On-line dynamic extraction and automated determination of readily bioavailable hexavalent chromium in solid substrates using micro-sequential injection bead-injection lab-on-valve hyphenated with electrothermal atomic absorption spectrometry
Xiangbao
Long
a,
Manuel
Miró
*b and
Elo Harald
Hansen
*a
aDepartment of Chemistry, Technical University of Denmark, Kemitorvet, Building 207, DK-2800 Kgs. Lyngby, Denmark. E-mail: ehh@kemi.dtu.dk; Fax: +45 4588 3136; Tel: +45 4525 2346
bDepartment of Chemistry, Faculty of Sciences, University of the Balearic Islands, Carretera de Valldemossa km. 7.5, E-07122 Palma de Mallorca, Illes Balears, Spain. E-mail: manuel.miro@uib.es; Fax: +34 971173426; Tel: +34 971173260
First published on 25th November 2005
Abstract
A novel and miniaturized micro-sequential injection bead-injection lab-on-valve (μSI-BI-LOV) fractionation system was developed for on-line microcolumn soil extraction under simulated environmental scenarios and accurate monitoring of the content of easily mobilisable hexavalent chromium in soil environments at the sub-low parts-per-million level. The flow system integrates dynamic leaching of hexavalent chromium using deionized water as recommended by the German Standard DIN 38414-S4 method; on-line pH adjustment of the extract by a 0.01 mol L−1 Tris–HNO3 buffer solution; isolation of the chromate leached from the matrix constituents onto a Q Sepharose strong anion-exchanger freshly packed into the microconduits of the μSI-assembly; air-segmented elution of the sorbed species by a 40 µL plug of 0.5 mol L−1 NH4NO3 (pH 8) eluent; and detection by electrothermal atomic absorption spectrometry (ETAAS). The effect of simulated acidic rain on the accessibility of chromate forms for plant uptake was also investigated. The proposed approach offers several advantages over conventional speciation/fractionation protocols in the batch mode, including immediate separation with concomitant preconcentration of the released chromate, minimization of Cr(VI) to Cr(III) interconversion risks, enhanced accuracy, and non-existence of re-adsorption/re-distribution problems along with a detailed pattern of the kinetics of the leaching process. The reliability of the proposed method was evaluated via spiking of a moderately polluted agricultural soil material (San Joaquin Soil-Baseline Trace Element Concentrations) with water-soluble Cr(VI) salts at different concentration levels. The potential of the μSI-BI-LOV set-up with renewable surfaces for flame-AAS determination of high levels of readily bioavailable chromate in contaminated soils is also addressed.
Introduction
Speciation analysis of physicochemical forms of chromium is currently a topic of major interest as a consequence of the increased anthropogenic levels of chromium in the environment due to industrial discharges—including metal electroplating, leather tanning, chromate ore processing, spray painting and wood treatment1—and the well-known different ecological significance of the two most relevant valence states, i.e., trivalent and hexavalent chromium.2,3 Trivalent chromium is an essential micronutrient in the diet of mammals to maintain effective glucose, lipid and protein metabolism.2,4 In contrast, hexavalent chromium is highly toxic and carcinogenic for a variety of organisms as a result of its elevated oxidation potential and the ability to penetrate biological membranes.2,5,6 In addition, epidemiological studies have shown that high exposures to Cr(VI) in the workplace cause dermal sensitisation and human respiratory diseases.7,8 Both chromium species also differ significantly with respect to environmental mobility in solid substrates, hexavalent chromium salts of alkaline metals being those most soluble in neutral and slightly acidic media.1,9 All these considerations make detection and quantification of hexavalent chromium rather than the total metal content a subject of major concern for risk assessment in ecology, water pollution and environmental management.Monitoring of pollutants in environmental solids is usually accomplished via extraction/digestion methods followed by chemical analysis of the extracts/digests. Extraction methods intended for speciation/fractionation analysis are based on exposing the solid sample to an extracting reagent able to dissolve the targeted compounds or pre-defined physico-chemical phases of ecological interest.10 In fact, fractionation methods have gained widespread acceptance in environmental studies because they can reveal relevant information regarding pollutant soil-phase associations as well as elucidating the mode of occurrence, magnitude of available reservoirs, and potential migration of elements in natural environments.11
Metal fractionation protocols are traditionally conceived as operationally defined single or sequential extraction methods performed under a pseudo-equilibrium regime.12–15 Yet, recent trends have been directed at designing flow-through multiple-step dynamic fractionation procedures mostly involving microcolumn extractions aimed at imitating field conditions more correctly than their batch counterparts.16 Automated flow-systems with on-line detection have, however, been used solely for fractionation of elements in highly contaminated solid substrates, the raw extracts generated being in all instances delivered directly to the hyphenated analytical instrument,17–21 mostly ICP-MS,17,18,20 without any prior sample treatment step. Hence, their applicability to highly salted matrices or to extracts containing ultra-trace metal contents is rather limited. Actually, these methods fail to monitor the most ecotoxicological significant fractions of trace metals, i.e., the water-soluble or exchangeable pools, which determine the readily available, and thus toxic, forms for biota uptake.
Amongst the various extraction approaches for determining chromium in soils and sediments22,23 the most commonly used protocol for quantitation of Cr(VI) involves alkaline digestion at a pH around 12 as endorsed by the USEPA, also known as method 3060A,24 followed by the spectrophotometric analysis of the digests via the diphenylcarbazide (DPC) method. The main pitfalls of the standard method are related to its operation under unrealistic environmental scenarios, mainly to minimize undesired interconversion between oxidation states,22,25 its low sensitivity for trace analysis,25 and the solubilisation of humic matter which makes the subsequent analysis of Cr(VI) by DPC questionable.26 In addition, the application of external energy sources such as ultrasonication,27,28 focused microwaves,29 and magnetic stirring or plate heating22,24 are aimed at releasing not only soluble but also sparingly soluble and partially insoluble Cr(VI).30 Therefore, these procedures lack the ability to ascertain the potentially harmful ecological and human health effects caused by the access of soluble soil-borne hexavalent chromium to groundwaters or plant uptake via surface runoff or irrigation waters.
In this paper, an automated and rugged micro-sequential injection lab-on-valve (μSI-LOV) microcolumn fractionation system hyphenated to ETAAS detection and integrating on-line matrix separation and additional analyte preconcentration is proposed for the first time for expeditious and accurate determination of the content of readily mobilisable forms of Cr(VI) in solid substrates of environmental origin utilizing distilled water, as recommended by the DIN 38414-S4 method,31 and artificial acid rain as well. The third generation of flow injection analysis, viz. μSI-LOV, particularly in the bead-injection (BI) fashion with renewable sorptive entities, has shown significant advantages for on-line handling and pre-treatment of liquid samples of relative complexity as regards to trace metal separation, concentration and automated quantification as recently reviewed.32,33 Yet, to the best of our knowledge the μSI-BI-LOV concept has not been exploited as an analytical tool for accommodating flow-through dynamic speciation/fractionation schemes of solid samples so far. The discontinuous flow nature of SI-LOV makes the hyphenation with discrete non-continuous detectors, such as ETAAS, uncomplicated, thus yielding improved sensitivity, as compared with the classical DPC spectrophotometric method, for reliable monitoring of ultratrace amounts of readily-leachable hexavalent chromium.
Experimental
Instrumentation
An atomic absorption spectrometer (PerkinElmer AAnalyst 600) with a Zeeman background correction, and a transversely heated graphite furnace equipped with pyrolytically coated graphite tubes was used for the determination of chromium. A wavelength of 357.9 nm with a spectral bandpass of 0.7 nm and an operating current of 25 mA were set for the chromium hollow cathode lamp (Perkin Elmer). The temperature program for the chromium analysis is shown in Table 1. The manufacturer's recommendations have been slightly modified for facilitating the progressive volatilisation of the eluate solvent. The signals were recorded in the integrated (peak area) mode.| Step | Temperature/°C | Ramp time/s | Holding time/s | Argon flow rate/mL min−1 |
|---|---|---|---|---|
| Drying 1 | 110 | 5 | 35 | 250 |
| Drying 2 | 130 | 5 | 45 | 250 |
| Pyrolysis | 1500 | 25 | 20 | 250 |
| Atomization | 2300 | 0 | 4 | 0 |
| Cleansing | 2450 | 1 | 3 | 250 |
The FIAlab-3000 sequential injection system (Bellevue, WA) was equipped with the integrated LOV central sample-processing unit mounted atop a six-port selection valve (SV), two high-precision bi-directional syringe pumps (SP1 and SP2) with a capacity of 10 mL and 2.5 mL, respectively, and a peristaltic pump. A diagram of the whole system is shown in Fig. 1. The LOV microbore assembly (diameter, 5 cm; thickness, 1 cm) made from hard PVC contains a central port which can communicate with the other six micro channels (1.66 mm i.d./12.0 mm length) through the central communication conduit (CC) in the SV. The microchannel connecting SP with CC and that of port 4 serve as containers for bead-microcolumns C1 and C2 in which PEEK stoppers (Upchurch Scientific, Oak Harbor, WA), which have a dimension slightly smaller than that of the channel, are used for retaining the beads while allowing the solution to flow freely. The soil column, bead container (syringe) and eluent solution were attached to the remaining peripheral ports of the LOV. The holding coil (HC) was connected with the central port and CC via microcolumn C1. The two-way valves at the heads of SP1 and SP2 facilitate the communication of each syringe with either an external reservoir (carrier or buffer) or with the central port in the LOV manifold via a PEEK T-connector. The manifold was built from PTFE tubing of 0.50 mm i.d./1.66 mm o.d., except the 170 cm long HC which was made from PTFE tubing of 1.50 mm i.d./2.10 mm o.d. corresponding to a volume of 3.0 mL. The delivery line to the atomizer consists of a 0.6 mm i.d. tube with a total length of 106 cm. This tube, which is manipulated by the ETAAS autosampler arm, was optionally used as a waste line.
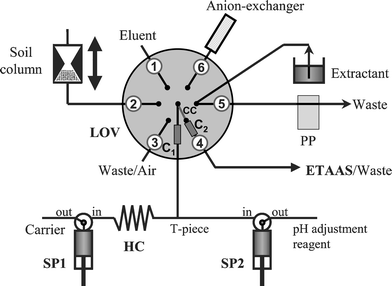 | ||
| Fig. 1 Schematic diagram of the μSI-BI-LOV-ETAAS system for dynamic fractionation of Cr(VI) in environmental solids. Carrier, 0.01 mol L−1 Tris–HNO3 buffer at pH 8.0; on-line pH adjustment reagent: 0.02 mol L−1 Tris–HNO3 buffer at pH 8.0; eluent, 0.5 mol L−1 NH4NO3–NH4OH at pH 8.0; anion-exchanger, Q Sepharose; SP1/SP2, syringe pumps 1 and 2; C1 and C2, LOV microcolumn positions; HC, holding coil; CC, central communication channel; PP, peristaltic pump, ETAAS, electrothermal atomic absorption spectrometer. | ||
The flow-through port of the LOV (port 5) is utilized as the inlet for the leaching solution into the flow network, the outgoing channel being connected to a peristaltic pump (operated at 2.0 mL min−1), thereby permitting thorough washing of the conduits between different extractants. The LOV port at position 3 plays a double role, serving both as outlet for final bead disposal and as an inlet for air (i.e., for air-segmentation purposes).
The specially designed chemical-resistant PEEK extraction microcolumn has been described in detail elsewhere.34 Briefly, it contains a central bi-conical shaped sample container (as shown in Fig. 1) and it is furnished with filters, filter supports and caps at both ends. The formerly used PTFE membrane filters with 1.0 µm pore size were here replaced with 0.45 µm cellulose acetate filters (Minisartfilters, Sartoriuos, Göttinger) for efficient retention of particulate matter within the sample holder.
The operational procedures of the μSI-BI-LOV system were computer controlled by the associated FIAlab software and synchronised with the commands for the activation of the ETAAS program through an intelligent electronic interface.35
Reagents, solutions and sample
All reagents were at least of analytical grade. Doubly de-ionised water (18.2 MΩ cm) obtained from a Milli-Q system (Millipore Synthesis A10, France) was used throughout. All flasks and beakers for solution preparation were cleaned with a 25% (v/v) concentrated nitric acid solution followed by repeated washing with Milli-Q water.A 0.1 mol L−1 Tris-buffer stock solution was prepared by dissolving 12.114 g of Tris(hydroxymethyl)-aminomethane in 1000 mL water which was afterward adjusted to pH 8.0 by dropwise addition of 1.0 mol L−1 HNO3. It was further diluted to 0.01 mol L−1 and 0.02 mol L−1 to serve as carrier and on-line pH adjustment solutions, respectively. The eluent consisted of a 0.5 mol L−1 NH4NO3–NH4OH buffer at pH 8.0. Double deionised water and simulated acid rain at pH 3.5 (adjusted with diluted nitric acid) were selected as mild extractants for determination of readily bioavailable Cr(VI). For calibration purposes, working standard solutions of hexavalent chromium were obtained by step-wise dilution of a 1000 mg L−1 stock solution of K2CrO4 in water.
A commercially available strong anion exchanger Q Sepharose™ Fast Flow (Amersham Biosciences, Sweden), as contained in 20% ethanol solution and with a mean particle size of 90 µm, was used directly in the LOV with no need for any additional swelling protocol. The beads were contained in a dedicated syringe reservoir mounted atop of port 6.
A moderately-polluted agricultural soil (San Joaquin Soil-SRM 2709, Baseline Trace Element Concentrations) purchased from the National Institute of Standards and Technology (NIST) was used for evaluation of the performance of the on-line soil fractionation/solid-phase extraction system. The standard material, which was highly homogenized during the NIST preparation, contained a total chromium concentration of 130 ± 4 µg g−1. The bi-conical microcolumn was packed with soil amounts ranging from 20 to 100 mg as detailed under Results and Discussion.
Operating procedures
The complete operational sequence for Cr(VI) fractionation in soil with further on-line analysis of the extracts using the μSI-BI-LOV scheme is listed in Table 2, and summarized as follows:| Sequence and description | SP1 valvea | SP2 valvea | SP1 | SP2 | LOV position | Flow rate/µL s−1 | Volume/µL |
|---|---|---|---|---|---|---|---|
| a The position “out” means connection of SP with the external reservoir, while “in” means connection of SP with the manifold. | |||||||
| 1. System preconditioning and bead loading | |||||||
| (a) Aspiration of carrier and buffer | Out | Out | Aspirate | Aspirate | — | 100 (SP1) | 1230 (SP1) |
| 50 (SP2) | 600 (SP2) | ||||||
| (b) Rinsing of port 3 | In | Out | Dispense | — | 3 | 50 | 400 |
| (c) Filling port 3 tubing with air | In | Out | Aspirate | — | 3 | 100 | 200 |
| (d) Aspiration of eluent into HC | In | Out | Aspirate | — | 1 | 50 | 400 |
| (e) Collection of beads into C1 | In | Out | Aspirate | — | 6 | 5 | 25 |
| (f) Moving beads to C2 and cleansing with eluent | In | Out | Dispense | — | 4 | 10 | 400 |
| (g) Flushing beads and ETAAS line with carrier | In | Out | Dispense | Dispense | 4 | 50 (SP1) | 425 (SP1) |
| 20 (SP2) | 50 (SP2) | ||||||
| 2. Soil extraction | |||||||
| (a) Air segment aspiration | In | In | Aspirate | — | 3 | 50 | 70 |
| (b) Loading of extracting reagent into HC | In | In | Aspirate | — | 5 | 50 | 500 |
| (c) Mixing of extractant with packed soil | In | In | Dispense | — | 2 | 50 | 570 |
| (d) Collection of soil extract | In | In | Aspirate | — | 2 | 5 | 770 |
| 3. Cr(VI) separation | |||||||
| (a) Moving of the head of extractant to T-connector | In | In | Dispense | — | 3 | 50 | 200 |
| (b) On-line buffering and Cr(VI) separation/preconcentration | In | In | Dispense | Dispense | 4 | 50 (SP1) | 610 (SP1) |
| 50 (SP2) | 550 (SP2) | ||||||
| (c) Rinsing of sorbent microcolumn with carrier | In | In | Dispense | — | 4 | 50 | 250 |
| 4. Elution and measurement | |||||||
| (a) Introduction of air into HC | In | In | Aspirate | — | 3 | 50 | 750 |
| (b) Filling of ETAAS line with air | In | In | Dispense | — | 4 | 20 | 380 |
| (c) Aspiration of eluent | In | In | Aspirate | — | 1 | 10 | 40 |
| (d) Elution of analyte loaded beads | In | In | Dispense | — | 4 | 10 | 50 |
| (e) Stopped flow (delay 7 s) | |||||||
| (f) Activation of ETAAS program: tip of autosampler arm moves into the graphite tube | |||||||
| (g) Transportation of eluate into the atomizer | In | In | Dispense | — | 4 | 10 | 360 |
| (h) Autosampler arm moves back to the original position, ETAAS runs the temperature program | |||||||
| 5. Bead disposal | |||||||
| (a) Dispensing of carrier to C2 | In | In | Dispense | — | 4 | 50 | 200 |
| (b) Bead transportation from C2 to C1 positions | In | In | Aspirate | — | 4 | 100 | 200 |
| (c) Withdrawal of used beads | In | In | Dispense | Dispense | 3 | 50 | 300 |
The ETAAS program is synchronized with the LOV method, whereby the next extraction comprising the multiple-step fractionation protocol starts to be effected while the former extract is being pyrolyzed and atomized in the furnace.
Results and discussion
Investigation of chemical variables and operating parameters in the μSI-BI-LOV for separation and preconcentration of Cr(VI) species
In order to find the best operational and chemical conditions for separation, preconcentration and speciation analysis of ultra-trace levels of chromium in the μSI-BI-LOV flow system prior to ETAAS quantitation, a series of preliminary investigations was conducted using aqueous solutions (attached to port 2) in lieu of soil extracts. Amongst the various parameters affecting the performance of the sorbent bead-injection preconcentration in terms of sorption efficiency for Cr(VI), the bead material, sample acidity, loading flow rate, eluent type and stripping-out conditions and tolerance to potential interfering species are regarded as the most crucial ones.Selection of sorptive material for Cr(VI) preconcentration/separation
Bearing in mind the inability of the atomic absorption spectrometer for discriminating chromium species and its low tolerance to high concentrations of electrolytes, the target analyte should be isolated from trivalent chromium and other matrix ingredients prior to presentation to the detector. This is accomplished in this work by exploiting anion-exchangers as a sorptive medium for Cr(VI) taking into account its anionic nature in most natural environments.Among the various sorbents exploited in a permanent fashion for Cr(VI) enrichment, namely Dowex 1-X827–29 and Sepharose/Sephadex-type36 exchangers, the latter ones are preferable for handling in the microbore LOV unit as renewable surfaces. The reasons that make them ideal for manipulation in the third generation of flow injection analysis without risks of bead settlement in the integrated conduits mainly lie in their hydrophilic nature, perfectly spherical shape and narrow size distribution. Additional features of functionalised polysaccharide-type solid phases for implementation as temporary reactors in flow systems include high binding capacities, excellent flow properties, and high chemical and physical stabilities.
Initially, a strong anionic exchanger (QAE Sephadex), wherein the diethyl-(2-hydroxypropyl)aminoethyl moiety is chemically attached to a cross-linked dextran matrix was evaluated. This sorbent, being supplied as a dry powder, requires a swelling pre-conditioning protocol with a high electrolyte concentration (e.g., 2 mol L−1 NaNO3) prior to moistening with buffer solution at the application pH. Although QAE-Sephadex showed appropriate adsorption performance for Cr(VI) traces, its utilization as a microcolumn in LOV cavities is limited by the volume changes of the resin-bed as a consequence of sorbent shrinking/swelling upon application of solutions of different composition and/or ionic strength. Thus, a cross-linked 6% agarose furnished with diethyl-(2-hydroxypropyl)aminoethyl, so-named Q Sepharose Fast Flow, which is physically more resistant than Sephadex exchangers and can be directly manipulated in the flow network with no need for any ancillary treatment, was selected for Cr(VI) enrichment in the LOV.
Sample loading pH
For optimum retention of hexavalent chromium onto the packed-bead Q Sephadex microcolumn, the acid–base nature of the target metal species should be taken into consideration. Anionic forms are the prevalent species at pH >2.8, which explains the fact that no significant differences on the efficiency of the sorbent reactor for collection of Cr(VI) in standard solutions were observed within the pH range 3.5–8.5 (higher values not tested), which is in accordance with earlier observations made by Hashemi et al.36 using batchwise column preconcentration systems. Yet, the tolerance to potentially interfering monovalent anions in real-life samples was improved at pH ≥ 8.0 as a consequence of the stronger affinity of the resin for the predominant divalent chromate oxoanion. Moreover, slightly alkaline media are commonly recommended for stabilization of Cr(VI) solutions (see below).22,28 Buffering of the standards was accomplished by addition of Tris–HNO3 buffer (pH 8.0). The dependence of the buffer concentration on the analyte recovery was studied from 5 × 10−3 to 0.1 mol L−1. Significant chromate breakthrough (>20%) was detected for buffer concentrations above 0.05 mol L−1 Tris as a result of the pre-elution effect occasioned by the surplus of nitrate. A 0.01 mol L−1 Tris–HNO3 buffer solution was selected for the remaining studies as a compromise between Tris buffer capacity and retention efficiency for Cr(VI).Sample loading flow rate
As opposed to Sephadex-type exchangers,37 highly cross-linked Sepharose beads are able to endure high solution flow rates with negligible squeezing,38 whereby the transient sorptive entities are effectively trapped within the LOV microchannels with no possibility for escaping through the space between the PEEK stopper and the wall of the LOV unit. High loading rates are particularly desirable for achieving elevated concentration factors whenever trace elements are determined. The effect of loading flow rate on the preconcentration behaviour of Cr(VI) onto the ion-exchanger was investigated from 25 to 125 µL s−1. The analytical readouts revealed excellent repeatabilities and comparable sensitivity in the whole range of applied rates for Q Sepharose. In fact, flow rates as high as 100 µL s−1 are tolerated in the LOV-BI sorption mode because of the minor deterioration of the analytical sensitivity (≤3%) as compared with the lowest loading rate assayed, i.e., 25 µL s−1.Elution procedure
Elution of retained species from packed-bead ion-exchange columns is frequently performed by a sharpened increase of the ionic strength of the mobile phase. Yet, one of the fundamental requirements for appropriate quantification of the released species is the compatibility of the elution medium with the detection instrument. It is well recognized that atomic spectrometers such as ETAAS lack sufficient tolerance to directly analyze solutions of high salt content. Thus, for example, the analysis of chromium species in 0.5 mol L−1 NaCl has been reported to be cumbersome because of the severe background absorption or interferences occurring in the presence of high concentrations of chloride.39 Recovery of chromate and suitable quantification by means of ETAAS is also feasible via application of a pH gradient through the anion-exchanger.40 A concentration of 2 mol L−1 HNO3 was hence utilized as eluting solution to decrease the affinity of the target species for the sorptive material. However, a 20% signal reduction was detected by direct injection of standards prepared under these acidic eluting conditions as compared with those in Milli-Q water as a result of the contribution of non-spectral interferences for the selected ETAAS operational sequence. Improvement of the analytical performance in terms of elution yields and minimization of interfering effects was accomplished by exploiting an electrolyte buffer (NH4NO3–NH4OH) adjusted to the same alkaline condition as that of the carrier medium (i.e., pH 8.0), but at a higher ionic strength, which is in accordance with previous observations.28 In fact, according to the electrochemical reduction potential of the Cr(VI)/Cr(III) redox pair, stabilization of Cr(VI) occurs in bases, thus minimizing the possible oxidation of the organic groups of the matrix beads, which has been described for co-polymer type ion-exchange resins.41 In order to prevent the existence of a pH gradient during the elution/preconditioning/preconcentration steps that might induce the generation of Cr(III) ions by oxidation of the organic beads—that would be then unavailable for the resin moieties—alkaline media were chosen for the overall analytical protocol.To fulfil the restricted volumetric requirements of the graphite platform of the atomizer and the reliable accommodation of the eluate in the tube, discrete eluent volumes ≤50 µL should be utilized. However, in the proposed system, an air-segmented 50 µL plug of 0.8 mol L−1 buffer delivered at 10 µL s−1 into the loaded microcolumn rendered incomplete Cr(VI) stripping with carryover of 3%, as detected by a multiple elution protocol. This drawback was overcome by the precise fluidic control and flow programming of SI systems that foster the straightforward implementation of stopped-flow approaches. Actually, by halting a mere 40 µL of NH4NO3–NH4OH buffer within the LOV cavity containing the Cr(VI)-loaded beads for 7 s the carryover was reduced to 0.75%. Thus, these elution conditions were adopted for further investigations.
The concentration of the alkaline buffer proved to be critical for suitable signal-to-noise ratios. Quantitative recovery of sorbed species was obtained in a single eluent plug by buffer concentrations ≥0.5 mol L−1 NH4NO3–NH4OH whenever a stopped-flow strategy was executed, yet the electrolyte content of the eluate should be as low as possible for reliable ETAAS quantification, although one can take advantage of the fact that most of the salts are vaporized during the pyrolysis step. Therefore, a 0.5 mol L−1 NH4NO3–NH4OH buffer (pH = 8) was selected for the remainder of the optimization and fractionation protocols.
Under the optimized chemical and physical variables detailed above, the retention efficiency of the anion exchanger for hexavalent chromium, defined as the ratio of the retained amount of analyte onto the microcolumn to the total amount available in the sample, was as high as 90 ± 3%, serving as a superb solid-phase reactor for our purpose of speciation/separation/preconcentration of traces of soluble Cr(VI) in soil samples.
On-line dynamic fractionation schemes for trace levels of Cr(VI) in environmentally relevant solid samples
The open architecture of the central processing unit in a μSI-LOV analyzer mounted atop a multi-position selection valve allows random access to not only reagent reservoirs and liquid solutions, but offers the possibility of hyphenation to ancillary modules at peripheral ports for facilitating unit operations on-line. This is here demonstrated by the inclusion of an external soil-containing microcolumn.Therefore, dynamic soil extraction processes as occurring in natura can be simulated via multiple leaching schemes involving steady renewal of eluent solutions that capitalize on the application of discontinuous forward–backward flow as precisely coordinated and controlled by the syringe pump.
Configuration of the μSI-LOV-BI set-up for on-line soil extraction
As opposed to previous works dealing with on-line FI/SI microcolumn fractionation of environmental solids,17–21,34 the specially designed soil container is attached to one of the peripheral ports of the LOV in lieu of being implemented into the manifold, with the upper outlet open to the atmosphere. The upright disposition is intended to withhold the entire substrate in the lower conical cavity of the column and facilitate the stripping out of the extractant moistening the packed solid. During each single extraction step of the overall operational protocol, the extracting reagent was pumped forward into the soil column at a flow rate of 50 µL s−1 to ensure fluidized-bed like conditions for improved mixing between leachant and sample, while the extract was progressively pulled inward at a slower rate, namely, 7 µL s−1, for a more realistic simulation of water percolation through environmental soil bodies. The continuous on-line renewal of the eluent in intimate contact with the soil material prevents the problem of metal re-adsorption in freshly exposed surfaces as detected in the most labile fractions (i.e., water-soluble, exchangeable, and acid soluble) of batchwise sequential extraction procedures for trace metals.12,42 In addition, by application of a bi-directional flow, back-pressure or clogging effects due to soil compaction that are frequently observed in continuous-flow or uni-directional flow injection fractionation manifolds16 are not encountered in the μSI-LOV manifold.The microflow SI-arrangement can, in fact, be viewed as an FI–SI hybrid system due to the external syringe pump (SP2) assembled for on-line pH adjustment. The role of this ancillary liquid driver is not only to minimize competitive sorption of interfering anions for solid-phase extraction of Cr(VI), but also to prevent undesirable Cr(VI) to Cr(III) interconversion under the slight acidic medium of the aqueous extractants. Actually, batchwise extraction methods for Cr(VI) using distilled water or acidic reagents are prone to render biased results as a consequence of the accelerated reduction of the target analyte by dissolved organic matter and other reductants in the time span from digestion/extraction to analysis.22,43 This can be solved elegantly in our system by the combined action of immediate pH adjustment of the extract to alkaline conditions (pH 8.0) and the subsequent isolation/preconcentration of the hexavalent chromium from matrix ingredients on the anion-exchanger. Since the soil extract and buffer are mixed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 ratio, a concentration of 0.02 mol L−1 Tris–HNO3 buffer solution was employed in SP2 according to the results presented above.
∶1 ratio, a concentration of 0.02 mol L−1 Tris–HNO3 buffer solution was employed in SP2 according to the results presented above.
In addition, the irreversible accumulation of matrix components on the sorbent material as detected by the progressive darkening of the bead surfaces when used repeatedly was fully circumvented by exploitation of solid-phase extraction in a bead-injection renewable fashion, that is, the active microcolumn is discarded after each extract analysis and replaced by a fresh portion of ion-exchange resin.
Effect of coexisting ions
The tolerance of the solid-phase preconcentration method to potentially interfering species either in the sorption process or in the final determination by ETAAS was ascertained using a fixed concentration of 0.2 µg L−1 Cr(VI) standard solution and variable amounts of foreign species. To this end, the influence of the prevailing water soluble anionic species in soil extracts, such as Cl−, HCO3−, NO3−, SO42− and the most ubiquitous cationic species, such as Ca2+ and Mg2+, which might lead to non-spectroscopic interferences during analysis, was evaluated. A given concentration level of a chemical species was regarded as interferent whenever the analytical readout of the Cr(VI) standard was affected by more than 10%. The tolerated interferent/analyte ratios of anions prepared from the corresponding sodium salts are listed in Table 3. As can be seen, the ion-exchanger materials can endure rather high concentrations of monovalent anions such as Cl−, HCO3− and NO3−, which is in accordance with earlier observations.28| Foreign species | Tolerated interferent/Cr(VI) ratio |
|---|---|
| a Concentration of Cr(VI) = 0.2 µg L−1. | |
| Cl− | 5 × 106 |
| HCO3− | 1 × 107 |
| NO3− | 5 × 106 |
| SO42− | 5 × 104 |
As for the potential interfering effect of Ca2+ and Mg2+ on the detection instrument, it should be borne in mind that cationic species are not sorbed on the anionic-exchanger so that concentrations as high as 200 mg L−1 are admissible for accurate dynamic fractionation of Cr(VI). Higher concentrations could not be tested as a consequence of the competitive sorption of concomitant nitrate on the active sites of the agarose resin.
In order to investigate the effect of organic matter and soil matrix ingredients released during water extraction on the ETAAS measurements of Cr(VI) at the low µg L−1 level, the batchwise DIN-38414-S4 protocol was applied to the agricultural San Joaquin NIST soil. The application of a 3-level standard addition method to the filtered extract using the LOV system rendered comparable sensitivity to that of the external calibration, thus revealing the inexistence of multiplicative matrix interferences, which is the result of the on-line sample clean-up step effected via bead-injection analysis.
Application and validation
The performance of the proposed on-line LOV-BI microcolumn fractionation method was evaluated by using a NIST agricultural soil (San Joaquin SRM 2709) as a model of environmental solid moderately polluted with trace elements. The 100 mg solid sample contained within the conical microcolumn was continuously exposed to air-sandwiched 500 µL distilled water plugs in a single forward–reversed motion. The implementation of the air-segmentation approach was aimed at monitoring the location of the discrete extractant zone through the sample line and preventing undue dispersion of the extract volume into the carrier stream prior to isolation of Cr(VI) onto the ion-exchanger. A 1∶5 soil to extractant volume was adopted for each leaching step rather than the 1∶10 ratio of the DIN protocol in order to obtain a more detailed pattern, i.e., higher resolution, of the partitioning process without excessive dilution of the mobilised Cr(VI) in the extractant medium. The multiple-step dynamic extraction profile, so-called extractogram, which yields a thorough insight into the leaching kinetics of the targeted metal fraction under environmentally simulated water infiltration/percolation conditions, is shown in Fig. 2. As can be seen, the water-soluble forms of hexavalent chromium are promptly and quantitatively stripped from soil compartments in less than 8 fractions (≤4.0 mL), and therefore they should be accurately determined for reliable risk assessment of chromium pollution in soil as a consequence of the immediate accessibility for fauna and flora uptake. Furthermore, it should be borne in mind that the proposed on-line LOV-ETAAS hyphenated system with integrated chromate preconcentration circumvents the lack of sensitivity of the traditional DPC photometric method for quantification of the most ecotoxicological significant forms of chromium. The effect of increased acidity (acid rain) on the extractability of chromate was mimicked by replacing distilled water with a diluted nitric acid solution at pH 3.5, since artificial rainwater at this pH has been previously utilized for batch column leaching experiments.44 No appreciable leachability increase upon acidification was observed whenever both extractants were applied sequentially, as shown in Fig. 2. This result reveals the efficiency of distilled water for quantitative removal of soluble (surface bound) chromate in an on-line dynamic mode, and the ability of the soil material to raise the pH of the applied extractant, thus precluding the additional release of sparingly-soluble forms of Cr(VI).
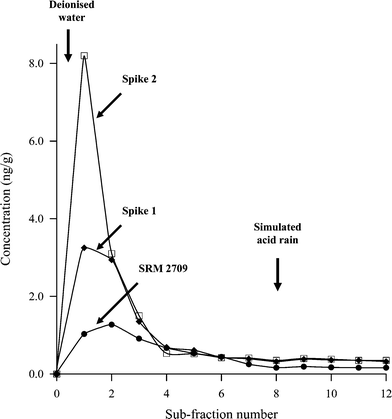 | ||
| Fig. 2 Extraction profiles of readily bioavailable Cr(VI) in SRM 2709 and spiked samples as obtained from the μSI-LOV microcolumn system using mild extractants. Soil amount, 100 mg; sub-fraction volume, 500 µL; spike 1, 5.0 ng g−1; spike 2, 8.0 ng g−1. | ||
Quantification of extractable chromium in the various fractions was performed by using an external calibration procedure against matched standards under the very same operational conditions as for the dynamic fractionation analysis, except that the soil containing microcolumn was replaced by a 2.5 m long open-tubular reactor (1.0 mm i.d., 2.0 mm o.d.) and the extractant at port 5 by the standard solutions. A straightforward calibration protocol based on using different volumes (≤1800 µL) of a single Cr(VI) standard was selected, because preliminary tests demonstrated the compatibility of mass calibration with the sorptive sample treatment. The mass calibration curve was linear from 0.02 to 0.6 ng Cr(VI) and fitted the equation Y = 0.6631X + 0.0112 (r = 0.9992) where Y and X stand for the integrated absorbance and injected amount of Cr(VI), respectively. For the microcolumn soil extraction, although some bubbles were formed eventually at the inlet of the column due to the decreased pressure during the backward flow of extractant, both the on-line separation step and the application of a mass-based least squares regression circumvented any interfering effect due to gases generated in the flow manifold. In contrast, bubble formation represents one of the most severe constraints of the DPC-photometric assay for Cr(VI) determination following on-line extraction, as recognized by Grate and Taylor.43
The water extractable content of Cr(VI) in the moderately polluted San Joaquin Soil obtained as a summation of the various extracts composing the multiple-step fractionation procedure amounted to 4.9 ± 0.3 ng Cr(VI) g−1 soil, which represents less than 5 × 10−3% of the certified value of total chromium in the sample. This is attributed to the high reduction potential of this agricultural soil due to the organic matter content. In fact, the batchwise EDTA-extractable chromate for this SRM standard is reported to be merely 0.1% of the total chromium in the sample.39 Yet, it must be borne in mind that EDTA is capable of dissolving soluble, sparingly-soluble and insoluble forms of hexavalent chromium by chelation of the counterions in insoluble salts (e.g., lead and barium chromates), thereby overestimating the pool size of readily available forms of Cr(VI).
For validation of the dynamic μSI-LOV fractionation system, strict comparison with the manual DIN38414-S4 standard method was not feasible because of the different operationally defined conditions and the influence of re-distribution phenomena in the equilibrium-based procedure. Besides, no certified solid material for readily bioavailable hexavalent chromium is currently commercially available. Reliability and ruggedness of the analytical method was evaluated via Cr(VI) spikes. Earlier researchers39,45 recommended effecting the spikes directly on the extracts rather than on the solid substrates due to soil redox reactions and immobilization processes that often occasion low or near-zero recoveries in batchwise analysis. However, fortification of the SRM substrate with soluble salts of Cr(VI) (viz., potassium chromate) did not pose any problem in the developed system whenever analyses were conducted without delay as a result of the drastic reduction of extraction time as compared with conventional end-over-end methods. Experimental results obtained by spiking variable amounts of soil with Cr(VI) levels ranging from 5 to 55 µg kg−1 are compiled in Table 4. Application of a second-order polynomial regression equation was needed for quantitation of the water-soluble Cr(VI) content in the first extract fraction of the spike of highest concentration. A statistical t-test46 was used for each spike to ascertain whether or not there was a significant difference between the concentration of Cr(VI) added and that found. Since the overall experimental values of |t| are below the critical value at the 0.05 significance level, i.e., 4.30, no significant differences were encountered for any set of data, thus indicating the nonexistence of multiplicative (non-spectroscopic) matrix interferences. For this particular soil, there is then no need to utilize the standard addition method that, whenever applied to flow-through fractionation schemes, not only demands highly repeatable extractograms but also implies tedious and time-consuming operational procedures as the sample containing microcolumn must be replaced for each addition.19 Reproducibility of the overall fractionation/solid-phase preconcentration method and soil homogeneity were assessed from data presented in Table 4. Maximum relative standard deviations of 5.3% and 6.8% were found for 100 and 20 mg soil packed columns, respectively. Yet, for handling poorly homogeneous soils, larger substrate amounts might be accommodated in the custom-built column to guarantee sample representativeness, as recently demonstrated.21,47
| Soil sample | Soil amount/mg | Concentration added/ng g−1 | Concentration found/ng g−1 | Recovery (%) | t exp |
|---|---|---|---|---|---|
| a Results are expressed as the mean of 3 extraction replicates ± SD | |||||
| SRM 2709 | 100 | — | 4.9 ± 0.3 | — | — |
| Spike 1 | 100 | 5.0 | 9.5 ± 0.5 | 96 ± 5 | 1.39 |
| Spike 2 | 100 | 8.0 | 13.9 ± 0.6 | 108 ± 4 | 2.89 |
| Spike 3 | 20 | 40 | 44 ± 3 | 98 ± 6 | 0.52 |
| Spike 4 | 20 | 55 | 64 ± 2 | 107 ± 3 | 3.55 |
The potential extension of the developed analyzer for fractionation of Cr(VI) in highly contaminated soils by on-line hyphenation with FAAS rather than ETAAS has also been investigated. To this end, an additional injection valve was implemented as an interface between the discontinuous μSI-LOV flow approach and the continuously operating detector for injection of the extracts into the FAAS nebuliser stream.21 Analyte breakthrough—calculated from the residual concentration of Cr(VI) during loading of the anion exchanger—did not occur up to 180 ng Cr(VI), corresponding to a minimum concentration of 9 µg g−1 water-soluble Cr(VI) for a 20 mg sample. Hence, the LOV microcolumn was proven to be suitable for separation purposes in on-line fractionation/speciation analysis of highly polluted substrates. As a result of the inherent versatility of the μSI-LOV-BI-AAS coupling, environmental solids with variable amounts of available Cr(VI) ranging from the sub-µg kg−1 to the mg kg−1 level, i.e., above the maximum permissible concentrations for agricultural use, may be automatically treated and further analyzed in the fully enclosed flow set-up.
Acknowledgements
Xiangbao Long is grateful for a three years' PhD stipend granted to him by the Technical University of Denmark. Manuel Miró is indebted to the Spanish Ministry of Education and Science for financial support through the “Ramon y Cajal” research program.References
- J. Barnhart, J. Soil Contam., 1997, 6, 561 Search PubMed.
- S. A. Katz and H. Salem, The Biological and Environmental Chemistry of Chromium, Wiley-VCH, New York, 1994 Search PubMed.
- D. E. Kimbrough, Y. Cohen, A. M. Winer, L. Creelman and C. Mabuni, Crit. Rev. Environ. Sci. Technol., 1999, 29, 1 Search PubMed.
- J. Versieck and R. Cornelis, Trace Elements in Human Plasma or Serum, CRC Press, Boca Raton, 1989 Search PubMed.
- A. Kortenkamp, Z. Ozolins, D. Beyersmann and P. O'Brien, Mutat. Res., 1989, 216, 19 CrossRef CAS.
- J. W. Hamilton and K. E. Wetterbahn, in Handbook on Toxicity of Inorganic Compounds, ed. H. H. Seiler, H. Sigel, A. Sigel, Marcel Dekker, New York, 1988 Search PubMed.
- R. B. Hayes, Biological and Environmental Aspects of Chromium, Elsevier, Amsterdam, 1982 Search PubMed.
- C. T. Dillon, P. A. Lay, A. M. Bonin, N. E. Dixon, T. J. Collins and K. Kostka, Carcinogenesis, 1993, 14, 1875 CrossRef CAS.
- B. R. James, Chemical Transformations of chromium in soils: Relevance to mobility, bio-availability and remediation, The Chromium File, n° 8, International Chromium Development Association, 2002, available at http://www.chromium-asoc.com/publications/crfile8feb02.htm.
- A. K. Das, R. Chakraborty, M. L. Cervera and M. de la Guardia, Talanta, 1995, 42, 1007 CrossRef CAS.
- V. H. Kennedy, A. L. Sanchez, D. H. Oughton and A. P. Rowland, Analyst, 1997, 122, 89R RSC.
- A. V. Filgueiras, I. Lavilla and C. Bendicho, J. Environ. Monit., 2002, 4, 823 RSC.
- J. Hlavay, T. Prohaska, M. Weisz, W. W. Wenzel and G. J. Stingeder, Pure Appl. Chem., 2004, 76, 415 CrossRef CAS.
- A. Sahuquillo, A. Rigol and G. Rauret, Trends Anal. Chem., 2003, 22, 152 CrossRef CAS.
- C. Gleyzes, S. Tellier and M. Astruc, Trends Anal. Chem., 2002, 21, 451 CrossRef CAS.
- M. Miró, E. H. Hansen, R. Chomchoei and W. Frenzel, Trends Anal. Chem., 2005, 24, 759 CrossRef CAS.
- D. Beauchemin, K. Kyser and D. Chipley, Anal. Chem., 2002, 74, 3924 CrossRef CAS.
- M. Jimoh, W. Frenzel, V. Müller, H. Stephanowitz and E. Hoffmann, Anal. Chem., 2004, 76, 1197 CrossRef CAS.
- L.-M. Dong and X.-P. Yan, Talanta, 2005, 65, 627 CrossRef CAS.
- M. Jimoh, W. Frenzel and V. Müller, Anal. Bioanal. Chem., 2005, 381, 438 CrossRef CAS.
- R. Chomchoei, M. Miró, E. H. Hansen and J. Shiowatana, Anal. Chem., 2005, 77, 2720 CrossRef CAS.
- M. Pettine and S. Capri, Anal. Chim. Acta, 2005, 540, 231 CrossRef CAS.
- M. Korolczuk and M. Grabarczyk, Talanta, 2005, 66, 1320 CrossRef CAS.
- United States Environmental Protection Agency (USEPA), Method 3060A, in Test Methods for Evaluating Solid Wastes, Physical/Chemical Methods, SW-846, 3rd Update, Office of Solid Waste and Emergency Response, Washington, DC, 1996 Search PubMed.
- R. J. Vitale, G. R. Mussoline, J. C. Petura and B. R. James, J. Soil. Contam., 1997, 6, 581 Search PubMed.
- M. Pettine and S. Capri, Anal. Chim. Acta, 2005, 540, 239 CrossRef CAS.
- J. L. Luque-García and M. D. Luque de Castro, Analyst, 2002, 127, 1115 RSC.
- J. Wang, K. Ashley, E. R. Kennedy and C. Neumeister, Analyst, 1997, 122, 1307 RSC.
- S. Morales-Muñoz, J. L. Luque-García and M. D. Luque de Castro, Anal. Chim. Acta, 2004, 515, 343 CrossRef CAS.
- R. J. Vitale, G. R. Mussoline, K. A. Rinehimer, J. C. Petura and B. R. James, Environ. Sci. Technol., 1997, 31, 390 CrossRef CAS.
- DIN 38414-S4, German Standard Methods for the Examination of Water, Wastewater and Sludge. Sludge and Sediment Group (group S): Determination of Leachability by Water, VCH-Verlag, Weinheim, 1984 Search PubMed.
- J-H. Wang, E. H. Hansen and M. Miró, Anal. Chim. Acta, 2003, 499, 139 CrossRef CAS.
- J.-H. Wang and E. H. Hansen, Trends Anal. Chem., 2003, 22, 225 CrossRef CAS.
- R. Chomchoei, E. H. Hansen and J. Shiowatana, Anal. Chim. Acta, 2004, 526, 177 CrossRef CAS.
- S. C. Nielsen and E. H. Hansen, Anal. Chim. Acta, 2000, 422, 47 CrossRef CAS.
- P. Hashemi, J. Boroumand and M. R. Fat'hi, Talanta, 2004, 64, 578 CrossRef CAS.
- J.-H. Wang and E. H. Hansen, Anal. Chim. Acta, 2000, 424, 223 CrossRef CAS.
- X.-B. Long, E. H. Hansen and M. Miró, Talanta, 2005, 66, 1326 CrossRef CAS.
- G.-X. Hu and R. L. Deming, Anal. Chim. Acta, 2005, 535, 237 CrossRef CAS.
- M. T. Siles-Cordero, E. I. Vereda-Alonso, A. García de Torres and J. M. Cano-Pavón, J. Anal. At. Spectrom., 2004, 19, 398 RSC.
- A. C. Sahayam, G. Venkateswarlu and S. C. Chaurasia, Anal. Chim. Acta, 2005, 537, 267 CrossRef CAS.
- J. L. Gómez-Ariza, I. Giráldez, D. Sánchez-Rodas and E. Morales, Anal. Chim. Acta, 1999, 399, 295 CrossRef CAS.
- J. W. Grate and R. H. Taylor, Field Anal. Chem. Technol., 1996, 1, 39 Search PubMed.
- P. Anderson, C. M. Davidson, A. L. Duncan, D. Littlejohn, A. M. Ure and L. M. Garden, J. Environ. Monit., 2000, 2, 234 RSC.
- R. J. Vitale, G. R. Mussoline and K. A. Rinehimer, Contam. Soils, 1996, 1, 221 Search PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Ltd, Harlow, 5th edn, 2005, ch. 3, pp. 39–40 Search PubMed.
- R. Chomchoei, M. Miró, E. H. Hansen and J. Shiowatana, Anal. Chim. Acta, 2005, 536, 183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |