The first insoluble polymer-bound palladium complexes of 2-pyridyldiphenylphosphine: highly efficient catalysts for the alkoxycarbonylation of terminal alkynes†
Simon
Doherty
*,
Julian G.
Knight
* and
Michael
Betham
School of Natural Sciences, Chemistry, Bedson Building, University of Newcastle upon Tyne, Newcastle upon Tyne, NE1 7RU, UK. E-mail: simon.doherty@ncl.ac.uk; j.g.knight@ncl.ac.uk; Fax: +44 (0) 191 222 6929; Tel: +44(0) 191 222 6537 Tel: +44(0) 191 2227068
First published on 14th November 2005
Abstract
Palladium complexes of 2-pyridyldiphenylphosphine anchored on polystyrene, polymethylmethacrylate and styrene–methylmethacrylate copolymer form highly active heterogeneous catalysts for the alkoxycarbonylation of terminal alkynes with activities approaching those obtained under homogeneous conditions.
The regioselective palladium catalysed inter- and intramolecular alkoxycarbonylation of terminal alkynes has proven to be a highly versatile process for the synthesis of acyclic and cyclic α,β-unsaturated esters (eqn (1)).1 Some time ago researchers at Shell discovered that catalysts formed by combining 2-pyridyldiphenylphosphine (2-pyPPh2), a source of palladium(II) and a sulfonic acid were highly active and selective for the methoxycarbonylation of propyne, giving turn over numbers as high as 40,000 molproduct molcat−1 h−1 with a selectivity for methyl methacrylate (MMA) of 99.95% under mild conditions.2
 | (1) |
The use of 2-pyPPh2 in the palladium catalysed alkoxycarbonylation of alkynes is an exceptional example of a ligand that plays a dual role achieving high selectivity through its ability to function as a bidentate P,N-coordinated ligand while a second P-coordinated N-protonated 2-pyPPh2 facilitates rapid proton transfer in the rate determining protonolysis step. The critical role of the pyridyl group becomes clearly evident upon replacement of 2-pyPPh2 by triphenylphosphine, which results in a dramatic decrease in catalyst activity to ca. 10 molproduct molcat−1 h−1 as well as a marked reduction in selectivity to 89%.2 Given the potential impact of this process for the commercial production of MMA, a large scale (2 × 106 tonnes per annum) industrial intermediate for the production of homopolymers and copolymers,3 as well as for the preparation of fine chemicals,4 there is likely to be interest in the immobilisation of 2-pyPPh2 to develop a continuous process which would combine high catalyst activity and selectivity with facile product separation, in which catalyst leaching is limited. While it is relatively straightforward to immobilise triphenylphosphine5 as well as bi-6 and terdentate phosphines,7 immobilisation of 2-pyPPh2 to a solid support presents a much greater challenge. Herein, we describe the first example of 2-pyPPh2 anchored to insoluble polymeric supports and a preliminary investigation of their application to the palladium-catalysed alkoxycarbonylation of terminal alkynes.
The 2-pyridylstyrylphenylphosphine monomer 3 was prepared according to Scheme 1. Lithiation of 2-bromopyridine followed by transmetalation with zinc chloride in pyridine gave the cyclic 2-pyridyl zinc chloride dimer 1,8 which was quenched with dichlorophenylphosphine to afford 2-pyridylphenylchlorophosphine 2. The polymerisable styryl group was introduced in the final step by reaction of 2 with styryl magnesium chloride. The copolymerisation of 3 with styrene was carried out in benzene using 2,2′-azobis(2-methyl)propionitrile (AIBN) as radical initiator to give polystyrene supported 2-pyridyldiphenylphosphine 4, as a white solid after precipitation by addition of methanol. Similarly, suspension copolymerisation of 3 with methyl methacrylate gave the corresponding 2-pyridyldiphenylphosphine loaded copolymer 5. Both polymers have a unimodal molecular weight distribution with polydispersities Mw/Mn of 2.1 and 2.0, respectively, and the 2-pyridyldiphenylphosphine loading was determined to be 1.20 and 1.49 mmol g−1, respectively, from elemental analysis.
 | ||
| Scheme 1 Reagents and conditions: (i) BuLi, ZnCl2, py; (ii) PPhCl2, THF–py; (iii) styrylMgCl, THF 70 °C; (iv) styrene, AIBN, C6H6 (v) MMA, AIBN, C6H6. | ||
The 2-pyridyl(p-methacryloyloxyphenyl)phenylphosphine monomer 9 was also targeted for the preparation of a 2-pyPPh2 anchored methacrylate homopolymer and details of the synthesis are outlined in Scheme 2. Initial attempts to reduce 2 with LiAlH4, NaH and DIBAL all proved unsuccessful either resulting in partial reduction or formation of phosphine oxide. Fortunately, the desired secondary phosphine 6 was successfully prepared by careful hydrolysis of the corresponding magnesium phosphide which was readily generated in situ from 2-pyridylphenylchlorophosphine and magnesium. A palladium-catalysed cross coupling between 4-iodophenol and 6 was used to introduce the 4-hydroxyphenyl substituent in 7, which was protected as its borane adduct and reacted with methacryloyl chloride to afford monomer 9, as a white crystalline air-stable solid. Methacrylate based homopolymer 10 was prepared by copolymerisation of 9 with methyl methacrylate in benzene using AIBN as radical initiator, isolated as an off-white powder by repeated precipitation with methanol and subsequently deprotected with DABCO in dichloromethane. The 2-pyridyldiphenylphosphine loading in 10 was determined to be 1.42 mmol g−1 from elemental analysis and the polydispersity of 3.8 was measured by gel permeation chromatography.
 | ||
| Scheme 2 Reagents and conditions: (i) Mg, H2O; (ii) 4-IC6H4OH, DMA, 10 mol% Pd(OAc)2, 70 °C, 12 h; (iii) BH3·SMe2, THF, 12 h; (iv) methacryloyl chloride, NEt3, CH2Cl2 −78–0 °C (v) C6H6, AIBN, MMA, 60 °C, 12 h (vi) CH2Cl2, DABCO, RT 12 h. | ||
Catalysts formed by combining polymers 4, 5 and 10 with palladium acetate were first tested in the methoxycarbonylation of phenylacetylene, the results of which are listed in Table 1. Since the effect on catalyst performance of the amount of acid and ligand has been well documented9 investigations were carried out to establish the optimal reaction conditions. The best performance in terms of activity and selectivity was most consistently obtained with L ∶ Pd and acid ∶ Pd mole ratios of 10 and 30, respectively, in agreement with the previous studies of Drent.2 Initial studies demonstrated that introduction of a 4-vinyl substituent into 2-pyPPh2 did not effect catalyst performance since catalysts formed from 2-pyPPh2 and 3 were both highly active and selective for production of the branched acryclic ester, methyl 2-phenylpropenoate, I (entries 1 & 2). The performance of supported catalysts generated from 4, 5 and 10 and palladium acetate were next investigated and each was found to be highly efficient, giving activities and selectivities approaching those of their homogeneous counterpart (entries 3–5). For each of the polymers, analysis of the reaction mixture after filtration revealed that the palladium content was too low to be detected by ICP-OES, confirming that leaching was negligible. In addition, the filtered solutions showed no activity indicating that any palladium in solution was either very low in concentration or was in an inactive form.
| Entry | Ligand/support | Substrateb | Activityc mol product mol cat−1 h−1 | Selectivityd |
|---|---|---|---|---|
| a Conditions: batch, intake: 30 ml methanol, 40 bar CO, 0.1 mmol ligand, 0.01 mmol Pd(OAc)2, 0.3 mmol CH3SO3H, 50 °C. b 10 mmol phenylacetylene or 2.0 bar propyne. c Calculated by GC analysis, average of three runs. d Determined by GC analysis and 1H NMR. | ||||
| 1 | 2-pyPPh2 | PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
306 | 97.4 |
| 2 | 3 | PhCCH |
300 | 97.1 |
| 3 | 4 | PhCCH |
273 | 97.5 |
| 4 | 5 | PhCCH |
225 | 97.3 |
| 5 | 10 | PhCCH |
260 | 98.0 |
| 6 | 11 | PhCCH |
20 | >99.5 |
| 7 | PPh3 | PhCCH |
11.7 | 81.8 |
| 8 | 12 | PhCCH |
2.0 | 80.3 |
| 9 | 13 | PhCCH |
4.3 | >99.5 |
| 11 | 2-pyPPh2 | MeCCH |
1171 | 97.8 |
| 12 | 3 | MeCCH |
1098 | 97.7 |
| 13 | 4 | MeCCH |
284 | 97.9 |
| 14 | 5 | MeCCH |
608 | 98.0 |
| 15 | 10 | MeCCH |
1022 | 97.8 |
| 16 | 11 | MeCCH |
55 | >99.5 |
| 17 | 13 | MeCCH |
17 | >99.5 |
The high activity and selectivity associated with alkoxycarbonylation of alkynes has been attributed to an active species containing two molecules of 2-pyPPh2, one of which functions as a bidentate P,N-ligand and is involved in the selectivity determining step while the other coordinates in a monodentate manner and facilitates proton transfer by acting as a cocatalyst in the protonolysis step of the catalytic cycle.2,10 In this regard, the performance of these polymer supported catalysts was rather surprising and suggests that copolymers 4, 5 and 10 have sufficient flexibility to form 2 ∶ 1 complexes at low palladium loadings. However, at this stage we cannot exclude the possibility that the copolymer precludes coordination of two ligands to a single metal centre and that the active species is actually mononuclear and coordinated by a single ligand. Further studies are clearly required to establish the distribution of 2-pyPPh2 ligands in these polymers and the composition of the active species. Accepting rapid protonolysis by a proximate pyridyl group to be an integral part of the palladium catalysed alkoxycarbonylation, we reasoned that the introduction of a pyridyl substituent on to the polymer backbone would manifest itself in an increase in catalyst activity. Polyvinylpyridine supported 2-pyPPh211 was prepared by copolymerisation of 4-vinylpyridine with 3 in the presence of AIBN. Contrary to expectation, replacement of the polystyrene backbone with polyvinylpyridine resulted in a dramatic decrease in activity from 273 to 20 molproduct molPd−1 h−1, even in the presence of a large excess of acid. At this stage we tentatively attribute this decrease in activty to coordination of the polyvinylpyridine matrix to the metal rather than to diffusional limitations or intrusion of the polymer matrix into the coordination sphere of the metal.11 Interestingly, Drent has reported a progressive decrease in catalyst activity with increasing number of pyridyl substituents attached to the phosphine while the number of 2-pyridyl groups had negligible effect on the regioselectivity.2 In this regard, the increase in selectivity to >99.5% with polyvinylpyridine supported 2-pyPPh2 is remarkable (entry 6). An increase in regioselectivity to >99.95% has previously been achieved by methyl substitution at the 6-position of the 2-pyridyl ring. Since regioselectivity appears to be controlled by steric factors it is tempting to attribute the increase in selectivity associated with polyvinylpyridine supported 11 to a change in the polymer microstructure, possibly as a result of the different swelling capacities in methanol.
In an attempt to glean further information about the polymer composition–catalyst activity/selectivity relationship the performance of polystyrene 12 and polyvinylpyridine 13 supported triphenylphosphine based catalysts was investigated and both were markedly less active than their polystyrene 4 and polyvinylpyridine 11 supported 2-pyPPh2 counterparts (entries 8 and 9). Moreover, while the selectivity of 80.3% achieved with polystyrene supported triphenylphosphine is similar to that of 81.8% obtained under homogeneous conditions (entry 7), its polyvinylpyridine supported counterpart 13 gave a selectivity >99.5%. This level of selectivity is particularly exceptional for a triphenylphosphine based catalyst and is similar to that obtained with polyvinylpyridine based 2-pyPPh211, described above. Thus, while the combination of PPh3 and a pyridine backbone in 13 does not give high activity it gives a selectivity comparable to that obtained with 2-pyPPh2 based catalyst systems.
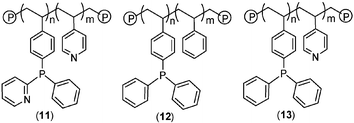
Interest in the selective production of methyl methacrylate prompted us to examine the efficiency of the same catalyst systems in the methoxycarbonylation of propyne, the results of which are listed in Table 1 (entries 11–17). Under standard conditions, the performance of a catalyst mixture generated with 3 was qualitatively similar to that obtained with 2-pyPPh2. In stark contrast to phenylacetylene, the performance of catalysts generated from polymers 4, 5 and 10 in the carbonylation of propyne showed a marked dependence on the co-monomer and the tether. For instance, the activty of 1022 molproduct molPd−1 h−1 obtained with methacrylate-based homopolymer 10 is comparable to that of the homogeneous 2-pyPPh2 system while the activities obtained with methacrylate-styrene copolymer 5 and the styrene homopolymer 4 are significantly lower, which is likely to be due to better diffusion of the reagents in the more polar polymeric network of 10. The performance of polyvinylpyridine supported 2-pyPPh213 and triphenylphosphine 11 were also examined under the same conditions and the activities of 17 and 55 molproduct molPd−1 h−1, respectively, correspond to a 20–50 fold decrease in activity compared with that of 1171 molproduct molPd−1 h−1 for 2-pyPPh2 under homogeneous conditions. However, catalyst systems based on 11 and 13 both gave a marked increase in selectivity to >99.5%, which parallels that observed in the methoxycarbonylation of phenylacetylene.
In conclusion, the first insoluble polymer supported 2-pyPPh2 ligands have been prepared and shown to form highly active and selective catalysts for the alkoxycarbonylation of terminal alkynes. In the case of polyvinylpyridine supported 2-pyPPh2, very high selectivities were obtained but at the expense of activity. Although the performance of the polystyrene and polymethacrylate based catalysts did not exceed that of the corresponding homogeneous system, in several cases it was very similar and there was no evidence of metal leaching, which renders these immobilized catalysts ideal candidates for multiple recycling in a range of platinum group metal catalysed transformations. Ultimately, we aim to identify robust processible catalysts that can either be extruded or coated onto high surface area substrates for use in a continuous process. Currently, studies are underway to (i) investigate the effect of pyridine ring substitution and basicity on catalyst performance, (ii) expand the range of substrates and reaction types available to these supported 2-pyPPh2 ligands, (iii) determine the nature and composition of the active species and (iv) systematically modify the co-monomer and the tether to delineate the factors that influence catalyst activity and selectivity.
We thank the Institute of applied catalysis (iAc) for a bursary to MB, the University of Newcastle upon Tyne for financial support and Johnson Matthey for generous loans of palladium salts.
Notes and references
- J. Tsuji, in Palladium reagents and catalysts, Wiley, New York, 1996, p. 471 Search PubMed; B. El Ali and H. Alper, in Transition metals for organic synthesis, ed. M. Beller, Vol I, Wiley-VCH, Weinheim, 1998 Search PubMed; B. El Ali and H. Alper, Synlett, 2000, 161 Search PubMed; C. S. Consorti, G. Ebeling and J. Dupont, Tetrahedron Lett., 2002, 43, 753 Search PubMed.
- E. Drent, P. Arnold and P. H. M. Budzelaar, J. Organomet. Chem., 1994, 475, 57 CrossRef CAS; E. Drent, P. Arnold and P. H. M. Budzelaar, J. Organomet. Chem., 1993, 455, 247 CrossRef CAS; E. Drent, W. W. Jager, J. J. Keijsper and G. G. M. Niele, in Applied homogeneous catalysis with organometallic compounds, ed. B. Cornils and W. A. Hermann, VCH, Weinheim, 1996, vol. II, p. 1119 Search PubMed.
- Kirk Othmer, Encyclopedia of Chemical Technology, Wiley, New York, 4th edn., 1995, vol. 16, p. 487, Chem Week, 1999, Nov. 10th 14 Search PubMed.
- A. Scrivanti and U. Matteoli, Tetrahedron Lett., 1995, 36, 9015 CrossRef CAS.
- For a leading reference see: N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 Search PubMed . For selected examples see: N. E. Leadbeater, K. A. Scott and L. Scott, J. Org. Chem., 2000, 65, 4770 CrossRef CAS; N. E. Leadbeater, K. A. Scott and L. Scott, J. Org. Chem., 2000, 65, 3231 Search PubMed.
- C. M. Crudden, D. Allen, M. D. Mikoluk and J. Sun, Chem. Commun., 2001, 1154 RSC; C. Bianchini, M. Frediani, G. Mantovani and F. Vizza, Organometallics, 2001, 20, 2660 CrossRef CAS; T. Mizugaki, M. Murata, M. Ooe, K. Ebitani and K. Kaneda, Chem. Commun., 2002, 52 RSC; Q. Yao, Angew. Chem., Int. Ed., 2000, 39, 3896 CrossRef CAS.
- C. Bianchini, M. Frediani and F. Vizza, Chem. Commun., 2001, 479 RSC.
- P. H. M. Budzelaar, J. H. G. Frijns and A. G. Orpen, Organometallics, 1990, 9, 1222 CrossRef CAS.
- A. Scrivanti, V. Beghetto, M. Zanato and U. Matteoli, J. Mol. Catal. A: Chem., 2000, 160, 331 CrossRef CAS; A. Scrivanti, V. Beghetto, E. Campagna and U. Matteoli, J. Mol. Catal. A: Chem., 2001, 168, 75 CrossRef CAS.
- A. Scrivanti, V. Beghetto, E. Campagna, M. Zanato and U. Matteoli, Organometallics, 1998, 17, 630 CrossRef CAS; A. Dervisi, P. G. Edwards, P. D. Newman, R. P. Tooze, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1999, 1113 RSC.
- R. A. Taylor, B. P. Santora and M. R. Gagné, Org. Lett., 2000, 2, 1781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: synthesis and characterisation of monomers and polymers and details of catalysis. See DOI: 10.1039/b512556a |
| This journal is © The Royal Society of Chemistry 2006 |