Iron hydrogenase active site mimic holding a proton and a hydride
Lennart
Schwartz
ab,
Gerriet
Eilers
c,
Lars
Eriksson
a,
Adolf
Gogoll
b,
Reiner
Lomoth
*c and
Sascha
Ott
*bc
aDepartments of Organic and Structural Chemistry, Arrhenius Laboratory, Stockholm University, 10691, Stockholm, Sweden
bDepartment of Chemistry, Organic Chemistry, BMC, Uppsala University, P.O. Box 599, 75124, Uppsala, Sweden. E-mail: sascha.ott@fki.uu.se; Fax: +46 (0)18 4717340
cDepartment of Physical Chemistry, BMC, Uppsala University, P.O. Box 579, 751 23, Uppsala, Sweden. E-mail: reiner.lomoth@fki.uu.se; Fax: +46 (0)18 4713654
First published on 20th December 2005
Abstract
The first model of the iron hydrogenase active site has been prepared which concomitantly carries a proton and a hydride; the title species was characterized by IR and NMR spectroscopy and is reduced at more positive potential than any other mimic of this kind.
Iron hydrogenases (Fe-H2ase) are enzymes which catalyze the reversible reaction of protons and electrons to molecular hydrogen.1 Whereas the crystal structure elucidation of two Fe-H2ases provided a detailed structural picture of their active sites (H-cluster, Chart 1a),2,3 small synthetic model complexes are valuable aids to compare and to understand the spectroscopic signatures of the enzyme.4–7 Furthermore, functional studies on the model complexes have contributed to the understanding of the enzyme's mechanism.8–10
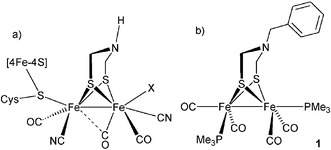 | ||
| Chart 1 | ||
The active site is comprised of two unusual low-valent diiron nuclei which are coordinated by a cysteine-linked [Fe4S4] cluster, carbon monoxide and cyanide ligands and by a non-proteic azadithiolate (adt = S-CH2-NH-CH2-S) bridging the two iron centers.11,12 As the reduction of protons or the oxidation of hydrogen follows an ionic mechanism, the H-cluster inevitably has to pass through a state where it carries a proton and a hydride. Using computational techniques, the central nitrogen atom of the dithiolate bridge was identified as a potential basic site,13 whereas the cyanide ligands increase the electron density at the iron centers and facilitate the formation of a hydride at this position. Although the hydride has to reside at least transiently in a terminal position to engage in an intramolecular reaction with the proton,14,15 a bridging hydride cannot be excluded as an intermediate state of the Fe-H2ase. In fact, a bridging hydride has recently been identified in the functionally related Ni-Fe H2ase.16 We therefore set out to synthesize a novel Fe-H2ase active site mimic which contains electron donating ligands at the diiron core as well as an adt linker (Chart 1b), anticipating that an unprecedented species carrying a hydride at the diiron core and a proton at the adt nitrogen may be realized. Such a structure can be regarded as a snapshot of the H-cluster just before the formation of the H–H bond.
Hence, a solution of readily synthesized [Fe2(μ-adt)(CO)6] 217–19 in hexane was treated with trimethylphosphine to afford complex 1 in excellent yield.20 Complex 1 is rather air sensitive in solution and attempts to purify the complex by column chromatography resulted in complete degradation of the product. However, addition of a little toluene and removal of the solvents and of excess trimethylphosphine at high vacuum spontaneously produced crystalline material suitable for X-ray analysis.† Single-crystal X-ray diffraction analysis of 1 (Fig. 1) reveals the usual edge-bridged bi-square-pyramidal geometry around the iron centers which are at a distance of 2.55 Å.21 The two trimethylphosphine ligands prefer an unsymmetric geometry around the iron centers in the solid state with one in an apical and the other in a basal position. This conformation is similar to that preferred by an ethyldithiolate-bridged diiron complex where a minimum of steric hindrance at the bridge has been suggested to facilitate this arrangement.20
![ORTEP view (ellipsoids at 50% probability level) of [Fe2(μ-SCH2N(CH2Ph)CH2S)(CO)4(PMe3)2] (1). Selected bond lengths (Å): Fe1–Fe2 2.5461(12), Fe1–S1 2.2663(18), Fe1–S2 2.2546(16), Fe2–S1 2.2560(17), Fe2–S2 2.2465(16), Fe1–P1 2.2174(18), Fe2–P2 2.2255(18), Fe1–C1 1.724(9), Fe1–C2 1.766(6), Fe2–C3 1.767(9), Fe2–C4 1.774(7).](/image/article/2006/CC/b514280f/b514280f-f1.gif) | ||
| Fig. 1 ORTEP view (ellipsoids at 50% probability level) of [Fe2(μ-SCH2N(CH2Ph)CH2S)(CO)4(PMe3)2] (1). Selected bond lengths (Å): Fe1–Fe2 2.5461(12), Fe1–S1 2.2663(18), Fe1–S2 2.2546(16), Fe2–S1 2.2560(17), Fe2–S2 2.2465(16), Fe1–P1 2.2174(18), Fe2–P2 2.2255(18), Fe1–C1 1.724(9), Fe1–C2 1.766(6), Fe2–C3 1.767(9), Fe2–C4 1.774(7). | ||
Addition of one equivalent of triflic acid to a solution of 1 in CH3CN results in the formation of [1H]+, as evidenced by the IR spectra (Fig. 2a, b) that show a shift of ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CO = 16 cm−1 towards higher energy. This shift is typical for a protonation of the adt nitrogen.8,17 Addition of triethanolamine (TEOA) reverses the adt protonation and complex 1 is quantitatively recovered. Upon addition of an excess of triflic acid, [1H]+ is transformed into a product characterized by the IR spectrum shown in Fig. 2c. This spectrum can be assigned to the doubly protonated species [Fe2(μ-Hadt)(μ-H)(CO)4(PMe3)2]2+, [1HHy]2+, with an additional average shift of CO
≈ 80 cm−1, reflecting the substantial decrease of electron density at the diiron site as formally expressed by the FeII–FeII valence state of the hydride complex.10,22[1HHy]2+ is the first Fe-H2ase active site mimic which carries a proton at the adt nitrogen as well as a hydride at the diiron core. Initial attempts to deprotonate [1HHy]2+ to recover 1 with an excess of triethanolamine failed and a new spectrum emerged which in shape resembled that of [1HHy]2+, however shifted by CO
≈ 16 cm−1 towards lower frequencies. It thus seems that the adt nitrogen is selectively deprotonated, leaving the hydride portion intact and the species formed is identified as the hydride complex [Fe2(μ-adt)(μ-H)(CO)4(PMe3)2]+, [1Hy]+. The formation of [1Hy]+ by deprotonation of [1HHy]2+ relies on the inertness of the hydride towards nitrogen bases. The sluggish deprotonation behavior with nitrogen bases has been reported for a related hydridic diiron complex,10 but could be overcome by the addition of cyanide or chloride.23 In the case of [1Hy]+ addition of tetrabutylammonium chloride results in quantitative recovery of 1 (Fig. 2d, dashed line). Complex 1 constitutes a dibasic species which, as such, should give rise to three different protonation states. However, owing to the remarkable difference in deprotonation kinetics between the adt nitrogen and the Fe–Fe bond, complex 1 can selectively be prepared in four protonation states, 1, [1H]+, [1Hy]+, and [1HHy]2+ (Scheme 1).24
CO = 16 cm−1 towards higher energy. This shift is typical for a protonation of the adt nitrogen.8,17 Addition of triethanolamine (TEOA) reverses the adt protonation and complex 1 is quantitatively recovered. Upon addition of an excess of triflic acid, [1H]+ is transformed into a product characterized by the IR spectrum shown in Fig. 2c. This spectrum can be assigned to the doubly protonated species [Fe2(μ-Hadt)(μ-H)(CO)4(PMe3)2]2+, [1HHy]2+, with an additional average shift of CO
≈ 80 cm−1, reflecting the substantial decrease of electron density at the diiron site as formally expressed by the FeII–FeII valence state of the hydride complex.10,22[1HHy]2+ is the first Fe-H2ase active site mimic which carries a proton at the adt nitrogen as well as a hydride at the diiron core. Initial attempts to deprotonate [1HHy]2+ to recover 1 with an excess of triethanolamine failed and a new spectrum emerged which in shape resembled that of [1HHy]2+, however shifted by CO
≈ 16 cm−1 towards lower frequencies. It thus seems that the adt nitrogen is selectively deprotonated, leaving the hydride portion intact and the species formed is identified as the hydride complex [Fe2(μ-adt)(μ-H)(CO)4(PMe3)2]+, [1Hy]+. The formation of [1Hy]+ by deprotonation of [1HHy]2+ relies on the inertness of the hydride towards nitrogen bases. The sluggish deprotonation behavior with nitrogen bases has been reported for a related hydridic diiron complex,10 but could be overcome by the addition of cyanide or chloride.23 In the case of [1Hy]+ addition of tetrabutylammonium chloride results in quantitative recovery of 1 (Fig. 2d, dashed line). Complex 1 constitutes a dibasic species which, as such, should give rise to three different protonation states. However, owing to the remarkable difference in deprotonation kinetics between the adt nitrogen and the Fe–Fe bond, complex 1 can selectively be prepared in four protonation states, 1, [1H]+, [1Hy]+, and [1HHy]2+ (Scheme 1).24
![Carbonyl region of the IR spectra obtained for solutions of 1 (3 mM) in CH3CN. Protonated states were generated under the conditions indicated in parentheses. a) 1. b) [1H]+ (3 mM HOTf, —); Spectrum after deprotonation with triethanolamine (TEOA, ). c) [1HHy]2+ (0.2 M HOTf). d) [1Hy]+ (0.2 M HOTf followed by TEOA, —); Spectrum after deprotonation with tetrabutylammonium chloride (TBACl, ).](/image/article/2006/CC/b514280f/b514280f-f2.gif) | ||
Fig. 2 Carbonyl region of the IR spectra obtained for solutions of 1 (3 mM) in CH3CN. Protonated states were generated under the conditions indicated in parentheses. a) 1. b) [1H]+ (3 mM HOTf, —); Spectrum after deprotonation with triethanolamine (TEOA, ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). c) [1HHy]2+ (0.2 M HOTf). d) [1Hy]+ (0.2 M HOTf followed by TEOA, —); Spectrum after deprotonation with tetrabutylammonium chloride (TBACl, ). ). c) [1HHy]2+ (0.2 M HOTf). d) [1Hy]+ (0.2 M HOTf followed by TEOA, —); Spectrum after deprotonation with tetrabutylammonium chloride (TBACl, ). | ||
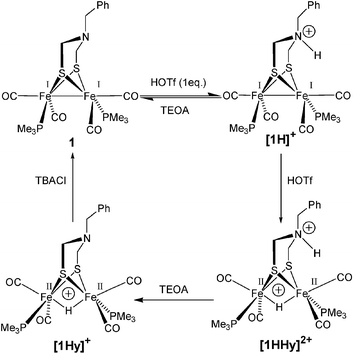 | ||
| Scheme 1 Protonation reactions of 1 in acetonitrile solution with triflic acid (HOTf). Deprotonation of the adt nitrogen with triethanolamine (TEOA) and of the hydride with tetrabutylammonium chloride (TBACl). | ||
NMR characterization of all four states shows that protonation of the adt nitrogen renders the two Fe(CO)2P(CH3)3 subunits in [1H]+ and [1HHy]2+ non-equivalent as the ring inversion of the six-membered metalloazaheterocycle is restricted. Hence, the trimethylphosphine protons which give rise to one doublet in the 1H NMR spectra of 1 and [1Hy]+ due to their coupling to the phosphorus center, are split into two doublets at δ = 1.56, 1.54 ppm for [1H]+ and at δ = 1.57, 1.63 ppm for [1HHy]2+. Similarly, the 31P NMR spectra of the adt-protonated [1H]+ and [1HHy]2+ feature two phosphorus signals at δ = 31.1, 19.8 ppm for [1H]+ and at δ = 25.3, 21.3 ppm for [1HHy]2+ whereas only one signal is observed in the spectra of 1 and [1Hy]+ (Fig. 3). The hydride is visible in the 1H NMR spectra of [1Hy]+ and [1HHy]2+ as a triplet (JH-P = 22 Hz) at δ = −15.6 ppm and as a doublet of doublets (JH–P = 21, 23 Hz) at similar chemical shift, respectively. Since the same coupling constant can be observed in the 31P NMR of the respective species, it is clear that the hydride in [1Hy]+ and [1HHy]2+ resides in a bridging position25 and that the complexes are stable even under these drastic acidic conditions. In contrast to the solid state structure, the phosphine ligands prefer a symmetrical basal position relative to the iron centers in [1Hy]+ and [1HHy]2+ as evidenced by the relatively large coupling constants of JH–P = 22 Hz.20
![31P NMR spectra of 1, [1H]+, [1Hy]+ and [1HHy]2+ in CD3CN at 25 oC. Prepared from 1 (6 mM) under conditions similar to those used in the IR experiments.](/image/article/2006/CC/b514280f/b514280f-f3.gif) | ||
| Fig. 3 31P NMR spectra of 1, [1H]+, [1Hy]+ and [1HHy]2+ in CD3CN at 25 oC. Prepared from 1 (6 mM) under conditions similar to those used in the IR experiments. | ||
Voltammetric investigation of complex 1 reveals an irreversible reduction at ca. −2.2 V vs. Fc+/0 and the potential for the first reduction is shifted to −1.0 V for [1HHy]2+. This dramatic shift of 1.2 V is a result of the fact that [1HHy]2+ concurrently carries a proton and a hydride.26
In summary, we have synthesized the first biomimetic model of the Fe-H2ase active site which can be protonated on either the Fe–Fe bond or the adt nitrogen as well as on both sites simultaneously. All four protonation states are well-defined and have been characterized by IR and NMR spectroscopy. Double protonated [1HHy]2+ is reduced at −1.0 V vs. Fc+/0, a potential considerably more positive than that of any other Fe-H2ase mimic ever reported. Reduction at such mild potential is a result of the two preceding protonations, one of which involves the adt nitrogen.26 From a structural point of view, [1HHy]2+ resembles an intermediate closer to the hydrogen formation event than other model complexes which only carry either a hydride or a proton.8,10 With regard to the enzyme, the question arises as to whether the catalytic cycle involves the formation of a double protonated species at the FeI–FeI level similar to our model system.
We gratefully acknowledge financial support by the Swedish Energy Agency, the Swedish Research Council and NEST, SOLAR-H (EU contr. nr. 516510).
Notes and references
- M. W. W. Adams, Biochim. Biophys. Acta, 1990, 1020, 115–145 CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS.
- Y. Nicolet, C. Piras, P. Legrand, E. C. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS.
- M. W. W. Adams and E. I. Stiefel, Curr. Opin. Chem. Biol., 2000, 4, 214–220 CrossRef CAS.
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268–275 RSC.
- M. Razavet, S. J. Borg, S. J. George, S. P. Best, S. A. Fairhurst and C. J. Pickett, Chem. Commun., 2002, 700–701 RSC.
- C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. D. Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610–613 CrossRef CAS.
- S. Ott, M. Kritikos, B. Åkermark, L. Sun and R. Lomoth, Angew. Chem., Int. Ed., 2004, 43, 1006–1009 CrossRef CAS.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2005, 249, 1664–1676 CrossRef CAS.
- F. Gloaguen, J. D. Lawrence, T. B. Rauchfuss, M. Benard and M.-M. Rohmer, Inorg. Chem., 2002, 41, 6573–6582 CrossRef CAS.
- M. Frey, ChemBioChem, 2002, 3, 152–160.
- Y. Nicolet, B. J. Lemon, J. C. Fontecilla-Camps and J. W. Peters, Trends Biochem. Sci., 2000, 25, 138–143 CrossRef CAS.
- H.-J. Fan and M. B. Hall, J. Am. Chem. Soc., 2001, 123, 3828–3829 CrossRef CAS.
- Y. Nicolet, A. L. D. Lacey, X. Vernede, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1602 CrossRef CAS.
- Z.-P. Liu and P. Hu, J. Phys. Chem., 2002, 117, 8177–8180 Search PubMed.
- S. Foerster, M. Stein, M. Brecht, H. Ogata, Y. Higuchi and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 83–93 CrossRef CAS.
- J. D. Lawrence, H. Li, T. B. Rauchfuss, M. Benard and M.-M. Rohmer, Angew. Chem., Int. Ed., 2001, 40, 1768–1771 CrossRef CAS.
- S. Ott, M. Kritikos, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2003, 42, 3285–3288 CrossRef CAS.
- S. Ott, M. Borgström, M. Kritikos, R. Lomoth, J. Bergquist, B. Åkermark, L. Hammarström and L. Sun, Inorg. Chem., 2004, 43, 4683–4692 CrossRef CAS.
- X. Zhao, I. P. Georgakaki, M. L. Miller, R. Mejia-Rodriguez, C.-Y. Chiang and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 3917–3928 CrossRef CAS.
- CCDC 283269 (1) and CCDC 283270 (2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
- R. Mejia-Rodriguez, D. Chong, J. H. Reibenspies, M. P. Soriaga and M. Y. Darensbourg, J. Am. Chem. Soc., 2004, 126, 12004–12014 CrossRef CAS.
- X. Zhao, Y.-M. Hsiao, C.-H. Lai, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 699–708 CrossRef CAS.
- Since no interconversion between [1H]+ and [1Hy]+ could be observed on the timescale of hours, it cannot readily be determined which mono-protonated species is the thermodynamically stable form.
- In addition, two-dimensional 1H-31P correlation spectroscopy (HMQC) of [1HHy]2+ revealed the coupling of the hydride to both phosphorus centers.
- Reference 10 describes a related diiron-hydride complex lacking the adt nitrogen which can be protonated at a cyanide ligand instead. This species, however, is reduced at −1 V vs. Ag/AgCl, corresponding to less than −1.4 V vs. Fc+/0.
Footnote |
| † [Fe2(μ-SCH2N(CH2Ph)CH2S)(CO)4(PMe3)2] (1). Trimethylphosphine (204 mg, 2.68 mmol) was added to a solution of complex 2 (160 mg, 0.335 mmol) in 5 ml hexane under nitrogen atmosphere. After 3 h of stirring, the solvent and unreacted trimethylphosphine were removed in vacuo. The resulting red-brown solid was re-dissolved in toluene and the solution was filtered through a plug of Celite. The solvent was removed, and the deep red solid was washed with cold hexane (174 mg, 91%). Single crystals suitable for X-ray analysis were obtained from toluene/hexane solution upon rapid concentration in vacuo. Anal. calculated for C19H29Fe2NO4P2S2: C, 39.81; H, 5.10; N, 2.44. Found: C, 39.83; H, 5.27; N, 2.38%. 1H NMR (400 MHz, CDCl3): δ = 7.30–7.26 (m, 3H, ArH), 7.18 (d, J = 6.8 Hz, 2H, ArH), 3.55 (s, 2H, NCH2Ph), 3.08 (s, 4H, SCH2N), 1.50 (d, J = 9.2 Hz, 18H, PMe3). 13C NMR (100.6 MHz, THF-d8): δ = 217.5, 217.3, 137.9, 130.3, 129.1, 128.3, 63.8, 54.2, 20.8, 20.4 (2C). 31P NMR (161.9 MHz, CD3CN): δ = 23.3. IR (CH3CN, cm−1): νCO = 1980, 1943, 1907, 1892 (sh). Crystallographic data of 1: Monoclinic, a = 10.7622(14) Å, b = 19.773(3) Å, c = 11.7983(15) Å, β = 90.390(15)°, vol = 2510.7(6) Å3, T = 293(2) K, P21/c, Z = 4, μ = 1.474 mm−1, Nmeasured = 16225, Nunique,all data = 4834, Nunique, (I ≥ 2σ(I)) = 3035, Rint = 0.1047, wR2 = 0.1650 (all data), R1 = 0.0492 (I ≥ 2σ(I)). CCDC 283269. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b514280f |
| This journal is © The Royal Society of Chemistry 2006 |