Coupling ligand recognition to protein folding in an engineered variant of rabbit ileal lipid binding protein
Nikolaos
Kouvatsos
,
Jill K.
Meldrum
,
Mark S.
Searle
* and
Neil R.
Thomas
*
Centre for Biomolecular Sciences, School of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: neil.thomas@nottingham.ac.uk; mark.searle@nottingham.ac.uk; Tel: +44 0115 951 3565 (N. R. Thomas); +44 0115 951 3567 (M. S. Searle)
First published on 27th September 2006
Abstract
We have engineered a variant of the β-clam shell protein ILBP which lacks the α-helical motif that caps the central binding cavity; the mutant protein is sufficiently destabilised that it is unfolded under physiological conditions, however, it unexpectedly binds its natural bile acid substrates with high affinity forming a native-like β-sheet rich structure and demonstrating strong thermodynamic coupling between ligand binding and protein folding.
Although the functions of many proteins are closely linked to their three-dimensional structure, numerous protein sequences have now been identified that lack intrinsic globular structure under physiological conditions. These “natively unfolded” states have led to a reassessment of the protein structure–function paradigm.1,2 The coupling of substrate recognition to protein folding, which potentially affords considerable binding specificity, has been described in a number of different contexts typically involving the binding of large flexible nucleic acid or peptide ligands.3–7 Here we report an engineered variant of rabbit ileal lipid binding protein (ILBP) which is unfolded under physiological conditions, but adopts a native-like conformation in the presence of small rigid bile acid ligands, demonstrating strong coupling between ligand binding and protein folding. Surprisingly, these ligands bind with affinities comparable to the natively folded protein with little apparent cost associated with the structural re-organisation of the disordered polypeptide chain. However, we observe much larger enthalpy changes for binding to the natively unfolded mutant which reflect the sum of the two coupled processes.
Members of the intracellular lipid binding protein family have a similar β-clam shell fold (Fig. 1), and bind a diverse range of natural ligands (both fatty acids and steroid-based bile acids; Fig. 2) within the inner cavity.8–10 A number of studies have implicated the helical capping motif in modulating interactions with membranes and other receptors, as well as the cavity size and its hydrophobicity.11–16 We have engineered an ILBP mutant (Δα-ILBP) in which the helical motif (residues 9–35) was replaced by a flexible Gly-Gly-Ser-Gly linker (Fig. 1). We envisaged that the more open binding cavity might permit the recognition of a wider range of non-natural substrates, making the mutant a promising candidate for development as a catalytic scaffold using display and selection strategies.17–19 Several “helix-less” versions of the structurally homologous intestinal fatty-acid binding protein (IFBP) have been shown to retain their structural integrity although the binding affinities for the natural ligands were reduced.11–16
 | ||
| Fig. 1 (a) β-clam-shell structure of rabbit ILBP showing the helical capping motif (arrows indicate the positions of residues 9 and 35 that define the helical motif). (b) Model of the folded Δα-ILBP mutant showing the substituted G-G-S-G linker. Ribbon representation using the program MOLMOL.26 | ||
 | ||
| Fig. 2 Structures of cholic acid substrates showing the substitution patterns at positions R1, R2 and R3, including the presence of a conjugated taurine or glycine residue at position R3. | ||
In contrast to these previous studies, NMR spectra of Δα-ILBP showed poor dispersion of resonances in both the amide NH region and aliphatic methyl region of the spectrum. Moreover, the observation of narrow line widths for many aromatic signals suggested significant side chain dynamics and an overall conformation typical of a significantly disordered polypeptide. This was confirmed by the observation of strong negative ellipticity at 200 nm in the far-UV CD spectrum of Δα-ILBP and correspondingly much weaker intensity at 216 nm (Fig. 3a), indicating that the mutant was significantly unfolded in aqueous buffer. In the presence of several equivalents of ligand, methyl resonances appeared upfield shifted as far as ∼ 0.1 ppm, indicative of core packing of aromatic side chains. CD spectra of the Δα-ILBP–ligand complexes showed strong negative intensity at 216 nm which becomes positive below 205 nm, providing evidence for ligand-induced folding to a native-like β-sheet-rich structure (Fig. 3a). In contrast, the CD spectrum of wt-ILBP is essentially unperturbed by the binding of ligands, indicative of a largely pre-organised ligand receptor.
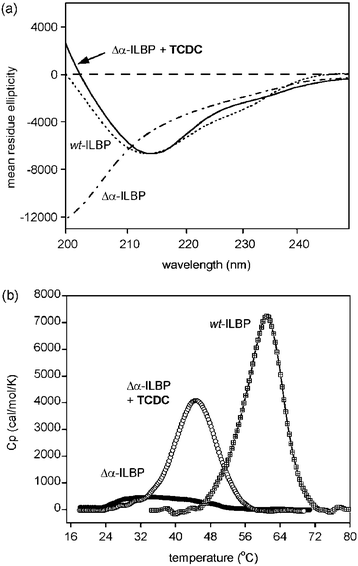 | ||
| Fig. 3 (a) Far UV CD data collected at 298 K for wt-ILBP, Δα-ILBP and the complex of Δα-ILBP with taurochenodeoxycholic acid (TCDC). (b) DSC data showing thermal unfolding curves for wt-ILBP (Tm = 60.5 ± 0.1 °C, ΔH = 367.1 ± 2.5 kJ mol−1) and Δα-ILBP in the absence and presence of TCDC (for the latter Tm = 44.5 ± 0.3 °C with an enthalpy change of 215.6 ± 1.2 kJ mol−1). Protein concentrations of 30 µM were used in phosphate buffer at pH 7.2 and a ligand concentration of 500 µM for studies of the Δα-ILBP complex. A scan rate of 90 °C h−1 was used.† | ||
The thermal unfolding transition for Δα-ILBP monitored by DSC in the absence of ligands is very broad and ill-defined over the 20–50 °C temperature range, suggesting the presence of a disordered ensemble of structures in solution. In contrast, ligand complexes of Δα-ILBP were associated with a co-operative unfolding transition and a large change in enthalpy at the Tm (Fig. 3b), demonstrating strong coupling between ligand binding and protein folding.
Equilibrium binding affinities for the complexation of a number of ligands with the natively unfolded Δα-ILBP were determined using isothermal titration calorimetry (ITC). The isotherms fit very well to a simple 1 : 1 binding model (Fig. 4). TCDC and GDC have the highest and lowest affinities of those ligands studied to date, with KA values for binding to Δα-ILBP of 3.8 ± 0.05 × 105 M−1 and 1.9 ± 0.04 × 104 M−1, respectively. Notably, 2–3 fold larger enthalpy and entropy changes are observed for binding to Δα-ILBP that appear to reflect the coupling between ligand complexation and folding. Although the ligand binding free energies are largely similar (ΔΔG ∼ 5–6 kJ mol−1), the enthalpy changes show a much more significant variation (−80 ± 1 kJ mol−1 for GDC, to −146 ± 2 kJ mol−1 for TDC) and are strongly compensated by the large entropic cost of ordering the polypeptide chain (ΔS = −185.5 J K−1 mol−1 and −402.5 J K−1 mol−1, respectively).
 | ||
| Fig. 4 Isothermal titration calorimetry (ITC) data for the binding of TCDC to 30 µM Δα-ILBP at 298 K in phosphate buffer following 5 µL injections; the inset shows the fit of the resultant binding isotherm to a 1 : 1 binding model. | ||
In contrast to the simple 1 : 1 binding mode observed for Δα-ILBP, ITC data for both the wild-type human-ILBP,9 and the rabbit protein studied here (data not shown), provide evidence for the co-operative binding of two ligand molecules within the central cavity. Our conclusions are consistent with earlier studies with other lipid binding proteins in which the helical capping motif appears to play an important role in modulating the cavity size, its hydrophobicity and ligand binding stoichiometry.11–16 In these studies, removing the helical domain decreases the binding stoichiometry. Strikingly, although Δα-ILBP appears to accommodate only a single bound ligand molecule, the KA values and binding free energies are comparable to those reported for a range of ligands binding to human-ILBP (∼ 104–105 M−1),9 despite the anticipated associated cost of assembling Δα-ILBP from the natively unfolded state.
In considering the folding of Δα-ILBP from its thermodynamically preferred disordered state we are led to the initial conclusion that a significant cost in free energy for the structural organisation is anticipated which must be paid by some fraction of the ligand binding energy. Given that we are unable to detect a significant population of folded Δα-ILBP molecules in solution, and based on the equilibrium stability of the natively folded wt-ILBP, we estimate that Δα-ILBP is destabilised relative to wt-ILBP by ≥ 25 kJ mol−1. This represents the upper-limit estimate of the cost of folding Δα-ILBP from the natively unfolded state. However, this energetic price is only paid if the ligand–protein complex is as compact and highly structured as the natively folded form of wt-ILBP. By analogy, Williams et al.20,21 have described how a subtle co-operative “loosening” or “tightening” of non-covalent interactions in a folded receptor can occur as a consequence of ligand binding. These delocalised effects on the protein structure are concluded to affect the thermodynamics of the interaction in a way that is not readily rationalised simply from an analysis of direct contacts at the protein–ligand interface.
In the current context, conformational selection from the ensemble of natively unfolded Δα-ILBP molecules, followed by induced-fit to optimise ligand interactions,22,23 plausibly leads to a ligand–protein complex in which the degree of non-covalent bonding and polypeptide flexibility may be significantly different to that observed for ligand complexes with wt-ILBP. Compensating enthalpy and entropy effects may largely negate the perceived energetic cost of assembling Δα-ILBP from an unfolded state. What is the evidence that this could be the case? The large variation in enthalpies (ΔΔH ∼ 66 kJ mol−1) and entropies (ΔΔS ∼ 217 J K−1 mol−1) for ligand binding to Δα-ILBP is greater than can be comfortably explained on the basis of the modest structural differences between substrate molecules. Further, such large differences in binding enthalpies and entropies are not apparent from binding studies of the same ligands with wt-ILBP,9 suggesting that their origin lies in the folding component of the binding and assembly of the Δα-ILBP complex. Thus, an alternative thermodynamic interpretation is that the various ligands are able to “order” the polypeptide chain of Δα-ILBP to different degrees, resulting in subtle differences in the degree or tightness of side chain packing. All of the spectroscopic data to date suggest that the Δα-ILBP complexes are structurally highly native-like and undergo a co-operative unfolding transition (see Fig. 3b). However, the Tm values are significantly lower than for wt-ILBP even without a bound ligand (Fig. 3), demonstrating a global destabilisation of non-covalent interactions within the Δα-ILBP complex.
In the general context, it has become clear that differences in the biological response to the binding of different ligands to protein receptors are related to the thermodynamics of the interaction. For example, fundamental differences are observed between the binding of agonists and antagonists to membrane-bound receptors.24,25 Such thermodynamic measurements appear to provide insights into ligand-induced changes in a receptor (tightening or loosening) that correlate with biological function.20,21 The experiments described here for the Δα-ILBP mutant have identified the thermodynamic coupling between ligand binding and order changes in a protein receptor, the structural basis of which we are continuing to investigate.
We thank the University of Nottingham for financial support to NK and JKM. We are grateful to Drs S. Stengelin and W. Kramer of Aventis Pharma, Germany for the provision of the plasmid containing the rabbit wt-ILBP gene.
Notes and references
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197 CrossRef CAS.
- J. E. Kohn and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10841 CrossRef CAS.
- E. K. Koepf, H. M. Petrassi, G. Ratnaswamy, M. E. Huff, M. Sudol and J. W. Kelly, Biochemistry, 1999, 38, 14338 CrossRef CAS.
- A. Friendler, D. B. Veprintsev, L. O. Hansson and A. R. Fersht, J. Biol. Chem., 2003, 278, 24108 CrossRef.
- A. Friedler, B. S. DeDecker, S. M. V. Freund, C. Blair, S. Rudiger and A. R. Fersht, J. Mol. Biol., 2004, 336, 187 CrossRef CAS.
- K. Vamvaca, B. Vogeli, P. Kast, K. Pervushin and D. Hilvert, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12860 CrossRef CAS.
- G. P. Tochtrop, K. Richter, C. Tang, J. T. Toner, D. F. Covey and D. P. Cistola, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1847 CrossRef CAS.
- G. P. Tochtrop, J. L. Bruns, C. Tang, D. F. Covet and D. P. Cistola, Biochemistry, 2003, 42, 11561 CrossRef CAS.
- O. Toke, J. D. Monsey, G. T. DeKoster, G. P. Tochtrop, C. Tang and D. P. Cistola, Biochemistry, 2006, 45, 727 CrossRef CAS.
- K. Kim, D. P. Cistola and C. Frieden, Biochemistry, 1996, 35, 7553 CrossRef CAS.
- L. M. Curto, J. J. Caramelo, J. M. Delfino and M. Jose, Biochemistry, 2005, 44, 13847 CrossRef CAS.
- B. Corsico, D. P. Cistola, C. Frieden and J. Storch, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12174 CrossRef CAS.
- B. Corsico, H. L. Liou and J. Storch, Biochemistry, 2004, 43, 3600 CrossRef CAS.
- B. Corsico, G. R. Franchini, K.-T. Hsu and J. Storch, J. Lipid Res., 2005, 46, 1765 CAS.
- C. Lucke, F. Zhang, H. Ruterjans, J. A. Hamilton and J. C. Sacchettini, Structure, 1996, 4, 785 CrossRef CAS.
- H. K. Binz, P. Amstutz and A. Pluckthun, Nat. Biotechnol., 2005, 23, 1257 CrossRef CAS.
- G. Beste, S. F. Schmidt, T. Stibora and A. Skerra, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1898 CrossRef CAS.
- D. Qi, C.-M. Tann, D. Haring and M. D. Distefano, Chem. Rev., 2001, 101, 3081 CrossRef CAS.
- D. H. Williams, D. P. O'Brien, A. M. Sanderbrook and E. Stephens, J. Mol. Biol., 2004, 340, 373 CrossRef CAS.
- D. H. Williams, E. Stephens and M. Zhou, J. Mol. Biol., 2003, 329, 389 CrossRef CAS.
- D. Tobi and I. Bahar, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18908 CrossRef CAS.
- H. R. Bossard, News Physiol. Sci., 2001, 16, 171 Search PubMed.
- P. A. Borea, A. Delpiaz, K. Varani, P. Gilli and G. Gilli, Biochem. Pharmacol., 2000, 60, 1549 CrossRef CAS.
- P. A. Borea, K. Varani, S. Gessi, P. Gilli and A. Delpiaz, Farmaco, 1998, 53, 249 CrossRef CAS.
- R. Koradi, M. Billeter and K. Wuthrich, J. Mol. Graphics, 1996, 14, 51 CrossRef CAS.
Footnote |
| † All bile acid salts were purchased from Sigma. NMR data were collected at 600 MHz on a Bruker Avance600 spectrometer equipped with a triple resonance probe with z-field gradients. CD spectra were collected on an Applied Photophysics pi-star system with temperature regulation using a Neslab RTE-300 circulating water bath. A 1 mm pathlength cell was used with 5 µM solutions of protein in 20 mM phosphate buffered solution (PBS) at pH 7.2 in the presence of up to 1 mM of bile acid ligand. Spectra were collected over the wavelength range 200–260 nm in 4 nm steps. For ITC experiments 1.43 ml of 30–170 µM protein solutions in the same phosphate buffer were injected with 50–70 successive 4–6 µl volumes of concentrated ligand solutions of 1.3–6.3 mM at 240 s intervals. DSC data were collected on 30 µM protein solutions in PBS at pH 7.2 at ligand concentrations between 100–500 µM. Up-scan rates were 90 °C h−1 and down scans 60 °C h−1. All ITC (one site model) and DSC data were fitted using Origin 7.0 software (MicroCal Inc.). |
| This journal is © The Royal Society of Chemistry 2006 |