment is determined by crossing probabilities and the phase shift ΔΦAC.
The alignment parameters a2(2)(⊥) and a1(2)(//,⊥) arise, respectively, from coherent excitation of the two components of a |Ω| = 1 state via a perpendicular transition, and coherent excitation of Ω = 0+ and 1 states via simultaneous parallel and perpendicular transitions. Non-adiabatic dynamics again control the magnitudes of these parameters, which prove to be wavelength dependent because of changing degrees of excitation to the A, B and C states and a dependence of non-adiabatic dynamics on excitation and fragment recoil energies, but interpretation is complicated by the sheer number of possible crossing mechanisms and consequent phase shifts that contribute to their values. The complexity, even for a diatomic molecule, is daunting, but two strategies are being developed to unravel the dynamics. The first approach is to measure all possible alignment and orientation parameters as a function of excitation wavelength, and thus to over-determine a fit to extract all crossing probabilities and phase shifts. The second method involves use of wavepacket dynamics methodology developed by Balint-Kurti and coworkers,129,130 together with best available potential energy curves and ab initio computed couplings, to predict the alignment and orientation of the dissociation products.
Before concluding this Section it is worth pointing out that photofragment alignment is one, but not the only, reason for observing additional angular modulation in an ion image. Multiphoton induced fragmentation is another and, once again, photolysis studies of the BrCl molecule provide a particularly striking illustration. Fig. 11 shows an image of the Cl+ ions observed following one colour excitation of BrCl at 316.85 nm (31568.4 cm−1). Two photon excitation at this wavelength leads to population of the v = 3 level of the first (5sσ) Rydberg state built on a 2Π1/2 ion core. This state has Ω = 1. Absorption of one further photon by these excited molecules leads to formation of BrCl+ (X2Π1/2) and, to a lesser extent, BrCl+ (X2Π3/2) ions, with v+ = 3 (where v+ is used explicitly to label a vibrational state of the ion, rather than the neutral molecule). One photon excitation of these ions induces dissociation to Br + Cl+ products, which are responsible for the two innermost rings in this image. The products of these dissociations exhibit angular distributions that peak at 0°, consistent with the view that these Cl+ ions arise from a predominantly parallel (π* ← π) excitation of the BrCl+ parent. The outer rings show a strikingly different angular anisotropy, peaking at ∼±35° to the vertical. As Fig. 11 shows, specifically for the case of the outermost intense ring, these angular distributions are well described by an expanded version of eqn (1) involving terms up to P6(cos θ). The best-fit β2, β4 and β6 coefficients so derived are consistent with three photon excitation (resonance enhanced at the two photon energy by the Ω = 1 Rydberg state) to one or more super-excited states of BrCl with Ω = 1, and dissociation to Br + Cl** products.115,131 Cl** represents a Rydberg state of Cl, the precise identity of which can be determined from the (calibrated) image radius. In the case of the outermost ring in Fig. 11, for example, it is the 2P3/2 state with configuration 3s23p4(3P)4s, which is ionised (thereby forming the detected Cl+ ions) by further photon absorption.
![(a) Raw image of the Cl+ ions resulting from multiphoton excitation of BrCl at 316.85 nm (ṽ
= 31568.4 cm−1). εphot is vertical in the plane of the displayed image. The two inner rings, which peak at 0° and exhibit parallel recoil angular anisotropy, are attributable to one photon dissociation of BrCl+ ions formed by three photon ionisation, resonance enhanced at the two photon energy by the v
= 3 level of the first [2Π1/2]5sσ Rydberg state. The outer rings show a strikingly different angular anisotropy, peaking at ∼±35° to the vertical. (b) shows the angular distribution associated with the outermost of the intense rings, together with best fits using both eqn (1)
() and an expanded version of this equation (—) involving terms up to P6(cos θ). The values of the β2, β4 and β6 coefficients derived in the latter fitting show these Cl+ ions to be formed as a result of a resonance enhanced three photon Ω
= 0+
→→
Ω
= 1 →
Ω
= 1 dissociation process, yielding Cl** atoms in their 3s23p4(3P)4s Rydberg state, which are then ionised by further photon absorption.](/image/article/2006/CP/b509304j/b509304j-f11.gif) | ||
| Fig. 11 (a) Raw image of the Cl+ ions resulting from multiphoton excitation of BrCl at 316.85 nm (ṽ
= 31568.4 cm−1). εphot is vertical in the plane of the displayed image. The two inner rings, which peak at 0° and exhibit parallel recoil angular anisotropy, are attributable to one photon dissociation of BrCl+ ions formed by three photon ionisation, resonance enhanced at the two photon energy by the v
= 3 level of the first [2Π1/2]5sσ Rydberg state. The outer rings show a strikingly different angular anisotropy, peaking at ∼±35° to the vertical. (b) shows the angular distribution associated with the outermost of the intense rings, together with best fits using both eqn (1)
() and an expanded version of this equation (—) involving terms up to P6(cos θ). The values of the β2, β4 and β6 coefficients derived in the latter fitting show these Cl+ ions to be formed as a result of a resonance enhanced three photon Ω
= 0+
→→
Ω
= 1 →
Ω
= 1 dissociation process, yielding Cl** atoms in their 3s23p4(3P)4s Rydberg state, which are then ionised by further photon absorption. | ||
Clearly, high resolution imaging studies of carefully chosen target systems performed in tandem with high level electronic structure theory offers a means of exploring many of the more intimate details of molecular photofragmentations, including the non-adiabatic couplings that must underpin most such dissociation processes but which, hitherto, have largely defied detailed investigation. Arguably, however, the potential impact of imaging methods in the study of bimolecular collision processes will be even greater. This is the topic of the next Section.
4. Bimolecular processes
Energy transfer in bimolecular collisions is central to gas-phase chemistry. The energy transfer may involve the excitation of the internal modes of the colliding species (inelastic collisions) or it may involve bond-breaking and bond-making (reactive collisions). The outcome of a collision is determined by the path taken over the underlying PES. The nature of this surface can be probed in detail by measuring the nascent product internal state distributions (integral cross-sections) and product centre-of-mass (CM) velocity distributions (differential cross-sections). Consequently the experimental determination of these parameters provides a rigorous test of ab initio or empirical PESs.Ion-imaging was first applied to the study of reactive and inelastic scattering processes in the early 1990s, and is now becoming recognised as a powerful technique for determining differential cross-sections (DCSs). The power of the method derives from the ability to image all the recoil velocity components of a product simultaneously, and thereby directly derive the doubly differential cross-section d2σ/dEd(cos θ), where E is the kinetic energy release and θ is the scattering angle. The velocity distribution is cylindrically symmetric about the relative velocity of the reactants (unless there is preferential molecular alignment prior to the collision), and experiments are typically configured so that the imaging plane lies parallel to this relative velocity vector. For crossed molecular beam studies the measured laboratory-frame velocities are then readily converted into CM velocities via a Newton (velocity vector) diagram. Examples of Newton diagrams for bimolecular collisions together with experimental images are shown in Fig. 12 for crossed molecular beam studies of Cl + n-butane which are discussed below.
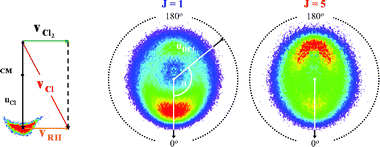 | ||
| Fig. 12 HCl(v = 0, J = 1 and 5) product images from the reaction of Cl with n-butane, with the appropriate Newton diagram for the reaction overlaid. CM indicates the position of the centre of mass, the vectors vCl2 and vRH represent the Cl2 and RH molecular beam velocities respectively, vCl and uCl are the velocities of the Cl atoms in the laboratory and CM frame respectively, uHCl is the velocity of the product HCl in the centre-of-mass frame and θ is the scattering angle. (adapted from ref. 160). | ||
Several experimental geometries have been employed in ion-imaging studies of bimolecular scattering. The traditional crossed molecular beam set-up involves two skimmed molecular beams intersecting at a set angle, typically 90°, with the probe laser overlapping the intersection region. The plane defined by the two crossed molecular beams is typically parallel to the imaging plane, thereby ensuring that the symmetry axis of the product Newton spheres (the relative velocity vector) will also be parallel to the imaging plane. Such crossed molecular beam set-ups have been used to study the rotationally inelastic scattering of NO from Ar,48,132–138 He and D2;139 of HCl from Ar,140 Ne, Kr, N2 and CH4;141 and of CO by Ne.142 Similar experimental arrangements have been employed in studies of non-adiabatic transfer between the 2P3/2 and 2P1/2 spin–orbit states of Cl atoms in collision with D2,143 and in studies of the reactive scattering of O(1D) atoms with D2;144 of Cl(2P3/2) atoms with alcohols;145,146 and of F atoms with CH4, CD4 and various of the partially deuterated isotopomers.78,147–153 For scattering of highly reactive species (e.g. O(1D), Cl(2P3/2) and F in the aforementioned collisions) a precursor is expanded through a nozzle and the reactant generated by photolysis (e.g. O(1D) from O3, Cl(2P3/2) from (ClCO)2) or high-voltage discharge (F from F2) prior to the skimmer. This set-up has also been used in a study of Penning ionisation which imaged Ne+ and NeAr+ created by the collision of Ne* with Ar,45 where the Ne* was electronically excited by a high voltage discharge to an energy above the ionisation potential of Ar. In an alternative arrangement, first used by Welge and coworkers,154 a precursor and reactant are expanded in two parallel molecular beams, the latter directed along the TOF axis (Fig. 13). The precursor beam is crossed by a photolysis laser which, if there is strong recoil anisotropy, is arranged to be linearly polarised along the direction required to maximise the photofragment flux intersecting the central molecular beam. The probe laser is delayed with respect to the photolysis laser to allow time for the photofragments to reach the central beam. The probe laser is also displaced slightly upstream to compensate for the distance travelled by the reactants along the TOF axis in the time between the two laser pulses. Thus the relative velocity of the reactants is perpendicular to the direction of the molecular beam, and hence parallel to the imaging plane. Examples of reactions studied using this set-up are O atoms with alkanes,155–158 H with D2,159 and Cl(2P3/2) with various small organic molecules;160–162 in the latter experiment unskimmed molecular beams were used. In some cases it is possible to coexpand the precursor and reactant in the same molecular beam. This arrangement, which is discussed at the end of this section, has been used to study the reaction of H with HI163 and Cl(2P3/2) with ethane164,165 and n-butane.166
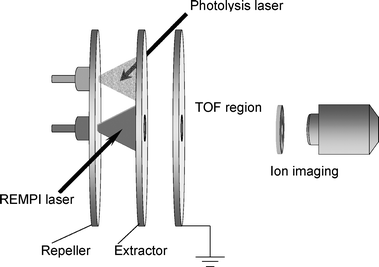 | ||
| Fig. 13 Dual (precursor and reactant) beam arrangement for imaging studies of bimolecular collision processes. The pump laser creates atomic fragments by photolysing an appropriate precursor entrained in the (upper) pulsed molecular beam. The resulting atomic reagents (e.g. Cl atoms) expand outwards and cross the molecular beam containing the other reactant (e.g. RH, lower beam). Products (e.g. HCl) that result are photoionised, state-selectively, by 2 + 1 REMPI and detected with a 2-D position sensitive imaging detector after passing through a TOF mass spectrometer. (adapted from ref. 160, with permission). | ||
From Fig. 12 it is obvious that images from crossed molecular beam experiments allow easy (albeit qualitative) assessment of angular and energy distributions. However, it is not usually sufficient simply to use the inverse-Abel transform common to unimolecular studies. Typically a density-to-flux transformation is performed using a forward simulation method to correct for experimental biases. Such biases, which are common to all crossed beam experiments using laser probing methods, can arise from a variety of sources: e.g. the spread in the beam velocities, the finite duration of the beam pulses and the size of the probe volume relative to the interaction volume. The gas pulses have a much wider temporal profile (many μs) than the laser pulses used for detection (a few ns), while the volume probed by the laser is much smaller than the volume in which collisions occur at the intersection of the two molecular beams. Thus different scattering velocities are detected with different efficiencies. For example, a scattering event occurring before the laser pulse arrives may be detected only if the product has a small laboratory velocity or if its velocity is directed along the laser path. The spread in molecular beam velocities means that the measured data must be modelled as a sum of many discrete scattering spheres, each associated with a particular set of molecular velocities. This is especially true for crossed molecular beam studies, and usually each of the molecular beam velocities is measured by direct imaging of the reactant or a suitable molecule (e.g. NO) seeded in the beam. In the geometry illustrated in Fig. 13 the divergence of the molecular beam parallel to the imaging plane is extremely small, and the photofragment reactant is often generated with a well-defined recoil speed. However, the spread in beam velocities perpendicular to the imaging plane will manifest itself as a distribution in the collision energies. The experimental biases are taken into account by a simulation that is typically combined with trial differential cross-sections that are varied until the simulated image matches the experimental image.
4.1 Inelastic scattering studies
The rotationally inelastic scattering of NO by Ar provided the first example of state-to-state differential cross-sections measured using ion-imaging.132 Rotational energy transfer in the scattering of open shell molecules, such as NO, is of considerable theoretical interest because of the participation of the electronic degrees of freedom. The collisions between NO and a closed shell atom can occur on two PESs of A′ and A″ symmetry; on the A′ surface, the unpaired electron occupies an orbital in the plane of the three atoms, whereas on the A″ surface the unpaired electron occupies an orbital perpendicular to the three atoms. Electron spin interactions within the NO give rise to two scattering channels, one of which is spin–orbit conserving (2Π1/2 → 2Π1/2) and one of which is spin–orbit changing (2Π1/2 → 2Π3/2). In theoretical treatments of the inelastic scattering the A′ and A″ surfaces are usually transformed to sum and difference PESs. The spin–orbit state changing collisions in particular are sensitive to the difference PES. The small difference between the A′ and A″ PESs makes the accurate prediction of the differential cross-sections extremely challenging, and comparison with experiment provides a stringent test of the accuracy of the PESs. In all the inelastic scattering experiments the NO scattered into different rotational states is ionised state-selectively by 1 + 1 REMPI via the A2Σ+ state. In the first ion-imaging studies of NO with Ar132,133 two maxima in the rotationally inelastic scattering angular distributions, known as rotational rainbows, were observed—as expected for a heteronuclear diatomic scattering from an atom.Later ion-imaging135 and VMI studies134 of this system, with increased resolution and sensitivity, have shown good agreement (for both spin–orbit state conserving and spin–orbit state changing collisions) with calculated differential cross-sections based on coupled cluster CCSD(T) PESs.167 At a CM collision energy (ECM) of 0.066 eV,135 low J products are observed to be strongly forward scattered, with sideways and backward scattering becoming significant at J > 4.5, and undulations at all J—which become wider and shift to higher angle with increasing J. These undulations have been related to the ellipsoidal nature of the atom-molecule interaction. Effects of reagent vibrational excitation have been studied also;48 only a slight shift to smaller scattering angles was noted for NO(v = 5) compared to NO(v = 0) scattered from Ar at ECM = 0.181 eV, consistent with a slight increase in the angular anisotropy in the intermolecular potential. Lorenz et al.48 were able to deduce the preferred sense of rotation of NO following collision with Ar at ECM = 0.066 eV, by comparing images obtained with right circularly polarised (RCP) and left circularly polarised (LCP) probe laser radiation. The scattered NO molecules in these experiments were probed via the R21 branch within the A2Σ+ ← X2Π1/2 transition. Analysis of the appropriate difference images (i.e. (MR − ML)/(MR + ML)) yields a direct measure of product orientation. NO molecules in high j states displayed substantial rotational orientation. Such observations have been rationalised by assuming that some of the collision encounters scatter product into lower J states via chattering (i.e. multiple) collisions with the recoiling Ar atom. The sense of rotation of the lower J state products was found to vary with deflection angle in a complicated way, which is reproduced well by quantum calculations on the coupled cluster CCSD(T) PES for Ar-NO. This oscillatory pattern in the preferred sense of rotation is thought to arise from interference between product trajectories. The rotational alignment of NO scattered from Ar137 has also been measured from images recorded with the probe laser linearly polarised either parallel (Z) or perpendicular (X) to the detection axis. As in the photodissociation studies described earlier, analysis of the angular distribution in the normalised alignment image (MZ − MX)/(MZ + MX) can yield alignment parameters, some of which are dependent on the azimuthal deflection and are not available with other detection methods.
When a diatomic molecule collides with an atom at relatively low collision energies, such as in the Ar–NO examples above, any transferred energy can only be partitioned into translational and rotational excitation of the diatomic. Thus each rotationally resolved image of the diatomic molecule contains the projection of a single scattering sphere, with its CM velocity constrained by energy and momentum conservation. This offers a route to forming translationally ‘cold’ molecules, as Chandler and coworkers168 have demonstrated for the specific case of the J = 7.5 products in the crossed-beam inelastic scattering of Ar atoms with NO(X2Π1/2, v = 0, J = 0.5) molecules. After due attention to reactant beam preparation and appropriate selection of collision energy, these authors succeeded in demonstrating J = 7.5 products with a LAB-frame rms velocity ≤14.8 ± 1.1 m s−1, corresponding to a translational temperature of just 406 ± 28 mK.168
When a diatomic molecule collides with another molecular species, several internal modes of the (unobserved) partner may be populated in association with any particular rotational state of the diatomic. Each image is thus the projection of a number of scattering spheres. In the study of the scattering of HCl from N2 and CH4 by Wade et al.,141 for example, the image resolution was not sufficient to extract pair-correlated DCSs. Nonetheless, it was clear that HCl products that had experienced a large change in J (i.e. high ΔJ) correlated with high ΔJ N2 or CH4 partners, while low ΔJ HCl products correlated with low ΔJ in the collider. It was also clear from the images that the DCSs for HCl–N2 and HCl–CH4 collisions were different from one another, and from those of HCl scattered from Ne, Ar and Kr. The DCSs of the latter were compared to those predicted from the potential energy surface H6(3,4,0) for HCl-Ar by Hutson,169 scaled to account for mass and velocity differences, and very good agreement was found.141
Measuring the inelastic scattering of HCl is more difficult than NO, mainly because detection of the former requires use of a 2 + 1 REMPI scheme rather than the 1 + 1 REMPI process used for monitoring NO. Much higher photon densities are required to drive a 2 + 1 REMPI transition; these are achieved by focussing the laser more tightly. The smaller size of the probed volume means that less product is detected, resulting in lower signal-to-noise. The H(D) atom is the only species, other than NO, that traditionally has been detected by 1 + 1 REMPI in ion-imaging studies.
4.2 Reactive scattering processes
Two colour 1 + 1′ REMPI schemes (via Lyman-α excitation to the 2p state) were used to detect D atom products in two early ion imaging studies of reactive processes: H + D2 → HD + D159 and O(1D) + D2 → OD + D.144 In the former experiment, the precursor (HI) and reagent (D2) molecular beams were parallel, with the latter directed along the TOF axis (Fig. 13). The precursor beam was photolysed at 266 nm, yielding two sub-groups of H atom velocities that can intersect the D2 molecular beam. Slower H atoms are produced in conjunction with I* atoms, via a perpendicular transition (yielding a recoil distribution that varies as cos2θ), whereas faster H atoms are produced in combination with I atoms via a parallel transition (sin2θ distribution). Thus two collision energies (ECM ∼1.29 and 0.54 eV) could be explored, simply by varying the polarisation axis of the linearly polarised photolysis light. The distance and timing between the photolysis and probe lasers were set so as to ensure that the relative velocity of the reaction was parallel to the imaging plane, and the inverse Abel transform was applied to the resulting image to obtain a 2-D section through the D atom velocity distribution. This early ion-imaging experiment did not use velocity mapping and distinct rings in the D atom velocity distribution associated with the individual quantum states of the HD co-product were not resolved. Nonetheless, the images clearly revealed that fast D atoms (formed in association with HD co-products with low internal excitation) were forward scattered, whereas the scattering of slower D atoms shifted to wider angles. The results at the lower collision energy were in good agreement with quasi-classical trajectory calculations based on the LSTH (Liu–Siegbahn–Truhlar–Horowitz) PES, but the agreement at the higher collision energy was less impressive.As already commented, relatively few species can be detected by 1 + 1 REMPI. To circumvent this problem, Suits and coworkers have investigated H atom abstraction from alkanes (by reaction with O atoms)155–157 and from alcohols (by Cl atoms)145,146 by so-called universal detection of the alkyl and hydroxyalkyl co-products. Universal detection in this case involved direct one-photon ionisation of the radical by 157 nm (7.9 eV) photons from a F2 excimer laser. This photon energy is sufficient to ionise radicals created by removal of a secondary or tertiary H atom, but not of a primary H atom (though this limitation could be circumvented by using VUV radiation from a tuneable synchrotron source). Cl(2P3/2), O(3P) and O(1D) reagents were generated by 193 nm photolysis of (ClCO)2, SO2 and N2O, respectively. For the Cl atom plus alcohol (methanol, ethanol and isopropanol) reactions, the (ClCO)2 and alcohol molecular beams were directed at 90° to one other, and the imaging detector was parallel to the plane of the molecular beams. In contrast, the O atom reaction studies employed a geometry analogous to that shown in Fig. 13. SO2 photolysis generates O(3P) atoms with a wide range of recoil velocities. The average collision energy could thus be tuned, simply by varying the time delay between the photolysis and detection lasers. Detection of residual product from earlier collisions (with faster O(3P) atoms) that was still in the detection volume when the probe laser pulse arrived caused only modest distortions, which could be simulated. For both experimental set-ups, the forward scattering region was obscured by photodissociation from the parent alcohol or alkane. This is a major disadvantage of universal detection, which severely limits the reliability of the data in the forward direction. Image analysis revealed predominately sideways/backwards scattering for Cl with methanol at ECM = 0.38 eV,145,146 backwards scattering for Cl with ethanol at ECM = 0.26 eV with increasing sideways scattering at higher ECM (0.42 eV),146 and backwards scattering for Cl with isopropanol at ECM = 0.52 eV.146 The images for O(3P) reactions with cyclohexane, n-butane and isobutene155,156 revealed backwards scattering, which broadened as ECM increased from 0.20 to 0.64 eV. The reaction of O(1D) with n-pentane at ECM = 0.6 eV showed isotropic/forward scattering, whereas O(3P) with n-pentane at a similar collision energy showed sideways/backwards scattering.157 The contrasting angular distributions reflect a change in reaction mechanism. The reactions of ground-state Cl atoms with alcohols, and of O(3P) atoms with alkanes, proceed via direct mechanisms with a small barrier, whereas the O(1D) + n-pentane reaction is generally assumed to proceed via a long-lived intermediate. That said, it has been suggested that the highly vibrationally excited OH products observed in O(1D) + alkene reactions may arise via a direct mechanism,170 as has been shown for the simpler (but related) O(1D) + H2 reaction.171,172 The images also revealed that most of the reaction exoergicity (∼60–70%, rising to ∼80% in the case of O(1D) + n-pentane) was partitioned into internal modes of the alkyl (or hydroxyalkyl) radical products.155–157 In reaching these conclusions, the authors had to assume that the radical ionisation efficiency is independent of its degree of internal excitation. Obviously, if radicals with excited internal modes are ionised preferentially, then the fractions of the available energy partitioned into internal energy quoted above will be overestimates. Moreover, the observed angular distributions could be biased due to unequal sampling of the trajectories in the reactive scattering process. Such a biasing has been suggested to account for the discrepancy between the results obtained using universal ionisation and Doppler-resolved polarised LIF of the OH products from the reaction of O(3P) atoms with isobutane and cyclohexane.173
Several limitations of universal detection schemes involving direct one-photon ionisation have been mentioned above: the lack of state-to-state information, background due to parent photodissociation, and possible biasing in favour of products with greater levels of internal excitation. Its main advantage is that it allows study of a very wide variety of reactions, with high sensitivity. This should be contrasted with the various REMPI detection schemes. As already commented, relatively few species may be probed conveniently by 1 + 1 REMPI, while 2 + 1 REMPI schemes generally suffer from substantially lower detection sensitivities. Nonetheless, as Fig. 14 illustrates, 2 + 1 REMPI detection has recently been used to great effect by Liu and coworkers to detect CD3 products from the reaction of F with CD4 at ECM = 0.233 eV.147 Their apparatus comprised two rotatable double skimmed molecular beams and a fixed, gated detector—to allow slice imaging. Different vibrational states of the CD3 products formed in the intersection region of the two molecular beams were ionised by 2 + 1 REMPI at the peak of the associated Q branches, giving partial rotational state resolution. As Fig. 14(a) shows, the image of the CD3(v = 0) products showed well-resolved rings, reflecting the formation of different vibrational states of the coincident DF product. The narrow widths of the rings reveal that the partner DF products are formed with little rotational excitation. A density-to-flux transformation (and corrections for imperfect time slicing) give the pair-correlated DCSs directly (see Fig. 14b), which are very reminiscent of the vibrational level resolved DCSs for the reaction of F atoms with H2 or D2 obtained using traditional crossed molecular beam and detection methods.174,175 Integration of the total signal associated with each ring yields the pair-correlated vibrational state population distributions—which show a distinct anti-correlation between the vibrational populations of the two products. The umbrella mode of the CD3 and the stretch of the DF are both expected to couple with the reaction co-ordinate, and it was suggested that ‘chattering’ of the D atom between the F and CD3 may allow energy flow between the two nascent products resulting in the observed anti-correlation. In another study by the same group, the probe laser wavelength was scanned to obtain a rotationally integrated image for each CH3 vibrational state arising in the F + CH4 reaction.78 In addition to time-gated detection of the central region of the scattering sphere, the probe laser position was scanned to generate an effective laser sheet across the interaction volume (as in Fig. 3a), thereby simplifying the density-to-flux transformation and reducing the scope for errors. The angle between the molecular beams was varied to change the collision energy, thereby enabling derivation of the excitation function (i.e. the dependence of the integral cross-section on the collision energy). This was shown to rise sharply above the threshold region, maximise at ECM ∼0.1 eV, and then decline with further increase in collision energy. Vibrational state resolution showed that, once energetically accessible, the HF(v = 3) product channel rapidly dominates but that, upon increasing ECM further, its relative importance falls relative to that of the HF(v = 2) channel. The yield of HF(v = 1) products was found to be small at all collision energies. The collision energy dependences of the images of methyl fragments from F + CH4 and CHD3 reactions have been argued as being indicative of reactive scattering resonances, observable because the excitation functions are quantum state resolved.152,153 The quality and resolution of data achieved in the best velocity map imaging experiments has now reached the point where even studies of bimolecular collision products can reveal new spectroscopic details of the probed product—as exemplified by recent imaging studies of the CD3 fragments arising in the F + CHD3 reaction.176
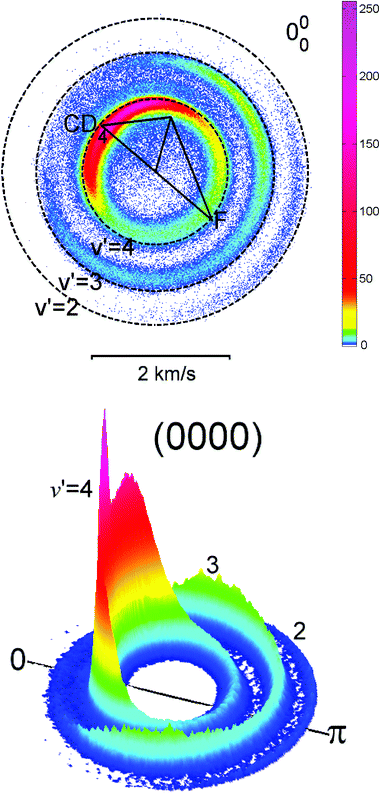 | ||
| Fig. 14 (a) Raw image of CD3(v = 0) fragments arising in the F + CD4 reaction at ECM = 0.233 eV. The successive rings correspond to the labelled vibrational states of the DF co-product. (b) the corresponding CD3 product state resolved flux-velocity contour map after application of the necessary density-to-flux corrections. (Adapted from ref. 147). | ||
This type of study has now been extended to include investigations of the reactive scattering of both OH radicals177,178 and Cl atoms179 with CH4 and/or CD4 as a function of collision energy and, in the case of the Cl + CH4 reaction, exploration of the role of low frequency distortion and bending vibrations in enhancing the reaction cross section.180 The relative reactivities of Cl and Cl* atoms were investigated. In both cases, if reagents have internal energy, either as vibrational motion of methane or spin–orbit excitation of Cl atoms, the HCl and CH3 radical products can be formed with kinetic energies higher than observed for products of the reaction of ground state reagents. Images of the velocities of the reaction products thus distinguish these various possible channels, and allow estimation of the degree of vibrational enhancement, although no evidence was found for reaction of Cl* atoms. The specific form of the angular distribution of the CH3(v = 0) + HCl(v = 1) product pair from the Cl + CH4 reaction, and its dependence on collision energy, has been proposed as another signature for a reactive resonance.181
Kitsopoulos and coworkers have obtained DCSs for forming HCl products in the reaction of Cl(2P3/2) atoms with various small organic molecules with full rotational state resolution.160–162 The Cl atoms were generated by the Cl2 photolysis at 355 nm and reaction ensued as the Cl atoms crossed the path of the central molecular beam. The high reactant number densities enabled by use of unskimmed molecular beams, in the geometry shown in Fig. 13, ensured HCl densities sufficient for 2 + 1 REMPI detection (in a number of different product rotational states). Most of the detected products were generated by scattering events outside the small volume probed by the focused probe laser. It was thus necessary to scan the time delay between the photolysis and probe lasers in order to minimise experimental bias towards forward and backward scattered product; on average, the latter take longer to reach the probed volume. In addition, the wavelength of the probe laser was scanned across the Doppler profile of the absorption line in order that all product velocity sub-groups (along the probe axis) were sampled with equal efficiency. Monte Carlo simulations indicated that the experimental data had only, at most, a weak bias towards backscattered products. Simulations also revealed that most detected HCl products were formed by collisions involving Cl atoms with laboratory frame velocities close to the line joining the foci of the photolysis and probe lasers.161 To a first approximation, the angular and speed distributions of the state-selected HCl products could be extracted directly from the experimental images by radial and angular integration, respectively. Examples of images of HCl (v = 0, J) products from the reaction of Cl atoms with n-butane,160 dimethylether,161 methanol161 and chloromethane162 are shown in Fig. 15. All of the HCl product angular distributions are broad, with significant scattering into both the forward and backward directions. However, there is a clear distinction between the images for the reaction of Cl atoms with CH3Cl (and CH3Br), and those for reaction with n-butane, dimethylether and methanol. The former show enhanced scattering in the backward direction, with little J-dependence, while in the latter cases the forward scattering component is dominant at low J but declines in relative importance with increasing rotational excitation of the HCl. In all cases, the broad scattering distributions are likely to result from direct mechanisms via a loose transition state. In the cases of the Cl + alkane (other than CH4), methanol and dimethylether reactions, the transition state lies lower in energy than the reagents, and the energy of the (low) barrier varies only weakly with the angle of approach of the reagents. The Cl + CH3Cl and CH3Br reactions have higher barriers, limiting the range of impact parameters and causing rebound dynamics to predominate. The speed distributions derived from the images indicate that, on average, ∼40% of the available energy is partitioned into product translation. Such a value is consistent with that derived for other H abstractions by reaction with Cl atoms, and implies significant excitation of the polyatomic radical product. Calculations by Rudić et al.182,183 suggest that most of this energy is partitioned as product rotation rather than vibrational excitation.
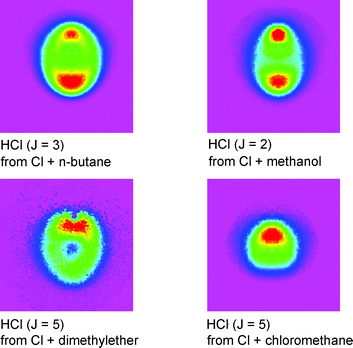 | ||
| Fig. 15 Examples of images of HCl (v = 0, J) products from the reaction of Cl atoms with: (a) n-butane, (J = 3 products, from ref. 160); (b) methanol, (J = 2, ref. 161); (c) dimethyl ether, (J = 5, ref. 161); and (d) chloromethane, (J = 5, ref. 162). The Cl + dimethyl ether image in (c) has a small section missing from the upper (backward scattering) part due to imperfect subtraction of strong background due to photoionization and fragmentation of dimethyl ether. | ||
Thus far, we have concentrated on reaction product imaging studies that have employed crossed molecular beams. In the early 1990s, Zare and coworkers pioneered a single beam technique in which a non-reactive precursor and reagent were co-expanded through a single nozzle and reaction was initiated by photolysis of the precursor.184,185 In fact, the first reported ion-imaging study of a reactive collision was for the H + HI → H2 + I reaction;163 the same laser pulse was used both to photolyse HI and for 2 + 1 REMPI detection of H2 product. Single beam experiments have several disadvantages when compared with crossed molecular beam experiments. Firstly, reaction product may accumulate behind the nozzle as a result of pre-reaction in the precursor/reagent mixture—giving rise to a significant background signal. Further, the requirement that the precursor exhibit very low reactivity imposes quite severe constraints on the range of reactions that can be studied. Fortunately, the vibrational state population distribution of the H2 products from the H + HI reaction peaks at v = 1, and the vibrationally excited H2 could be detected with minimal background from thermal H2 in the molecular beam.163 To widen the scope of reactions that can be studied, these problems can be obviated by using two closely spaced nozzles, with outputs only a couple of mm apart, so that the molecular beams overlap within a very short distance from the nozzles.186 A second disadvantage of single beam studies is that the qualitative form of the DCS is not immediately apparent from the raw image. Furthermore, data analysis requires some knowledge of the internal energy distribution of all of the products. If the unobserved species is an atom, carrying only translational energy dictated by the conservation of momentum, the data can then be analysed exactly using the law of cosines184 (leading to the acronym photoloc187 for this kind of experiment). In general, photoloc analyses are only applicable if the detected species have a CM speed distribution described by a δ-function, which will only occur if the internal excitation within the unobserved species is a δ-function. In the first single-beam studies of polyatomic reactions, the internal excitation of the unobserved radical species was presumed to be zero, on the basis of the velocity dependence of the anisotropy parameter—thereby justifying use of a photoloc based analysis. The first example of this technique in combination with ion-imaging detection involved study of H abstraction from ethane by photolytically generated Cl(2P3/2) atoms, with 2 + 1 REMPI detection of the HCl(v = 0, J = 1) products.164 The subsequent photoloc analysis suggested broad scattering, but the assumption of zero internal excitation of the C2H5 co-product has since been shown to be erroneous. As mentioned above, a number of subsequent ion-imaging studies have shown the alkyl moiety is more than a mere spectator and that it carries away some considerable fraction of the available energy as internal excitation. Brouard et al. have recently applied a basis set analysis method,188 originally developed for analysing Doppler-resolved LIF lineshapes,189 to obtain the joint distribution in CM scattering angle and fractional kinetic energy release from single beam data. Application of these methods to data obtained from the reaction of Cl(2P3/2) atoms with ethane165 and n-butane166 at ECM ∼0.24 and 0.32 eV, respectively, suggests that ∼22% and ∼30% of the available energy is deposited into the internal modes of the ethyl and butyl, respectively—results in good accord with crossed molecular beam data for the same systems. In the case of n-butane, the channels for abstraction of primary and secondary H atoms were distinguished—thereby leading to an estimation of the contribution of each channel to each rotational state of the HCl product. The basis set analysis method has the disadvantage of being less transparent than the more direct analysis of crossed molecular beam data but, on the upside, there is less scope for bias in the detection of different velocities, provided the pump–probe delay times are sufficiently short to avoid fly-out. Thus the transformation between density and flux is less prone to error.
5. Conclusions and future prospects
The foregoing is intended to provide a reasonably complete overview of a selected sub-set of the many applications whereby imaging methods have been used to probe the dynamics of gas phase chemical reactions. Our survey has been limited to applications involving ion detection; to survey the many complementary studies focussing on electron imaging would require as much space again. Thus we have not discussed the benefits and additional insights that can be obtained by adding an imaging capability to the traditional REMPI-photoelectron spectroscopy (PES) experiment,298 or the application of imaging methods in studies of electron photodetachment from anions190 and anion clusters.191,192 We have not described any of the beautiful new insights into photoionisation dynamics that have resulted from imaging very slow photoelectrons released in threshold ionisation processes.193–195 Similarly, we have glossed over the many new insights that ultra-fast photoelectron imaging methods are providing to our understanding of radiationless transitions (isomerisations, internal conversion and/or intersystem crossing processes) within molecules,196–198 and not done justice to the pioneering ultra-fast, time-resolved studies of Hayden and co-workers in which parent and fragment ions, and electrons, arising in a REMPI process were imaged, in coincidence.37,199 Given the increasing availability of fast time and position sensitive (delay line) detectors, we envisage many extensions of this type of coincidence study.Instead, we have concentrated on describing some of the many new insights into chemical reaction dynamics afforded by ion imaging methods. Clearly, the technique has come a very long way since the time of Chandler and Houston’s landmark study.28 Image resolution and fidelity have improved enormously and, as this Review illustrates, ion imaging methods have now been applied to an impressively diverse range of gas phase processes—both unimolecular and bimolecular. For completeness, we also draw attention to pioneering imaging studies of the products of ion—surface scattering processes.200 Experimental imaging methods continue to develop, both in the achieved resolution and in their range of application. The ultimate resolution should be approachable by solving the ion-recoil problem, reducing space charge effects and circumventing the problem of insufficient rotational cooling using, for example, electrostatic state-selection or double resonance excitation. Slicing techniques are offering new solutions to probing systems that lack cylindrical symmetry—as illustrated, for example, by the recent study of the wavelength and recoil velocity dependence of the orbital orientation of the O(1D) atoms arising in the near UV photolysis of O3.201 Pulsed-field slicing, in particular, opens the way to studies of reactions involving charged species, and introduces a t = 0 for TOF mass selection in cases where pulsed laser ionization is not used. Photodissociation studies of progressively larger molecules will have to address not just the resolution issue, but also the problem of rapid intramolecular relaxation to the electronic ground state—as pointed out by Lee and coworkers,202 who also demonstrated the utility of multiple mass imaging using a radial cylindrical energy analyzer. Dissociation from the highly vibrationally excited ground state molecules that generally arise in such intramolecular relaxation processes can be much slower than the 0–20 ns dissociation-ionization time delay typically used in most current imaging studies. With some effort, time delays approaching 10 μs should be possible using VMI, and simultaneous two-mass, 2-D imaging may be achievable using a double-exposure CCD camera. Reports describing the extension of the Rydberg tagging method to species other than H atoms, and its combination with VMI, are starting to appear.203,204 Clearly, both the technology and the range of possible applications for imaging the dynamics of gas phase reactions have some way yet to go!
Acknowledgements
The participating groups are grateful to the Commission of the European Communities for funding via the IHP Network HPRN-CT-2002-00183 (PICNIC), to Prof. B. J. Whitaker (Leeds) for his support and encouragement as Coordinator of this Network, and to Professor Kopin Liu for providing Fig. 14. The Bristol authors also wish to acknowledge financial support from EPSRC (Portfolio Partnership award LASER), the EU (Marie-Curie Fellowship MEIF-CT-2003-500999 (PHOSPHOR) to NHN), and the important contributions made to the local imaging programme by, among others, M. Beckert, R. N. Dixon, C. Murray, K. N. Rosser, A. D. Webb, E. Wrede and E. R. Wouters. TNK is grateful to the Alexander von Humboldt Foundation, for support through the Friedrich von Bessel Award. DHP also wishes to acknowledge the Dutch National Science Foundation—FOM programme “Molecular Atmospheric Processes” for support, and contributions from the Nijmegen imaging group members including M. Coroiu, A. Eppink, D. Radenovic, A. van Roij, K. Vidma, A. Zawadska and Y. Zhang.References
- G. Porter, Proc. R. Soc. London, Ser. A, 1950, 9, 60.
- F. J. Lipscomb, R. G. W. Norrish and B. A. Thrush, Proc. R. Soc. London, Ser. A, 1956, 233, 455 Search PubMed.
- J. K. Cashion and J. C. Polanyi, J. Chem. Phys., 1959, 30, 317 CAS.
- N. Basco and R. G. W. Norrish, Nature, 1961, 189, 455 CAS.
- R. N. Zare and P. J. Dagdigian, Science, 1974, 185, 739 CrossRef CAS.
- J. L. Kinsey, Annu. Rev. Phys. Chem., 1977, 28, 349 CrossRef CAS.
- J. J. Valentini, Spectrometric Techniques, Academic, London, 1985 Search PubMed.
- M. N. R. Ashfold and J. D. Howe, Annu. Rev. Phys. Chem., 1994, 45, 57 CrossRef CAS , and references therein.
- R. W. Diesen, J. C. Wahr and S. E. Adler, J. Chem. Phys., 1969, 50, 3635 CrossRef CAS.
- S. J. Riley and K. R. Wilson, Faraday Discuss. Chem. Soc., 1972, 53, 132 RSC.
- A. M. Wodtke and Y. T. Lee, in Molecular Photodissociation Dynamics, eds. M. N. R. Ashfold and J. E. Baggott, Royal Society of Chemistry, London, 1987, pp. 31–59 Search PubMed.
- R. Bersohn, in Molecular Photodissociation Dynamics, eds. M. N. R. Ashfold and J. E. Baggott, Royal Society of Chemistry, London, 1987, pp. 1–30 Search PubMed.
- M. N. R. Ashfold, I. R. Lambert, D. H. Mordaunt, G. P. Morley and C. M. Western, J. Phys. Chem., 1992, 96, 2938 CrossRef CAS.
- L. J. Butler and D. M. Neumark, J. Phys. Chem., 1996, 100, 12801 CrossRef CAS.
- G. Hancock, P. J. Pearson, G. A. D. Ritchie and D. F. Tibbetts, Phys. Chem. Chem. Phys., 2003, 5, 5386 RSC , and references therein.
- A. J. Orr-Ewing and R. N. Zare, Annu. Rev. Phys. Chem., 1994, 45, 315 CrossRef CAS.
- L. Schnieder, W. Meier, K. H. Welge, M. N. R. Ashfold and C. M. Western, J. Chem. Phys., 1990, 92, 7027 CrossRef CAS.
- M. N. R. Ashfold, D. H. Mordaunt and S. H. S. Wilson, in Photochemistry, ed. D. C. Neckers, D. H. Volman and G. von Bünau, Wiley, New York, 1996, 21, 217 Search PubMed.
- E. Wrede, L. Schnieder, K. H. Welge, F. J. Aoiz, L. Banares, J. F. Castillo, B. Martinez-Haya and V. J. Herrero, J. Chem. Phys., 1999, 110, 9971 CrossRef CAS.
- R. N. Dixon, D. W. Hwang, X. F. Yang, S. Harich, J. J. Lin and X. Yang, Science, 1999, 285, 1249 CrossRef CAS.
- F. J. Aoiz, L. Banares, J. F. Castillo, V. J. Herrero, B. Martinez-Haya, P. Honvault, J. M. Launay, X. Liu, J. J. Lin, S. A. Harich, C. C. Wang and X. Yang, J. Chem. Phys., 2002, 116, 10692 CrossRef CAS.
- J. A. Mueller, S. A. Rogers and P. L. Houston, J. Phys. Chem. A, 1998, 102, 9666 CrossRef CAS.
- C. Lin, M. F. Witinski and H. F. Davis, J. Chem. Phys., 2003, 119, 251 CrossRef CAS.
- Z. Xu, B. Koplitz and C. Wittig, J. Chem. Phys., 1989, 90, 2692 CrossRef CAS.
- R. N. Dixon, J. Chem. Phys., 1986, 85, 1866 CrossRef CAS.
- G. E. Hall and P. L. Houston, Annu. Rev. Phys. Chem., 1989, 40, 375 CrossRef CAS.
- M. Brouard, I. Burak, D. Minayev, P. O’Keeffe, C. Vallance, F. J. Aoiz, L. Banares, J. F. Castillo, D. H. Zhang and M. A. Collins, J. Chem. Phys., 2003, 118, 1162 CrossRef CAS , and references therein.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1986, 87, 1445.
- A. J. R. Heck and D. W. Chandler, Annu. Rev. Phys. Chem., 1995, 46, 335 CrossRef CAS.
- P. L. Houston, Acc. Chem. Res., 1995, 28, 453 CrossRef CAS.
- Imaging in Chemical Dynamics, eds. A. G. Suits and R. E. Continetti, American Chemical Society, Washington, DC, 2001 Search PubMed.
- Imaging in Molecular Dynamics, Technology and Applications, ed. B. J. Whitaker, Cambridge University Press, Cambridge, 2003 Search PubMed.
- C. Vallance, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2591 CrossRef CAS.
- D. H. Parker and A. T. J. B. Eppink, ch. 2 in ref. 32.
- A. T. J. B. Eppink, S.-M. Wu and B. J. Whitaker, ch. 3 in ref. 32.
- H. Helm, D. P. Debruijn and J. Los, Phys. Rev. Lett., 1984, 53, 1642 CrossRef CAS.
- J. A. Davies, R. E. Continetti, D. W. Chandler and C. C. Hayden, Phys. Rev. Lett., 2000, 84, 5983 CrossRef CAS.
- A. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477 CrossRef CAS.
- C. Bordas, F. Paulig, H. Helm and D. L. Huestis, Rev. Sci. Instrum., 1997, 67, 2257.
- M. Beckert, S. J. Greaves and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2003, 5, 308 RSC.
- P. W. Harland, B. R. Cameron, M. Bart and M. O. Pearce, Rev. Sci. Instrum., 2002, 73, 2398 CrossRef CAS.
- R. S. E. Mabbs and A. Sanov, Analyst, 2003, 128, 765 RSC.
- A. V. Davis, R. Wester, A. E. Bragg and D. M. Neumark, J. Chem. Phys., 2003, 118, 999 CrossRef CAS.
- A. L. Sanov and W. C. Lineberger, Phys. Chem. Chem. Phys., 2004, 6, 2018 RSC.
- R. F. Delmdahl, B. L. G. Bakker and D. H. Parker, J. Chem. Phys., 2000, 113, 7728 CrossRef CAS.
- E. L. Reichert, G. Thurau and J. C. Weisshaar, J. Chem. Phys., 2002, 117, 653 CrossRef CAS.
- D. H. Parker, in Photoionization and Photodetachment, Advanced Series in Physical Chemistry, World Scientific Publishing Co. Ltd., Singapore, 2000, vol. 10A and 10B Search PubMed.
- K. T. Lorenz, D. W. Chandler, J. W. Barr, W. W. Chen, G. L. Barnes and J. I. Cline, Science, 2001, 293, 2063 CrossRef CAS.
- M. Ahmed, D. S. Peterka and A. G. Suits, Abstr. Pap. Am. Chem. Soc., 1998, 215, U164.
- H. S. Chung, B. S. Zhao, S. H. Lee, S. G. Hwang, K. C. Cho, S. H. Shim, S. M. Lim, W. K. Kang and D. S. Chung, J. Chem. Phys., 2001, 114, 8293 CrossRef CAS.
- S. Wolf and H. Helm, Phys. Rev. A, 1997, 56, R4385 CrossRef CAS.
- N. Yonekura, C. Gebauer, H. Kohguchi and T. Suzuki, Rev. Sci. Instrum., 1999, 70, 3265 CrossRef CAS.
- Y. Yamakita, S. R. Procter, A. L. Goodgame, T. P. Softley and F. Merkt, J. Chem. Phys., 2004, 121, 1419 CrossRef CAS.
- A. I. Chichinin, T. S. Einfeld, C. M. Maul and K. H. Gericke, Chem. Phys. Lett., 2004, 390, 50 CrossRef CAS.
- E. Wrede, S. Laubach, S. Schulenberg, A. Brown, A. J. Orr-Ewing and M. N. R. Ashfold, J. Chem. Phys., 2001, 114, 2629 CrossRef CAS.
- E. Wrede, E. R. Wouters, M. Beckert, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 2002, 116, 6064 CrossRef CAS.
- A. Eppink and D. H. Parker, J. Chem. Phys., 1999, 110, 832 CrossRef CAS.
- D. A. Chestakov, S. M. Wu, G. R. Wu, D. H. Parker, A. T. J. B. Eppink and T. N. Kitsopoulos, J. Phys. Chem. A, 2004, 108, 8100 CrossRef CAS.
- D. H. Parker, R. F. Delmdahl, B. Bakker and H. P. Loock, J. Chin. Chem. Soc., 2001, 48, 327 CAS.
- R. L. Toomes, P. C. Samartzis, T. P. Rakitzis and T. N. Kitsopoulos, Chem. Phys., 2004, 301, 209 CrossRef CAS.
- A. E. Pomerantz and R. N. Zare, Chem. Phys. Lett., 2003, 370, 515 CrossRef CAS.
- P. M. Regan, D. Ascenzi, E. Wrede, P. A. Cook, M. N. R. Ashfold and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2000, 2, 5364 RSC.
- O. Kaufmann, A. Ekers, K. Bergmann, N. Bezuglov, K. Miculis, M. Auzinsh and W. Meyer, J. Chem. Phys., 2003, 119, 3174 CrossRef CAS.
- H. L. Offerhaus, C. Nicole, F. Lepine, C. Bordas, F. Rosca-Pruna and M. J. J. Vrakking, Rev. Sci. Instrum., 2001, 72, 3245 CrossRef CAS.
- M. J. J. Vrakking, private communication.
- S. D. Carpenter, C. P. Schick and P. M. Weber, Rev. Sci. Instrum., 1999, 70, 2262 CrossRef CAS.
- D. Townsend, M. P. Minitti and A. G. Suits, Rev. Sci. Instrum., 2003, 74, 2530 CrossRef CAS.
- J. J. Lin, J. G. Zhou, W. C. Shiu and K. P. Liu, Rev. Sci. Instrum., 2003, 74, 2495 CrossRef CAS.
- B. Baguenard, J. B. Wills, F. Pagliarulo, F. Lepine, B. Climen, M. Barbaire, C. Clavier, M. A. Lebeault and C. Bordas, Rev. Sci. Instrum., 2004, 75, 324 CrossRef CAS.
- B. Y. Chang, R. C. Hoetzlein, J. A. Mueller, J. D. Geiser and P. L. Houston, Rev. Sci. Instrum., 1998, 69, 1665 CrossRef CAS.
- M. J. Cooper, P. J. Jackson, L. J. Rogers, A. J. Orr-Ewing, M. N. R. Ashfold and B. J. Whitaker, J. Chem. Phys., 1998, 109, 4367 CrossRef CAS.
- T. P. Rakitzis, Chem. Phys. Lett., 2001, 342, 121 CrossRef.
- V. Dribinski, A. Ossadtchi, V. A. Mandelshtam and H. Reisler, Rev. Sci. Instrum., 2002, 73, 2634 CrossRef CAS.
- M. J. J. Vrakking, Rev. Sci. Instrum., 2001, 72, 4084 CrossRef CAS.
- S. Manzhos and H. P. Loock, Comput. Phys. Commun., 2003, 154, 76 CrossRef CAS.
- M. J. Bass, M. Brouard, A. P. Clark and C. Vallance, J. Chem. Phys., 2002, 117, 8723 CrossRef CAS.
- S. Manzhos and H. P. Loock, Rev. Sci. Instrum., 2004, 75, 2435 CrossRef CAS.
- W. Shiu, J. J. Lin, K. Liu, M. Wu and D. H. Parker, J. Chem. Phys., 2004, 120, 117 CrossRef CAS.
- C. R. Gebhardt, T. P. Rakitzis, P. C. Samartzis, V. Ladopoulos and T. N. Kitsopoulos, Rev. Sci. Instrum., 2001, 72, 3848 CrossRef.
- L. Dinu, A. Eppink, F. Rosca-Pruna, H. L. Offerhaus, W. J. van der Zande and M. J. J. Vrakking, Rev. Sci. Instrum., 2002, 73, 4206 CrossRef CAS.
- K. Tonokura and T. Suzuki, T. Chem. Phys. Lett., 1994, 224, 1 Search PubMed.
- T. Kinugawa and T. Arikawa, J. Chem. Phys., 1992, 96, 4801 CrossRef CAS.
- M. A. Young, J. Chem. Phys., 1995, 102, 7925 CrossRef CAS , and references therein.
- T. Shibata and T. Suzuki, Chem. Phys. Lett., 1996, 262, 115 CrossRef CAS.
- T. Shibata, H. Y. Li, H. Katayanagi and T. Suzuki, J. Phys. Chem. A, 1998, 102, 3643 CrossRef CAS.
- N. Hemmi and A. G. Suits, J. Phys. Chem. A, 1997, 101, 6633 CrossRef CAS.
- M. Ahmed, D. Blunt, D. Chen and A. G. Suits, J. Chem. Phys., 1997, 106, 7617 CrossRef CAS.
- D. S. Peterka, M. Ahmed, C. Y. Ng and A. G. Suits, Chem. Phys. Lett., 1999, 312, 108 CrossRef CAS.
- W. Li, L. Poisson, D. S. Peterka, M. Ahmed, R. R. Lucchese and A. G. Suits, Chem. Phys. Lett., 2003, 374, 334 CrossRef CAS.
- J. R. Greene, J. S. Francisco, J. H. Huang, D. D. Xu and W. M. Jackson, J. Chem. Phys., 2004, 121, 5868 CrossRef CAS.
- J. J. Larsen, N. J. Morkbak, J. Olesen, N. Bjerre, M. Machholm, S. R. Keiding and H. Stapelfeldt, J. Chem. Phys., 1998, 109, 8857 CrossRef.
- P. C. Samartzis, B. L. G. Bakker, D. H. Parker and T. N. Kitsopoulos, J. Phys. Chem. A, 1999, 103, 6106 CrossRef CAS.
- B. L. G. Bakker, D. H. Parker, P. C. Samartzis and T. N. Kitsopoulos, J. Chem. Phys., 2000, 112, 5654 CrossRef CAS.
- B. L. G. Bakker, D. H. Parker, P. C. Samartzis and T. N. Kitsopoulos, J. Chem. Phys., 2000, 113, 9044 CrossRef CAS.
- W. G. Roeterdink and M. H. M. Janssen, Chem. Phys. Lett., 2001, 345, 72 CrossRef CAS.
- S. Unny, Y. Du, L. C. Zhu, R. J. Gordon, A. Sugita, M. Kawasaki, Y. Matsumi and T. Seideman, Phys. Rev. Lett., 2001, 86, 2245 CrossRef CAS.
- H. Yamada, N. Taniguchi, M. Kawasaki, Y. Matsumi and R. J. Gordon, J. Chem. Phys., 2002, 117, 1130 CrossRef CAS.
- W. G. Roeterdink and M. H. M. Janssen, J. Chem. Phys., 2002, 117, 6500 CrossRef CAS.
- W. G. Roeterdink and M. H. M. Janssen, Phys. Chem. Chem. Phys., 2002, 4, 601 RSC.
- A. T. J. B. Eppink, B. J. Whitaker, E. Gloaguen, B. Soep, A. M. Coriou and D. H. Parker, J. Chem. Phys., 2004, 121, 7776 CrossRef CAS.
- J. A. Davies, J. E. LeClaire, R. E. Continetti and C. C. Hayden, J. Chem. Phys., 1999, 111, 1 CrossRef.
- K. Suto, Y. Sato, C. L. Reed, V. Skorokhodov, Y. Matsumi and M. Kawasaki, J. Phys. Chem. A, 1997, 101, 1222 CrossRef CAS.
- M. Ahmed, D. S. Peterka, P. Regan, X. H. Liu and A. G. Suits, Chem. Phys. Lett., 2001, 339, 203 CrossRef CAS.
- X. H. Liu, R. L. Gross and A. G. Suits, Science, 2001, 294, 2527 CrossRef CAS.
- W. Li, R. R. Lucchese, A. A. Doyuran, Z. L. Wu, H. Loos, G. E. Hall and A. G. Suits, Phys. Rev. Lett., 2004, 92, 083002 CrossRef.
- L. M. Yoder, J. R. Barker, K. T. Lorenz and D. W. Chandler, Chem. Phys. Lett., 1999, 302, 602 CrossRef CAS.
- R. K. Sampson and W. D. Lawrance, Aust. J. Chem., 2003, 56, 275 CrossRef CAS.
- S. M. Bellm and W. D. Lawrance, J. Chem. Phys., 2003, 118, 2581 CrossRef CAS.
- B. F. Parsons and D. W. Chandler, J. Phys. Chem. A, 2003, 107, 10544 CrossRef CAS.
- K. V. Vidma, A. V. Baklanov, E. B. Khvorostov, V. N. Ishchenko, S. A. Kochubei, A. T. J. B. Eppink, D. A. Chestakov and D. H. Parker, J. Chem. Phys., 2005, 122, 204301 CrossRef.
- J. H. Huang, D. D. Xu, J. S. Francisco and W. M. Jackson, J. Chem. Phys., 2003, 118, 3083 CrossRef CAS.
- N. Hansen, A. M. Wodtke, A. V. Komissarov, K. Morokuma and M. C. Heaven, J. Chem. Phys., 2003, 118, 10485 CrossRef CAS.
- O. P. J. Vieuxmaire, M. G. D. Nix, J. A. J. Fitzpatrick, M. Beckert, R. N. Dixon and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2004, 6, 543 RSC.
- N. H. Nahler, O. P. J. Vieuxmaire, J. R. Jones, M. N. R. Ashfold, A. T. J. B. Eppink, A. M. Coriou and D. H. Parker, J. Phys. Chem. A, 2004, 108, 8077 CrossRef CAS.
- O. P. J. Vieuxmaire, N. H. Nahler, J. R. Jones, R. N. Dixon and M. N. R. Ashfold, Mol. Phys., 2005, 103, 2437 CrossRef CAS.
- C. Chang, C.-Y. Luo and K. Liu, J. Phys. Chem. A, 2005, 109, 1022 CrossRef CAS.
- M. H. Kim, B. D. Leskiw and A. G. Suits, J. Phys. Chem. A Search PubMed , in press.
- F. Aguirre and S. T. Pratt, J. Chem. Phys., 2003, 118, 6318 CrossRef CAS.
- F. Aguirre and S. T. Pratt, J. Chem. Phys., 2003, 118, 9467.
- J. Tellinghuisen, J. Phys. Chem. A, 2003, 107, 753 CrossRef CAS.
- S. Manzhos, C. Romanescu, H. P. Loock and J. G. Underwood, J. Chem. Phys., 2004, 121, 11802 CrossRef CAS.
- R. N. Dixon, J. Chem. Phys., 2005, 122, 194302 CrossRef.
- B. R. Johnson, C. Kittrell, P. B. Kelly and J. L. Kinsey, J. Phys. Chem., 1996, 100, 7743 CrossRef CAS.
- R. S. Mullikan, J. Chem. Phys., 1940, 8, 382 CrossRef CAS.
- R. Ogorzalek Loo, H. P. Haerri, G. E. Hall and P. L. Houston, J. Chem. Phys., 1989, 90, 4222 CrossRef.
- E. R. Wouters, M. Ahmed, D. S. Peterka, A. S. Bracker, A. G. Suits and O. S. Vasyutinskii, in ref. 31, pp. 238–284, and references therein.
- T. P. Rakitzis, G. E. Hall, M. L. Costen and R. N. Zare, J. Chem. Phys., 1999, 111, 8751 CrossRef CAS.
- A. J. Orr-Ewing and O. S. Vasyutinskii, 2005, unpublished results.
- G. G. Balint-Kurti, A. J. Orr-Ewing, A. Brown, J. A. Beswick and O. S. Vasyutinskii, J. Chem. Phys., 2002, 116, 10760 CrossRef CAS.
- A. Brown, G. G. Balint-Kurti and O. S. Vasyutinskii, J. Phys. Chem. A, 2004, 108, 7790 CrossRef CAS.
- O. P. J. Vieuxmaire, N. H. Nahler, J. R. Jones, M. N. R. Ashfold and R. N. Dixon, to be published.
- A. G. Suits, L. S. Bontuyan, P. L. Houston and B. J. Whitaker, J. Chem. Phys., 1992, 96, 8618 CrossRef CAS.
- L. S. Bontuyan, A. G. Suits, P. L. Houston and B. J. Whitaker, J. Phys. Chem., 1993, 97, 6342 CrossRef CAS.
- H. Kohguchi, T. Suzuki and M. H. Alexander, Science, 2001, 294, 832 CrossRef CAS.
- M. S. Elioff and D. W. Chandler, J. Chem. Phys., 2002, 117, 6455 CrossRef CAS.
- A. A. Dixit, P. J. Pisano and P. L. Houston, J. Phys. Chem. A, 2001, 105, 11165 CrossRef CAS.
- J. I. Cline, K. T. Lorenz, E. A. Wade, J. W. Barr and D. W. Chandler, J. Chem. Phys., 2001, 115, 6277 CrossRef CAS.
- E. A. Wade, K. T. Lorenz, D. W. Chandler, J. W. Barr, G. L. Barnes and J. I. Cline, Chem. Phys., 2004, 301, 261 CrossRef CAS.
- M. S. Westley, K. T. Lorenz, D. W. Chandler and P. L. Houston, J. Chem. Phys., 2001, 114, 2669 CrossRef CAS.
- K. T. Lorenz, M. S. Westley and D. W. Chandler, Phys. Chem. Chem. Phys., 2000, 2, 481 RSC.
- E. A. Wade, K. T. Lorenz, J. L. Springfield and D. W. Chandler, J. Phys. Chem. A, 2003, 107, 4976 CrossRef CAS.
- K. T. Lorenz, D. W. Chandler and G. C. McBane, J. Phys. Chem. A, 2002, 106, 1144 CrossRef CAS.
- B. F. Parsons and D. W. Chandler, J. Chem. Phys., 2005, 122, 174306 CrossRef.
- M. Ahmed, D. S. Peterka and A. G. Suits, Chem. Phys. Lett., 1999, 301, 372 CrossRef CAS.
- M. Ahmed, D. S. Peterka and A. G. Suits, Chem. Phys. Lett., 2000, 317, 264 CrossRef CAS.
- M. Ahmed, D. S. Peterka and A. G. Suits, Phys. Chem. Chem. Phys., 2000, 2, 861 RSC.
- J. J. Lin, J. Zhou, W. Shiu and K. Liu, Science, 2003, 300, 966 CrossRef CAS.
- J. G. Zhou, J. J. Lin, W. C. Shiu, S. C. Pu and K. P. Liu, J. Chem. Phys., 2003, 119, 2538 CrossRef CAS.
- J. G. Zhou, J. J. Lin, W. C. Shiu and K. P. Liu, J. Chem. Phys., 2003, 119, 4997 CrossRef CAS.
- J. G. Zhou, J. J. Lin and K. P. Liu, J. Chem. Phys., 2003, 119, 8289 CrossRef CAS.
- J. G. Zhou, W. C. Shiu, J. J. Lin and K. P. Liu, J. Chem. Phys., 2004, 120, 5863 CrossRef CAS.
- W. Shiu, J. J. Lin and K. P. Liu, Phys. Rev. Lett., 2004, 92, 103201 CrossRef.
- J. G. Zhou, J. J. Lin and K. P. Liu, J. Chem. Phys., 2004, 121, 813 CrossRef CAS.
- L. Schnieder, K. Seekamp-Rahn, F. Liedeker, H. Steuwe and K. H. Welge, Faraday Discuss., 1991, 91, 259 Search PubMed.
- X. H. Liu, R. L. Gross and A. G. Suits, J. Chem. Phys., 2002, 116, 5341 CrossRef CAS.
- X. H. Liu, R. L. Gross, G. E. Hall, J. T. Muckerman and A. G. Suits, J. Chem. Phys., 2002, 117, 7947 CrossRef CAS.
- R. L. Gross, X. H. Liu and A. G. Suits, Chem. Phys. Lett., 2003, 376, 710 CrossRef CAS.
- B. Zhang and K. Liu, J. Phys. Chem. A, 2005, 109, 1022 CrossRef CAS.
- T. N. Kitsopoulos, M. A. Buntine, D. P. Baldwin, R. N. Zare and D. W. Chandler, Science, 1993, 260, 1605 CrossRef CAS.
- R. L. Toomes and T. N. Kitsopoulos, Phys. Chem. Chem. Phys., 2003, 5, 2481 RSC.
- C. Murray, A. J. Orr-Ewing, R. L. Toomes and T. N. Kitsopoulos, J. Chem. Phys., 2004, 120, 2230 CrossRef CAS.
- R. L. Toomes, A. J. van den Brom, T. N. Kitsopoulos, C. Murray and A. J. Orr-Ewing, J. Phys. Chem. A, 2004, 108, 7909 CrossRef CAS.
- M. A. Buntine, D. P. Baldwin, R. N. Zare and D. W. Chandler, J. Chem. Phys., 1991, 94, 4672 CrossRef CAS.
- P. C. Samartzis, D. Smith, T. P. Rakitzis and T. N. Kitsopoulos, Chem. Phys. Lett., 2000, 324, 337 CrossRef CAS.
- M. J. Bass, M. Brouard, C. Vallance, T. N. Kitsopoulos, P. C. Samartzis and R. L. Toomes, J. Chem. Phys., 2003, 119, 7168 CrossRef CAS.
- M. J. Bass, M. Brouard, C. Vallance, T. N. Kitsopoulos, P. C. Samartzis and R. L. Toomes, J. Chem. Phys., 2004, 121, 7175 CrossRef CAS.
- M. H. Alexander, J. Chem. Phys., 1999, 111, 7426 CrossRef CAS.
- M. S. Elioff, J. J. Valentini and D. W. Chandler, Eur. Phys. J. D, 2004, 31, 385 CrossRef CAS.
- J. M. Hutson, J. Phys. Chem., 1992, 96, 4237 CrossRef CAS.
- I. Anton Garcia, PhD thesis, University La Rioja, Logroño, 2004.
- F. J. Aoiz, L. Banares, M. Brouard, J. E. Castillo and V. J. Herrero, J. Chem. Phys., 2000, 113, 5339 CrossRef CAS.
- F. J. Aoiz, L. Banares, J. E. Castillo, M. Brouard, W. Denzer, C. Vallance, P. Honvault, J. M. Launay, A. J. Dobbyn and P. J. Knowles, Phys. Rev. Lett., 2001, 86, 1729 CrossRef CAS.
- H. Tsurumaki, Y. Fujimura and O. Kajimoto, J. Chem. Phys., 2000, 112, 8338 CrossRef CAS.
- D. M. Neumark, A. M. Wodtke, G. N. Robinson, C. C. Hayden and Y. T. Lee, J. Chem. Phys., 1985, 82, 3045 CrossRef CAS.
- D. M. Neumark, A. M. Wodtke, G. N. Robinson, C. C. Hayden, K. Shobatake, R. K. Sparks, T. P. Schafer and Y. T. Lee, J. Chem. Phys., 1985, 82, 3067 CrossRef CAS.
- B. L. Zhang, J. H. Zhang and K. P. Liu, J. Chem. Phys., 2005, 122, 104310 CrossRef.
- B. Zhang, W. Shiu, J. J. Lin and K. Liu, J. Chem. Phys., 2005, 122, 131102 CrossRef.
- B. Zhang, W. Shiu and K. Liu, J. Phys. Chem. A, 2005, 109, 8983 CrossRef CAS; B. Zhang, W. Shiu and K. Liu, J. Phys. Chem. A, 2005, 109, 8989 CrossRef CAS.
- J. Zhou, B. Zhang, J. J. Lin and K. Liu, Mol. Phys., 2005, 103, 1757 CAS.
- J. Zhou, J. J. Lin, B. Zhang and K. Liu, J. Phys. Chem. A, 2004, 108, 7832 CrossRef CAS.
- B. L. Zhang and K. P. Liu, J. Chem. Phys., 2005, 122, 101102 CrossRef.
- S. Rudić, C. Murray, D. Ascenzi, H. Anderson, J. N. Harvey and A. J. Orr-Ewing, J. Chem. Phys., 2002, 117, 5692 CrossRef CAS.
- S. Rudić, C. Murray, J. N. Harvey and A. J. Orr-Ewing, J. Chem. Phys., 2004, 120, 186 CrossRef CAS.
- N. E. Shafer, A. J. Orr-Ewing, W. R. Simpson, H. Xu and R. N. Zare, Chem. Phys. Lett., 1993, 212, 155 CrossRef CAS.
- W. R. Simpson, A. J. Orr-Ewing and R. N. Zare, Chem. Phys. Lett., 1993, 212, 163 CrossRef CAS.
- S. Rudić, C. Murray, D. Ascenzi, H. Anderson, J. N. Harvey and A. J. Orr-Ewing, J. Chem. Phys., 2002, 117, 5692 CrossRef CAS.
- N. E. Shafer, A. J. Orr-Ewing and R. N. Zare, J. Phys. Chem., 1995, 99, 7591 CrossRef CAS.
- M. Brouard, P. O’Keefe and C. Vallance, J. Phys. Chem. A, 2002, 106, 3629 CrossRef CAS.
- M. Brouard, S. P. Duxon, P. A. Enriquez and J. P. Simons, J. Chem. Phys., 1992, 97, 7414 CrossRef CAS.
- E. Surber, R. Mabbs and A. Sanov, J. Phys. Chem. A, 2003, 107, 8215 CrossRef CAS.
- A. Sanov and W. C. Lineberger, Phys. Chem. Chem. Phys., 2004, 6, 2018 RSC , and references therein.
- A. Osterwalder, M. J. Nee, J. Zhou and D. M. Neumark, J. Chem. Phys., 2004, 121, 6317 CrossRef CAS.
- C. Nicole, I. Sluimer, F. Rosca-Pruna, M. Warntjes, M. Vrakking, C. Bordas, F. Texier and F. Robicheaux, Phys. Rev. Lett., 2000, 85, 4024 CrossRef CAS.
- A. Wetzels, A. Gurtler, L. D. Noordam, F. Robicheaux, C. Dinu, H. G. Muller, M. J. J. Vrakking and W. J. van der Zande, Phys. Rev. Lett., 2002, 89, 273003 CrossRef CAS.
- A. Osterwalder, M. J. Nee, J. Zhou and D. M. Neumark, J. Chem. Phys., 2004, 121, 6317 CrossRef CAS.
- T. Suzuki and B. J. Whitaker, Int. Rev. Phys. Chem., 2001, 20, 313 CrossRef CAS , and references therein.
- K. L. Reid, Annu. Rev. Phys. Chem., 2003, 54, 397 CrossRef CAS , and references therein.
- A. Stolow, A. E. Bragg and D. M. Neumark, Chem. Rev., 2004, 104, 1719 CrossRef CAS , and references therein.
- O. Gessner, E. T. H. Chrysostom, A. M. D. Lee, D. M. Wardlaw, M. L. Ho, S. J. Lee, B. M. Cheng, M. Z. Zgierski, I. C. Chen, J. P. Shaffer, C. C. Hayden and A. Stolow, Faraday Discuss., 2004, 127, 193 RSC , and references therein.
- M. Maazouz, P. L. Maazouz and D. C. Jacobs, J. Chem. Phys., 2002, 117, 10917 CrossRef CAS.
- S. K. Lee, D. Townsend, O. S. Vasyutinskii and A. G. Suits, Phys. Chem. Chem. Phys., 2005, 7, 1650 RSC.
- S. T. Tsai, C. K. Lin, Y. T. Lee and C. K. Ni, Rev. Sci. Instrum., 2001, 72, 1963 CrossRef CAS.
- T. P. Softley, Int. Rev. Phys. Chem., 2004, 23, 1 CrossRef CAS.
- H. A. Cruse and T. P. Softley, J. Chem. Phys., 2004, 121, 4089 CrossRef CAS.
- A. J. R. Heck, D. W. Neyer, R. N. Zare and D. W. Chandler, J. Phys. Chem., 1995, 99, 17700 CrossRef CAS.
- T. P. Rakitzis, P. C. Samartzis, R. L. Toomes, L. Tsigaridas, M. Coriou, D. Chestakov, A. T. J. B. Eppink, D. H. Parker and T. N. Kitsopoulos, Chem. Phys. Lett., 2002, 364, 115 CrossRef CAS.
- T. P. Rakitzis, P. C. Samartzis, R. L. Toomes and T. N. Kitsopoulos, J. Chem. Phys., 2004, 121, 7222 CrossRef CAS.
- M. Beckert, E. R. Wouters, M. N. R. Ashfold and E. Wrede, J. Chem. Phys., 2003, 119, 9576 CrossRef CAS.
- M. J. Cooper, E. Wrede, A. J. Orr-Ewing and M. N. R. Ashfold, J. Chem. Soc., Faraday Trans., 1998, 94, 2901 RSC.
- Y. J. Jee, M. S. Park, Y. S. Kim, Y. J. Jung and K. H. Jung, Chem. Phys. Lett., 1998, 287, 701 CrossRef CAS.
- P. C. Samartzis, T. N. Kitsopoulos and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2000, 2, 453 RSC.
- Y. J. Jee, Y. J. Jung and K. H. Jung, J. Chem. Phys., 2001, 115, 9739 CrossRef CAS.
- T. P. Rakitzis and T. N. Kitsopoulos, J. Chem. Phys., 2002, 116, 9228 CrossRef CAS.
- Y. S. Kim, Y. J. Jung and K. H. Jung, J. Chem. Phys., 1997, 107, 3805 CrossRef CAS.
- E. Wrede, S. Laubach, S. Schulenburg, A. J. Orr-Ewing and M. N. R. Ashfold, Chem. Phys. Lett., 2000, 326, 22 CrossRef CAS.
- W. P. Hess, D. W. Chandler and J. W. Thoman, Chem. Phys., 1992, 163, 277 CrossRef CAS.
- T. Gougousi, P. C. Samartzis and T. N. Kitsopoulos, J. Chem. Phys., 1998, 108, 5742 CrossRef CAS.
- D. D. Xu, J. S. Francisco, J. Huang and W. M. Jackson, J. Chem. Phys., 2002, 117, 2578 CrossRef CAS.
- T. K. Kim, M. S. Park, K. W. Lee and K. H. Jung, J. Chem. Phys., 2001, 115, 10745 CrossRef CAS.
- S. H. Lee and K. H. Jung, Chem. Phys. Lett., 2001, 350, 306 CrossRef CAS.
- Y. J. Jung, M. S. Park, Y. S. Kim, K. H. Jung and H. R. Volpp, J. Chem. Phys., 1999, 111, 4005 CrossRef CAS.
- H. Katayanagi, N. Yonekuira and T. Suzuki, Chem. Phys., 1998, 231, 345 CrossRef CAS.
- K. S. Lee, K. W. Lee, T. K. Kim, R. Ryoo and K. H. Jung, J. Chem. Phys., 2005, 122, 034308 CrossRef.
- M. S. Park, K. W. Lee and K. H. Jung, J. Chem. Phys., 2001, 114, 10368 CrossRef CAS.
- K. W. Lee, Y. J. Jee and K. H. Jung, J. Chem. Phys., 2002, 116, 4490 CrossRef CAS.
- B. L. G. Bakker and D. H. Parker, Chem. Phys. Lett., 2000, 330, 293 CrossRef CAS.
- N. Hansen and A. M. Wodtke, Chem. Phys. Lett., 2002, 356, 340 CAS.
- P. C. Samartzis, T. Gougousi and T. N. Kitsopoulos, Laser Chem., 1997, 17, 185 Search PubMed.
- P. C. Samartzis, I. Sakellariou, T. Gougousi and T. N. Kitsopoulos, J. Chem. Phys., 1997, 107, 43 CrossRef CAS.
- P. C. Samartzis, B. L. G. Bakker, T. P. Rakitzis, D. H. Parker and T. N. Kitsopoulos, J. Chem. Phys., 1999, 110, 5201 CrossRef CAS.
- A. S. Bracker, E. R. Wouters, A. G. Suits and O. S. Vasyutinskii, J. Chem. Phys., 1999, 110, 6749 CrossRef CAS.
- C. R. Gebhardt, T. P. Rakitzis, P. C. Samartzis, V. Ladopoulos and T. N. Kitsopoulos, Rev. Sci. Instrum., 2001, 72, 3848 CrossRef.
- D. H. Parker, B. L. G. Bakker, P. C. Samartzis and T. N. Kitsopoulos, J. Chem. Phys., 2001, 115, 1205 CrossRef CAS.
- A. Sugita, K. Suto, M. Kawasaki and Y. Matsumi, Chem. Phys. Lett., 2001, 340, 83 CrossRef CAS.
- A. I. Chichinin, T. Einfeld, C. Maul and K. H. Gericke, Rev. Sci. Instrum., 2002, 73, 1856 CrossRef CAS.
- M. J. Bass, M. Brouard, A. P. Clark, C. Vallance and B. Martinez-Haya, Phys. Chem. Chem. Phys., 2003, 5, 856 RSC.
- T. P. Rakitzis, P. C. Samartzis, R. L. Toomes, T. N. Kitsopoulos, A. Brown, G. G. Balint-Kurti, O. S. Vasyutinskii and J. A. Beswick, Science, 2003, 300, 1936 CrossRef CAS.
- C. Romanescu, S. Manzhos, D. Boldovsky, J. Clarke and H. P. Loock, J. Chem. Phys., 2004, 120, 767 CrossRef CAS.
- M. S. Park, Y. J. Jung, S. H. Lee, D. C. Kim and K. H. Jung, Chem. Phys. Lett., 2000, 322, 429 CrossRef CAS.
- E. R. Wouters, M. Beckert, L. J. Russell, K. N. Rosser, A. J. Orr-Ewing, M. N. R. Ashfold and O. S. Vasyutinskii, J. Chem. Phys., 2002, 117, 2087 CrossRef CAS.
- L. J. Rogers, M. N. R. Ashfold, Y. Matsumi, M. Kawasaki and B. J. Whitaker, Chem. Phys. Lett., 1996, 258, 159 CrossRef CAS.
- Y. Tanaka, M. Kawasaki, Y. Matsumi, H. Fujiwara, T. Ishiwata, L. J. Rogers, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 1998, 109, 1315 CrossRef CAS.
- V. Skorokhodov, Y. Sato, K. Suto, Y. Matsumi and M. Kawasaki, J. Phys. Chem., 1996, 100, 12321 CrossRef CAS.
- M. Kawasaki, K. Suto, Y. Sato, Y. Matsumi and R. Bersohn, J. Phys. Chem., 1996, 100, 19853 CrossRef.
- N. Hansen and A. M. Wodtke, J. Phys. Chem. A, 2003, 107, 10608 CrossRef CAS.
- T. Einfeld, A. Chichinin, C. Maul and K. H. Gericke, J. Chem. Phys., 2002, 116, 2803 CrossRef CAS.
- T. S. Einfeld, C. Maul, K. H. Gericke and A. Chichinin, J. Chem. Phys., 2002, 117, 1123 CrossRef CAS.
- T. S. Einfeld, C. Maul, K. H. Gericke and A. Chichinin, J. Chem. Phys., 2002, 117, 4214 CrossRef CAS.
- D. Townsend, S. K. Lee and A. G. Suits, J. Phys. Chem. A, 2004, 108, 8106 CrossRef CAS.
- S. H. Lee, Y. J. Jung and K. H. Jung, Chem. Phys., 2000, 260, 143 CrossRef CAS.
- S. Deshmukh, J. D. Myers, S. S. Xantheas and W. P. Hess, J. Phys. Chem., 1994, 98, 12535 CrossRef CAS.
- K. Tonokura, L. B. Daniels, T. Suzuki and Y. Yamashita, J. Phys. Chem. A, 1997, 101, 7754 CrossRef CAS.
- Y. Liu and L. J. Butler, J. Chem. Phys., 2004, 121, 11016 CrossRef CAS.
- R. L. Gross, X. H. Liu and A. G. Suits, Chem. Phys. Lett., 2002, 362, 229 CrossRef CAS.
- V. Dribinski, A. B. Potter, A. V. Demyanenko and H. Reisler, J. Chem. Phys., 2001, 115, 7474 CrossRef CAS.
- A. B. Potter, V. Dribinski, A. Demyanenko and H. Reisler, Chem. Phys. Lett., 2001, 349, 257 CrossRef CAS.
- M. A. Buntine, D. P. Baldwin and D. W. Chandler, J. Chem. Phys., 1992, 96, 5843 CrossRef CAS.
- F. Aguirre and S. T. Pratt, J. Chem. Phys., 2004, 121, 9855 CrossRef CAS.
- D. H. Parker, R. F. Delmdahl, B. B. L. G. Bakker and H. P. Loock, J. Chin. Chem. Soc., 2001, 48, 327 CAS.
- S. Manzhos, H. P. Loock, B. L. G. Bakker and D. H. Parker, J. Chem. Phys., 2002, 117, 9347 CrossRef CAS.
- L. J. Rogers, M. N. R. Ashfold, Y. Matsumi, M. Kawasaki and B. J. Whitaker, J. Chem. Soc., Faraday Trans., 1996, 92, 5181 RSC.
- M. Kawasaki, A. Sugita, C. Ramos, Y. Matsumi and H. Tachikawa, J. Phys. Chem. A, 2004, 108, 8119 CrossRef CAS.
- L. R. Valachovic, M. F. Tuchler, M. Dulligan, T. Droz-Georget, M. Zyrianov, A. Kolessov, H. Reisler and C. Wittig, J. Chem. Phys., 2000, 112, 2752 CrossRef CAS.
- A. Sanov, T. Droz-Georget, M. Zyrianov and H. Reisler, J. Chem. Phys., 1997, 106, 7013 CrossRef CAS.
- D. P. Baldwin, M. A. Buntine and D. W. Chandler, J. Chem. Phys., 1990, 93, 6578 CrossRef CAS.
- A. J. R. Heck, R. N. Zare and D. W. Chandler, J. Chem. Phys., 1996, 104, 3399 CrossRef CAS.
- A. J. R. Heck, R. N. Zare and D. W. Chandler, J. Chem. Phys., 1996, 104, 4019 CrossRef CAS.
- W. M. Jackson, R. J. Price, D. D. Xu, J. D. Wrobel, M. Ahmed, D. S. Peterka and A. G. Suits, J. Chem. Phys., 1998, 109, 4703 CrossRef CAS.
- J. H. Zhang, B. Jiang, X. M. Yang and J. C. Xie, Chem. Phys. Lett., 2002, 364, 80 CrossRef CAS.
- J. Wei, A. Kuczmann, J. Riedel, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2003, 5, 315 RSC.
- J. Wei, J. Riedel, A. Kuczmann, F. Renth and F. Temps, Faraday Discuss., 2004, 127, 267 RSC.
- A. J. van den Brom, M. Kapelios, T. N. Kitsopoulos, N. H. Nahler, B. Cronin and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2005, 7, 892 RSC.
- D. C. Radenovic, A. J. A. van Roij, D. A. Chestakov, A. T. J. B. Eppink, J. J. ter Meulen, D. H. Parker, M. P. J. van der Loo, G. C. Groenenboom, M. E. Greenslade and M. I. Lester, J. Chem. Phys., 2003, 119, 9341 CrossRef CAS.
- M. Ahmed, D. S. Peterka and A. G. Suits, J. Chem. Phys., 1999, 110, 4248 CrossRef CAS.
- H. P. Loock, B. L. G. Bakker and D. H. Parker, Can. J. Phys., 2001, 79, 211 CrossRef CAS.
- H. F. Xu, S. L. Liu, X. X. Ma, D. X. Dai, J. C. Xie and G. H. Sha, Acta Physica Sinica, 2002, 51, 240 Search PubMed.
- H. F. Xu, Y. Guo, S. L. Liu, X. X. Ma, D. X. Dai and G. H. Sha, J. Chem. Phys., 2002, 117, 5722 CrossRef CAS.
- M. H. M. Janssen, J. W. G. Mastenbroek and S. Stolte, J. Phys. Chem. A, 1997, 101, 7605 CrossRef CAS.
- A. T. J. B. Eppink and D. H. Parker, J. Chem. Phys., 1998, 109, 4758 CrossRef CAS.
- A. T. J. B. Eppink and D. H. Parker, J. Chem. Phys., 1999, 110, 832 CrossRef CAS.
- Y. Tanaka, M. Kawasaki and Y. Matsumi, Bull. Chem. Soc. Jpn., 1998, 71, 2539 CAS.
- Y. S. Kim, W. K. Kang and K. H. Jung, J. Chem. Phys., 1996, 105, 551 CrossRef CAS.
- F. Aguirre and S. T. Pratt, J. Chem. Phys., 2003, 118, 1175 CrossRef CAS.
- P. A. Arnold, B. R. Cosofret, S. M. Dylewski, P. L. Houston and B. K. Carpenter, J. Phys. Chem. A, 2001, 105, 1693 CrossRef CAS.
- S. Unny, Y. Du, L. C. Zhu, K. Truhins, R. J. Gordon, A. Sugita, M. Kawasaki, Y. Matsumi, R. Delmdahl, D. H. Parker and A. Berces, J. Phys. Chem. A, 2001, 105, 2270 CrossRef CAS.
- B. R. Cosofret, H. M. Lambert and P. L. Houston, J. Chem. Phys., 2002, 117, 8787 CrossRef CAS.
- B. R. Cosofret, H. M. Lambert and P. L. Houston, Bull. Korean Chem. Soc., 2002, 23, 179 CAS.
- B. L. G. Bakker, A. T. J. B. Eppink, D. H. Parker, M. L. Costen, G. Hancock and G. A. D. Ritchie, Chem. Phys. Lett., 1998, 283, 319 CrossRef CAS.
- B. L. G. Bakker, D. H. Parker, G. Hancock and G. A. D. Ritchie, Chem. Phys. Lett., 1998, 294, 565 CrossRef CAS.
- J. M. Teule, G. C. Groenenboom, D. W. Neyer, D. W. Chandler and M. H. M. Janssen, Chem. Phys. Lett., 2000, 320, 177 CrossRef CAS.
- M. Brouard, A. P. Clark, C. Vallance and O. S. Vasyutinskii, J. Chem. Phys., 2003, 119, 771 CrossRef CAS.
- H. M. Lambert, E. W. Davis, O. Tokel, A. A. Dixit and P. L. Houston, J. Chem. Phys., 2005, 122, art. no. 174304.
- V. P. Hradil, T. Suzuki, S. A. Hewitt, P. L. Houston and B. J. Whitaker, J. Chem. Phys., 1993, 99, 4455 CrossRef CAS.
- K. Tonokura and T. Suzuki, Chem. Phys. Lett., 1994, 224, 1 CrossRef CAS.
- M. Ahmed, D. S. Peterka, A. S. Bracker, O. Vasyutinskii and A. G. Suits, J. Chem. Phys., 1999, 110, 4115 CrossRef CAS.
- A. V. Demyanenko, V. Dribinski, H. Reisler, H. Meyer and C. X. W. Qian, J. Chem. Phys., 1999, 111, 7383 CrossRef CAS.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3484.
- D. H. Parker and A. T. J. B. Eppink, J. Chem. Phys., 1997, 107, 2357 CrossRef CAS.
- B. Buijsse, W. J. van der Zande, A. T. J. B. Eppink, D. H. Parker, B. R. Lewis and S. T. Gibson, J. Chem. Phys., 1998, 108, 7229 CrossRef CAS.
- B. L. G. Bakker and D. H. Parker, J. Chem. Phys., 2000, 112, 4037 CrossRef CAS.
- D. H. Parker, Acc. Chem. Res., 2000, 33, 563 CrossRef CAS.
- H. M. Lambert, A. A. Dixit, E. W. Davis and P. L. Houston, J. Chem. Phys., 2004, 121, 10437 CrossRef CAS.
- R. J. Wilson, J. A. Mueller and P. L. Houston, J. Phys. Chem. A, 1997, 101, 7593 CrossRef CAS.
- J. D. Geiser, S. M. Dylewski, J. A. Mueller, R. J. Wilson, R. Toumi and P. L. Houston, J. Chem. Phys., 2000, 112, 1279 CrossRef CAS.
- P. L. Houston, B. R. Cosofret, A. Dixit, S. M. Dylewski, J. D. Geiser, J. A. Mueller, R. J. Wilson, P. J. Pisano, M. S. Westley, K. T. Lorenz and D. W. Chandler, J. Chin. Chem. Soc., 2001, 48, 309 CAS.
- B. R. Cosofret, S. M. Dylewski and P. L. Houston, J. Phys. Chem. A, 2000, 104, 10240 CrossRef CAS.
- M. Brouard, R. Cireasa, A. P. Clark, T. J. Preston, C. Vallance, G. C. Groenenboom and O. S. Vasyutinskii, J. Phys. Chem. A, 2004, 108, 7965 CrossRef CAS.
- R. F. Delmdahl, B. L. G. Bakker and D. H. Parker, J. Chem. Phys., 2000, 112, 5298 CrossRef CAS.
- R. F. Delmdahl, D. H. Parker and A. T. J. B. Eppink, J. Chem. Phys., 2001, 114, 8339 CrossRef CAS.
- A. T. J. B. Eppink, D. H. Parker, M. H. M. Janssen, B. Buijsse and W. J. van der Zande, J. Chem. Phys., 1998, 108, 1305 CrossRef CAS.
- N. Taniguchi, K. Takahashi, Y. Matsumi, S. M. Dylewski, J. D. Geiser and P. L. Houston, J. Chem. Phys., 1999, 111, 6350 CrossRef CAS.
- S. M. Dylewski, J. D. Geiser and P. L. Houston, J. Chem. Phys., 2001, 115, 7460 CrossRef CAS.
- T. Suzuki, H. Katayanagi, Y. X. Mo and K. Tonokura, Chem. Phys. Lett., 1996, 256, 90 CrossRef CAS.
- M. Ahmed, E. R. Wouters, D. S. Peterka, O. S. Vasyutinskii and A. G. Suits, Faraday Discuss., 1999, 113, 425 RSC.
- D. W. Neyer, A. J. R. Heck, D. W. Chandler, J. M. Teule and M. H. M. Janssen, J. Phys. Chem. A, 1999, 103, 10388 CrossRef CAS.
- T. Nishide and T. Suzuki, J. Phys. Chem. A, 2004, 108, 7863 CrossRef CAS.
- A. G. Smolin, O. S. Vasyutinskii, E. R. Wouters and A. G. Suits, J. Chem. Phys., 2004, 121, 6759 CrossRef CAS.
- P. C. Samartzis and T. N. Kitsopoulos, J. Phys. Chem. A, 1997, 101, 5620 CrossRef CAS.
- T. N. Kitsopoulos, C. R. Gebhardt and T. P. Rakitzis, J. Chem. Phys., 2001, 115, 9727 CrossRef CAS.
- H. Katayanagi, Y. X. Mo and T. Suzuki, Chem. Phys. Lett., 1995, 247, 571 CrossRef CAS.
- A. A. Dixit, Y. Lei, K. W. Lee, E. Quinones and P. L. Houston, J. Phys. Chem. A, 2005, 109, 1770 CrossRef CAS.
- Y. X. Mo, H. Katayanagi, M. C. Heaven and T. Suzuki, Phys. Rev. Lett., 1996, 77, 830 CrossRef CAS.
- T. Suzuki, H. Katayanagi, S. Nanbu and M. Aoyagi, J. Chem. Phys., 1998, 109, 5778 CrossRef CAS.
- T. P. Rakitzis, P. C. Samartzis and T. N. Kitsopoulos, J. Chem. Phys., 1999, 111, 10415 CrossRef CAS.
- T. P. Rakitzis, P. C. Samartzis and T. N. Kitsopoulos, Phys. Rev. Lett., 2001, 87, 123001 CrossRef CAS.
- M. H. Kim, W. Li, S. K. Lee and A. G. Suits, Can. J. Chem., 2004, 82, 880 CrossRef CAS.
- D. Townsend, S. K. Lee and A. G. Suits, Chem. Phys., 2004, 301, 197 CrossRef CAS.
- S.-M. Wu, X. Yang and D. H. Parker, Mol. Phys., 2005, 103, 1797 CrossRef CAS.
- D. A. Chestakov, D. H. Parker and A. V. Baklanov, J. Chem. Phys., 2005, 122, 084302 CrossRef CAS.
- Y. Sato, Y. Matsumi, M. Kawasaki, K. Tsukiyama and R. Bersohn, J. Phys. Chem., 1995, 99, 16307 CrossRef CAS.
- A. Sugita, M. Mashino, M. Kawasaki, Y. Matsumi, R. Bersohn, G. Trott-Kriegeskorte and K. H. Gericke, J. Chem. Phys., 2000, 112, 7095 CrossRef CAS.
- A. J. van den Brom, T. P. Rakitzis, J. van Heyst, T. N. Kitsopoulos, S. R. Jezowski and M. H. M. Janssen, J. Chem. Phys., 2002, 117, 4255 CrossRef CAS.
- T. P. Rakitzis, A. J. van den Brom and M. H. M. Janssen, Science, 2004, 303, 1852 CrossRef CAS.
- A. J. van den Brom, T. P. Rakitzis and M. H. M. Janssen, J. Chem. Phys., 2004, 121, 11645 CrossRef CAS.
- D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, J. Rheinecker, L. B. Harding and J. M. Bowman, Science, 2004, 306, 1158 CrossRef CAS.
- M. Kawasaki, Y. Sato, K. Suto, Y. Matsumi and S. H. S. Wilson, Chem. Phys. Lett., 1996, 251, 67 CrossRef CAS.
- T. Droz-Georget, M. Zyrianov, A. Sanov and H. Reisler, Ber. Bunsen-Ges. Phys. Chem., 1997, 101, 469 CAS.
- T. Droz-Georget, M. Zyrianov, H. Reisler and D. W. Chandler, Chem. Phys. Lett., 1997, 276, 316 CrossRef CAS.
- M. Zyrianov, T. Droz-Georget and H. Reisler, J. Chem. Phys., 1999, 110, 2059 CrossRef CAS.
- J. Liu, F. Wang, H. Wang, B. Jiang and X. Yang, J. Chem. Phys., 2005, 122, 104309 CrossRef.
- S. M. Clegg, B. F. Parsons, S. J. Klippenstein and D. L. Osborn, J. Chem. Phys., 2003, 119, 7222 CrossRef CAS.
- D. W. Neyer, A. J. R. Heck and D. W. Chandler, J. Chem. Phys., 1999, 110, 3411 CrossRef CAS.
- N. Hansen, A. M. Wodtke, A. V. Komissarov and M. C. Heaven, Chem. Phys. Lett., 2003, 368, 568 CrossRef CAS.
- V. K. Nestorov and J. I. Cline, J. Chem. Phys., 1999, 111, 5287 CrossRef CAS.
- T. Suzuki, V. P. Hradil, S. A. Hewitt, P. L. Houston and B. J. Whitaker, Chem. Phys. Lett., 1991, 187, 257 CrossRef.
- Y. X. Mo, H. Katayanagi and T. Suzuki, J. Chem. Phys., 1999, 110, 2029 CrossRef CAS.
- O. L. A. Monti, H. Dickinson, S. R. Mackenzie and T. P. Softley, J. Chem. Phys., 2000, 112, 3699 CrossRef CAS.
- B. F. Parsons, D. W. Chandler, E. C. Sklute, S. L. Li and E. A. Wade, J. Phys. Chem. A, 2004, 108, 9742 CrossRef CAS.
- E. A. Wade, J. I. Cline, K. T. Lorenz, C. Hayden and D. W. Chandler, J. Chem. Phys., 2002, 116, 4755 CrossRef CAS.
- A. V. Demyanenko, A. B. Potter, V. Dribinski and H. Reisler, J. Chem. Phys., 2002, 117, 2568 CrossRef CAS.
- A. B. Potter, V. Dribinski, A. V. Demyanenko and H. Reisler, J. Chem. Phys., 2003, 119, 7197 CrossRef CAS.
- M. Tsubouchi and T. Suzuki, Chem. Phys. Lett., 2003, 382, 418 CrossRef CAS.
- M. Tsubouchi, C. A. de Lange and T. Suzuki, J. Chem. Phys., 2003, 119, 11728 CrossRef CAS.
- V. Dribinski, A. B. Potter, I. Fedorov and H. Reisler, Chem. Phys. Lett., 2004, 385, 233 CrossRef CAS.
- V. Dribinski, A. B. Potter, I. Fedorov and H. Reisler, J. Chem. Phys., 2004, 121, 12353 CrossRef CAS.
- V. K. Nestorov, R. D. Hinchliffe, R. Uberna, J. I. Cline, K. T. Lorenz and D. W. Chandler, J. Chem. Phys., 2001, 115, 7881 CrossRef CAS.
- T. J. Obernhuber, U. Kensy and B. Dick, Phys. Chem. Chem. Phys., 2003, 5, 2799 RSC.
- A. G. Suits, R. L. Miller, L. S. Bontuyan and P. L. Houston, J. Chem. Soc., Faraday Trans., 1993, 89, 1443–3667 RSC.
- D. W. Chandler, J. W. Thoman, M. H. M. Janssen and D. H. Parker, Chem. Phys. Lett., 1989, 156, 151 CrossRef CAS.
- D. W. Chandler, M. H. M. Janssen, S. Stolte, R. N. Strickland, J. W. Thoman and D. H. Parker, J. Phys. Chem., 1990, 94, 4839 CrossRef.
- M. H. M. Janssen, D. H. Parker, G. O. Sitz, S. Stolte and D. W. Chandler, J. Phys. Chem., 1991, 95, 8007 CrossRef CAS.
- J. W. G. Mastenbroek, C. A. Taatjes, K. Nauta, M. H. M. Janssen and S. Stolte, J. Phys. Chem., 1995, 99, 4360 CrossRef CAS.
- A. J. van den Brom, M. Lipciuc and M. H. M. Janssen, Chem. Phys. Lett., 2003, 368, 324 CrossRef CAS.
- P. Quintana, R. F. Delmdahl, D. H. Parker, B. Martinez-Haya, F. J. Aoiz, L. Banares and E. Verdasco, Chem. Phys. Lett., 2000, 325, 146 CrossRef CAS.
- B. Martinez-Haya, P. Quintana, L. Banares, P. Samartzis, D. J. Smith and T. N. Kitsopoulos, J. Chem. Phys., 2001, 114, 4450 CrossRef CAS.
- B. Martinez-Haya, M. J. Bass, M. Brouard, C. Vallance, I. Torres and J. Barr, J. Chem. Phys., 2004, 120, 11042 CrossRef CAS.
- H. A. Cruse and T. P. Softley, J. Chem. Phys., 2005, 122, 124303 CrossRef CAS.
| This journal is © the Owner Societies 2006 |