Piezoelectric immunosensor for bisphenol A based on signal enhancing step with 2-methacrolyloxyethyl phosphorylcholine polymeric nanoparticle†
Jong-Won
Park
ac,
Shigeru
Kurosawa
*a,
Hidenobu
Aizawa
a,
Yasuhiro
Goda
b,
Madoka
Takai
c and
Kazuhiko
Ishihara
c
aInstitute for Environmental Management Technology, National Institute of Advanced Industrial Science and Technology (AIST), 1-1Higashi, Tsukuba 305-8565, Japan. E-mail: shigeru-kurosawa@aist.go.jp; Fax: +81-29-861-4746; Tel: +81-29-861-4746
bJapan EnviroChemicals, Ltd, 3-8 Doshomachi, 2-chome, Chuo-ku, Osaka 541-0045, Japan
cDepartment of Materials Science, School of Engineering, The University of Tokyo, 7-3-1, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 9th November 2005
Abstract
An immunoassay in which BPA competed with a BPA-horseradish peroxidase conjugate for binding to anti-BPA antibodies, coupled to a piezoelectric (PZ) immunosensor, was able to detect 0.1 ng mL−1 BPA. To enhance the sensitivity of the assay, we tested nanoparticles approximately 200 nm in diameter, coupled to anti-BPA antibodies, to increase the mass change on the surface of the immunosensor and thereby increase the frequency shift detected. This second step, using nanoparticles coated with anti-BPA antibodies, improved the sensitivity of the assay by approximately eight times at BPA concentrations below 10 ng mL−1. Field emission-scanning electron microscopy (FE-SEM) showed that polymeric 2-methacrolyloxyethyl phosphorylcholine (MPC) nanoparticles coupled to antibodies remained monodisperse on the surface of the immunosensor and therefore produced stable signals in the immunosensors. Since the frequency shift detected in the assay mainly originated from the mass change on the surface of the PZ crystal, the colloidal stability of the antibody-conjugated particles used in the enhancement step played an extremely important role in achieving a stable and highly sensitive signal.
Introduction
Using the specific affinity between an antibody and its corresponding antigen, immunoassay techniques represent a promising technology and possible methodology for evaluating real samples. There are numerous advantages of using immunoassays in the field rather than formal analysis in a fixed laboratory that include speed, portability, ease of use, relatively low cost per sample, and the range of contaminants that can be analyzed. Although immunoassay methods, such as enzyme-linked immunosorbent assays (ELISA), radioimmunoassay (RIA), immunoagglutination assays (IAA), and fluorescent immunoassays (FIA) play an important role in medical diagnostics, food, and environmental analyses,1–4 the immunosensor, which integrates immunoassay principles and an electronic device, has been extensively developed because of its advantages of high selectivity and specificity, relatively low construction cost, potential for miniaturization, facility of automation and simple and portable equipment construction.5–9 With these several advantages, the immunosensor has been investigated as an alternative analytical tool for environmental analyses.Most immunosensors could be used to perform all four types of immunoassays: direct,10–12 displacement,13 and competitive.14–16 Normally, small molecular analytes such as endocrine disruptors, which have molecular weights below 1000, have only one epitope for antibody binding. In the case of the direct immunoassay, the analyte in solution directly binds to the receptor on the surface. This makes it difficult to use in an immunosensor because the sensor response originating from the binding of extremely low molecular weight analytes was not detectable. Furthermore, although the displacement immunoassay can be regarded as a good assay format, it is difficult to optimize its experimental conditions for detection because a low molecular analyte has only one hapten. On the other hand, competitive immunoassays rely on the competition between the labeled and unlabeled antigens for a fixed and limited number of antibody-combining sites. With this principle, small molecular weight analytes can be detected through the competitive binding with conjugated analytes, which were labeled by large molecular weight materials such as proteins. Furthermore, if the signal is not strong enough when certain small molecular analytes are detected by the immunosensor, one can add a second antibody, perhaps with a conjugated microsphere.17,18 There should then be a large change in the signal, with amplification brought about by the microspheres. Richards et al. and Ward et al. disclosed the mass-amplified QCM assay in US patents 4847193 and 5501986.19,20 The procedures involve the use of massive sol particles to modify the protein; the binding of sol particle-labeled material greatly increases the frequency shift. This scheme can be applied to an immunosensor as a signal enhancing step. Therefore, the integrating protocols of the competitive immunoassay and signal enhancing step are the best for detecting low molecular weight analytes with extremely low detection limits using an immunosensor.
The PZ immunosensor is one of the popular transducers among the immunosensing methods.21–26 Due to its simplicity, convenience, and low cost, this method has been important for biodetection since Shons and Dorman made the first PZ immunosensor to detect the bovine serum IgG antibody in 1972.27 The PZ immunosensor consists of a PZ crystal with an antigen or antibody immobilized on its surface. The principle of the PZ immunosensor detection relies on recording the frequency decrease corresponding to the mass increase on the chip surface during the biological interaction. The increase in mass at the sensor results in a decrease of the frequency as shown by Sauerbrey,28 who first introduced the quartz crystal microbalance principle. This relationship of frequency to mass has been outlined by the following equation,

Bisphenol A (BPA) [2,2-bis(4-hydroxyphenyl)propane], as a low molecular weight antigen, was selected and detected using the piezoelectric (PZ) immunosensor. BPA is an important intermediate in the industrial manufacture of engineering and superengineering plastics, such as polycarbonate and polyethersulfones.29 Consequently, it can be found in the lacquer lining of metal food and beverage cans, in drums, reinforced pipes, adhesives, dental sealants, nail polishes, baby bottles and food packaging materials among other items.30–32 BPA may be released into the environment during the manufacture, use or disposal of products containing this substance. Autoclaving and thermal treatments can increase the leaching of BPA due to hydrolysis of its polymers. The use of alkalis can also accelerate this process. Perez et al. identified that besides BPA, some other related hydroxylated diphenylalkanes are also estrogenic.33 BPA can disturb the hormone balance in a living body; i.e., it is considered as an endocrine disrupter34,35 and the limit for BPA exposure was set as 2.5 ppm day−1 by the Japanese government.36 Endocrine disrupters are attracting increasing attention in the field of environmental science, and the quantitative detection of BPA molecules with a high sensitivity and selectivity has become an extremely important issue.
In this study, we used the 9 MHz fundamental frequency PZ crystal which has A = 0.196 cm2, ρq = 2.65 g cm−3, and μq = 2.95 × 1011 dyn cm−2. Thus, if a substance with the weight of 1 ng is homogeneously attached to the active area of a crystal, a frequency change of 1.04 Hz will occur. The anti-BPA monoclonal antibody immobilized PZ immunosensor was constructed after the investigation of the antibody adsorption behavior, and then the optimal conditions for the competitive immunoreaction with an extended detection limit were considered. After the determination of the optimal conditions, once a suitable competitive immunoreaction was identified, the cross-reactivity with BPA was verified. Furthermore, the relationship between the PZ immunosensor and ELISA for BPA detection was also considered using environmental BPA samples prepared by solid phase extraction protocols. Next, the effect of the signal enhancement, meaning a 2nd immunoreaction, was demonstrated using 2-methacryloyloxyethyl phosphorylcholine (MPC) polymeric nanoparticles (MPC-PNP), which were prepared and evaluated for immunoagglutination ability for C-reactive protein (CRP) detection in our previous report,37 and conventional polystyrene nanoparticles for the signal/experimental error (S/E) and signal/signal (S/S) ratios for BPA detection using the PZ crystal immunosensor.
Experimental
Instrumentation
All the PZ crystals were AT-cut with a basic resonance frequency of 9 MHz. The crystal consisted of a quartz wafer, which was placed between two gold electrodes (8 mm × 8 mm × 0.185 mm), that were purchased from Nihon Dempa Kogyo Co., Ltd (Tokyo, Japan). The laboratory-made transistor–transistor logic (TTL) electronic circuit was used as the oscillating circuit.38 The oscillator was supplied with 5 V by a dc voltage regulator. The frequency change from the oscillator was monitored by a universal counter (Iwatsu-SC7201, Iwatsu Inc., Tokyo, Japan). The data collection and processing system was programmed using Labview software (version 6.0.2i, National Instruments Corp., USA). It could accept the data from the counter and display real time in a plot of the frequency shift on the computer monitoring a GPIB interface (National Instruments Corp., Texas, USA). The temperature (25 ± 0.1 °C) was strictly controlled for all experimental steps by an incubator (Yamato Scientific Co., Ltd, Tokyo, Japan).Measurements
All bare PZ crystals had their gold surfaces thoroughly cleaned for 5 min in a freshly prepared 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of H2O2 and H2SO4 before the introduction of the thiol compounds. After rinsing with water, acetone, and ethanol, the PZ crystals were sufficiently dried under an N2 atmosphere. All the bare PZ crystals were then measured for their initial frequency for 5 min, which was determined as F0.
:3 mixture of H2O2 and H2SO4 before the introduction of the thiol compounds. After rinsing with water, acetone, and ethanol, the PZ crystals were sufficiently dried under an N2 atmosphere. All the bare PZ crystals were then measured for their initial frequency for 5 min, which was determined as F0.
After each experimental step, such as the surface activation to measure the frequency shift, the PZ crystal was carefully rinsed with distilled water and thoroughly dried under N2 gas. To determine the mass change on the surface, the oscillating frequency (F1) was measured in the air state after each experimental step. Thereafter, the measured oscillating frequency was calculated by a comparison with the initial frequency (F0) of each PZ crystal and it was translated into the mass change using Sauerbrey's equation. In the same manner, to calculate the frequency shift for further steps such as antibody immobilization or antigen–antibody binding, the corresponding oscillating frequency (Fn) was calculated using the oscillating frequency from the previous step (Fn−1). All the experimental results were obtained using six PZ crystals and the standard deviation was expressed as experimental error.
Surface activation on gold surface
To immobilize the anti-BPA monoclonal antibody, which was kindly supplied from Japan EnviroChemicals, Ltd (Osaka, Japan), the gold electrode surfaces on the PZ crystals were chemically activated. All the PZ crystals were immersed in the solution of 10 mM cysteamine (Wako Pure Chemical Co., Ltd, Osaka, Japan) for 60 min; they were then placed in a 1% glutaraldehyde (Wako Pure Chemical Co., Ltd) solution for 60 min to attach the aldehyde group on the gold electrodes. The anti-BPA antibody solution (30 µL) was injected onto the chemically modified gold electrode of the PZ crystal, and then it was immobilized for 90 min by covalent binding at room temperature. After antibody immobilization, the remaining aldehyde groups were blocked with 20 mM glycine, which was also purchased from Wako Pure Chemical Co., Ltd. Also, the water soluble poly[2-methacryloyloxyethyl phosphorylcholine (MPC)-co-n-butyl methacrylate (BMA) (PMB)], chemical structure shown in Fig. 1(a), used as a stabilizer was treated to avoid any non-specific binding from a competitive reaction and to achieve the immunological activity of immobilized antibody.39 | ||
| Fig. 1 Chemical structure of water soluble (a) PMB for stabilizers and (b) PMBN for MPC-PNP which consisted of a hydrophilic part (MPC unit), a hydrophobic part (BMA unit), and an active ester part (MEONP unit) with different molecular ratios, respectively. | ||
Competitive immunoreaction
The competitive immunoreaction was performed using a mixture (30 µL) of various concentrations of BPA and 103 ng mL−1 of BPA-horseradish peroxidase (BPA-HRP) for 60 min at 25 °C. For the all-antigen–antibody reaction, the reaction media used a 10% methanol mixture with phosphate buffered saline (PBS, 10 mM, pH 7.4). Furthermore, to consider the relationship between the PZ crystals and ELISA detection methods, the BPA samples that were collected from sea water (Tokyo Bay, Japan) were prepared by simple solid phase extraction protocols (Japan EnviroChemicals, Ltd). Samples were filtered through specified glass fiber filters (GL-Pak Glass Nexus, #5010-26021, GL Science Inc., Tokyo, Japan), preconditioned with dichloromethane (10 mL), methanol (5 mL), and distilled water (5 mL) and washed with distilled water (5 mL) and distilled water∶methanol = 1∶1 (5 mL). The analytes were eluted with dichloromethane (6 mL) and the solvent was evaporated with nitrogen. The samples for ELISA and the PZ immunosensor were finally prepared by adding methanol to the filtrate to a final methanol concentration of 10% (v/v). The results using the PZ crystal were obtained by the same competitive immunoreaction format with a 1 µg mL−1 fixed concentration of BPA-HRP. Moreover, the corresponding ELISA results using the same extracted samples were achieved by experiments using the supersensitive BPA ELISA kit® (ECOLOGIENA series, Japan EnviroChemicals, Ltd, Osaka, Japan).
Cross reactivities
A competitive immunoreaction was also performed using four structural BPA analogues, which were obtained from Japan EnviroChemicals, Ltd, in order to determine their respective I50 values. I50 is the concentration of the analyte at which half of the maximal signal intensity is reached. The data from the PZ immunosensor and ELISA methods were obtained from standard curves of the related compounds and calculated using the following formula:| Cross reactivity (CR, %) = (I50, BPA)/(I50,BPA analogues) × 100 |
Signal enhancement
According to previously reported methods,37 poly[MPC-co-n-BMA-co-p-nitrophenyloxycarbonyl polyethylene glycol methacrylate (MEONP) (PMBN)] (Fig. 1(b)) which has active ester groups for bioconjugation on the side chains was synthesized, and two types of anti-BPA antibody conjugated nanoparticles, i.e., MPC-PNP (diameter = 205 nm) and polystyrene nanoparticles (Polysciences Inc., Warrington, USA) were prepared. Also, the affinity test of the anti-BPA antibody conjugated particles were carefully investigated using BPA-HRP by immunoagglutination techniques at 570 nm (data not shown). After the competitive immunoreaction between BPA and BPA-HRP, the prepared anti-BPA antibody conjugated particles, which had their concentrations adjusted to 10 µg mL−1 by PBS solution, were injected onto the PZ crystals, and then a secondary immunoreaction, i.e., signal enhancement step, was done for 60 min at 25 °C. After carefully rinsing the samples with distilled water, the binding conditions of the antibody conjugated particles on the surface were determined by field emission scanning electron microscopy (FE-SEM, DS-720, Topcon, Co., Ltd, Tokyo, Japan). Also, the frequency shift based on the binding of particles was calculated as already described.Results and discussions
Adsorption behavior of anti-BPA monoclonal antibody
The gold surface of the PZ crystal was treated to immobilize the antibody by cysteamine and glutaraldehyde. The antibody could be immobilized via covalent binding between the amino groups of the antibody and aldehyde groups of the activated gold surface. It is difficult to prove the immobilization status of an antibody on the surface because the current surface survey tools could not satisfactorily evaluate soft biomolecules, such as an antibody, on the surface. On the other hand, if the adsorption behavior, which could be calculated from the amounts of antibody adsorbed, could be investigated, it would be a useful consideration for estimating the immobilization status. For all concentration studies, the frequency shift due to the anti-BPA antibody immobilization reached a steady-state value after 90 min (data not shown). Each point in Fig. 2 was collected after the immobilization time of 90 min. The frequency shift according to the antibody immobilization was measured and calculated using Sauerbrey's equation to obtain the mass change on the surface. Fig. 2 shows the amounts of the antibody immobilized onto the activated gold surface (a) and its reciprocal plots (b). As Fig. 2(a) shows, the general shape of the isotherm is consistent with those measured for antibody adsorption on other surfaces, and the results fit a Langmuir isotherm, which is commonly used to describe protein adsorption.40,41 Therefore, the maximum immobilizing amount and association constant could be calculated from the equation of the Langmuir isotherm shown as below.
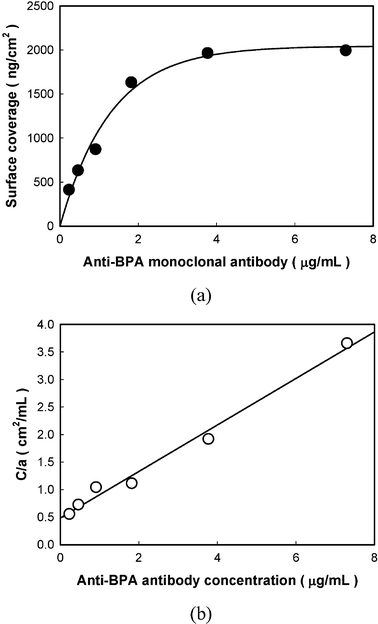 | ||
| Fig. 2 Adsorption isotherm for the anti-BPA monoclonal antibody on aldehyde functionalized surface; (a) surface coverage according to anti-BPA antibody immobilization, (b) linear reciprocal plots of C/aversus antibody concentration. | ||
The reciprocal plot between C/amax and C gave simple straight lines according to the Langmuir isotherm equation where C is the concentration of the antibody, a is the amount immobilized, amax is the maximum amount immobilized, and A is the association constant. The values of amax and A could be calculated and are 2041 ng cm−2 and 0.876 (10−3 mL ng−1), respectively, from the slope of the reciprocal line in Fig. 2(b) when concentrations above 3.77 µg mL−1 of the anti-BPA antibody were injected. Based on the assumption that the surface coverage represented the saturation state of antibody immobilization, the optimal concentration of the anti-BPA antibody to immobilize on the PZ crystal was determined to be 3.77 µg mL−1.
Optimal conditions for the competitive immunoreaction
For a small molecule, having a molecular weight below 1000 Daltons, it was quite difficult to directly obtain quantitative information using normal immunoassay systems. Therefore, the competitive immunoreaction, which is applied with BPA conjugated protein, is a very useful method to achieve the quantitative analysis of BPA. In this study, we used BPA-HRP and considered its concentration effect during the competitive immunoreaction. To avoid the non-specific binding of BPA-HRP, the surface of the PZ crystals were thoroughly blocked with 20 mM glycine and 0.2% MPC copolymer after the anti-BPA antibody was immobilized. Fig. 3 indicates the frequency shift according to BPA-HRP binding with the anti-BPA antibody, which was previously immobilized at a concentration of 3.77 µg mL−1. It shows that the frequency shift denoting the binding between BPA-HRP and the anti-BPA antibody increased as the BPA-HRP concentration increased, and it was saturated at 30 ng mL−1 of BPA-HRP. This indicated that antigen binding sites in the immobilized anti-BPA antibody completely captured the epitope of BPA-HRP and no more BPA molecules were bound with anti-BPA antibody. If 30 ng mL−1 or higher concentrations of BPA-HRP are used in the competitive immunoreaction using PZ crystals, BPA-HRP and BPA can be competitively bound with the anti-BPA antibody. | ||
| Fig. 3 The frequency shift depending on the direct binding between BPA-HRP and anti-BPA antibody. | ||
The binding of BPA-HRP with the immobilized anti-BPA antibody was saturated after 30 ng mL−1 (see Fig. 3), therefore, to select the optimal concentration of the fixed standard BPA-HRP concentration, 30 and 300 ng mL−1 BPA-HRP were investigated in the competitive immunoreaction. Fig. 4(a) and (b) show the calibration curve of the BPA detection and the detection limits when 30 and 300 ng mL−1 BPA-HRP respectively were competitively reacted with various concentrations of BPA. The end of the titration range of the two calibration curves, which signifies the limitation of the BPA detection, was 1 ng mL−1 and 0.1 ng mL−1, respectively. The small molecular weight antigen obstructs the binding of the antigen conjugated macromolecule with the immobilized antibody when two different molecules were competitively bound with the antibody, therefore, the calibration curve for the antigen was indicated as a negatively decreasing curve. Therefore, careful selection of the antigen conjugated macromolecule is very important in the competitive reaction because an excess concentration of the antigen conjugated macromolecule was aggressively bound with antibody when it was competitively reacted with a low concentration of antigen. Therefore, the excess BPA-HRP that induced the low concentration of BPA could not be detected using the competitive reaction in our experiment. As a conclusion, 30 ng mL−1 of BPA-HRP was the most suitable for BPA detection when the competitive immunoreaction was used.
 | ||
| Fig. 4 The dose-responses for various concentrations of BPA when 300 ng mL−1 (a) and 30 ng mL−1 (b) of BPA-HRP were used in the competitive immunoreaction. | ||
A correlation study was performed by analyzing 10 environmental pollutant samples, prepared by solid phase extraction protocols, with the PZ crystal and ELISA methods, which adopted the same competitive reaction. As shown in Fig. 5, the correlation coefficient between PZ crystal and ELISA was 0.96 (n = 10) for the entire range, the respective slope of the equation for the regression line was 0.84, and the y-intercept was 8.55 µg L−1. This implied that the PZ crystal method, not requiring the process of the 2nd antibody binding or chromogenic reaction, could be regarded as a good alternative method to ELISA when the competitive immunoreaction was used to detect BPA in environmental samples.
 | ||
| Fig. 5 The relationship between PZ immunosensor and ELISA method using environmental samples. | ||
Cross reactivity
The cross reactivity (CR) of the anti-BPA monoclonal antibody was evaluated using various structural BPA analogues in order to determine their respective I50 values. The I50 of BPA was assigned a value of 100%, and the CRs for other compounds were reported according to their I50 relative to this value. Most compounds were selected because they can be present in the environment in addition to BPA. Table 1 shows the CR of the anti-BPA monoclonal antibody for BPA analogues using the PZ immunosensor and ELISA methods. Compounds with a structure similar to BPA showed some varied CR even though the anti-BPA antibody was a monoclonal antibody. It was difficult to distinguish which molecular part of BPA acted as a hapten, however, R1 and R2 parts of the molecular structure clearly affected the BPA recognition in the anti-BPA monoclonal antibody. The anti-BPA monoclonal antibody indicated a 100% recognition ability for BPA, which has methyl groups in the R1 and R2 positions. On the other hand, the CRs were 10.6% and ND when the R1 and R2 positions were substituted by H, respectively. Also, the CRs decreased when other molecules were introduced into the R2 position. Therefore, the methyl groups in the R1 and R2 positions are regarded as a very important part for the recognition of BPA by the anti-BPA antibody. The CRs in the PZ immunosensor and ELISA method did not show any significant recognition for these compounds, therefore, the PZ immunosensor can be regarded as an alternative to ELISA.|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | CR by ELISA (%) | CR by PZ immunosensor (%) |
| CR is determined by expressing the ratio of the I50 of the chemical assigned to be 100% (BPA) to the I50 of the other compounds and expressed as a percent. | ||||
| BPA | CH3 | CH3 | 100 | 100 |
| A | H | CH3 | 6.0 | 10.6 |
| B | H | H | 1.8 | ND |
| C | CH3 | CH2OH | 1.7 | 3.5 |
| D | CH3 | C2H5 | 15.6 | 5.7 |

Signal enhancement
In the ELISA technique, the secondary reaction was usually used to introduce an enzyme or fluorescent molecules for the quantitative analysis of the analytes.1,3 For the PZ immunosensor, this step is not required because the signal can be directly received because a certain mass change occurs on its surface. Therefore, if needed, the 2nd immunoreaction can be used to increase the sensitivity and lead to improved detection limits in the PZ immunosensor. The anti-BPA antibody conjugated MPC-PNP and polystyrene nanoparticles were used in the signal enhancement step, which could be regarded as the secondary immunoreaction since the competitive binding between BPA and BPA-HRP was completed. The two particles applied had almost the same diameter of about 202 nm to avoid a steric difference when each particle was bound with BPA-HRP on the surface. The anti-BPA antibody conjugated MPC-PNP and polystyrene nanoparticles were bound with the remaining epitopes of BPA-HRP after competitive binding and their binding situations were determined by the FE-SEM pictures (see Fig. 6(a) and (b), respectively). As shown in the FE-SEM pictures, their binding situations were quite different. MPC-PNP was clearly bound in a monodisperse state, on the other hand, polystyrene nanoparticles were slightly aggregated when bound. This was quite dependent on the colloidal stability of the two particles in the solution.37 In our previous study, it was indicated that MPC-PNP showed an excellent colloidal stability although the biomolecules conjugated with it because it was strongly solvated with water molecules, and polar groups like bound water molecules resist coagulation among the MPC-PNPs, besides, polystyrene nanoparticles were easily coagulated in the solution because they have a highly hydrophobic surface. The frequency shift mainly originated from the mass change on the surface of the PZ crystal when particles were bound with BPA-HRP. After all, the colloidal stability of antibody conjugated particles plays an important role in the achievement of a stable highly-sensitive signal when these were used in the signal enhancing step. | ||
| Fig. 6 FE-SEM pictures of anti-BPA antibody conjugated MPC-PNP (a) and polystyrene nanoparticles (b) which were bound with BPA-HRP through the signal enhancing step. | ||
Table 2 shows the S/E and S/S ratios which were calculated from the raw data from the competitive immunoreaction and the signal enhancing step using MPC-PNP and polystyrene nanoparticles. The S/E increased with decreasing BPA concentration because the signals were negatively increased in the all reactions. These results were similar in the competitive immunoreaction and MPC-PNP, however, the polystyrene nanoparticles were definitely different. Also, the S/E of the reactions using MPC-PNP was higher than for those using the competitive immunoreaction in all the BPA concentration ranges. For example, the limit of detection of 0.01 ng mL−1, considered from the linear range, was achieved for the MPC-PNP, while 0.1 ng mL−1 was considered as the detection limit using the competitive immunoreaction. This means that the anti-BPA antibody conjugated MPC-PNP could be used to amplify the analytical sensitivity through the signal enhancement step, although polystyrene nanoparticles were not appropriate. As mentioned above, the difference between the two particle experiments mainly depended on the colloidal stability of the particles. The initially coagulated antibody conjugated polystyrene nanoparticles were bound with BPA-HRP, and then the frequency shift was very unstable because the frequency shift of the PZ crystal was mainly dominated by the mass change on the surface. Also, using these particles, the S/S showed the amplified magnitude of frequency shift which was obtained from the competitive immunoreaction. The sensitivities of the two signal enhancing steps were almost 8 times higher than that of the competitive reaction at below 10 ng mL−1 BPA. This means that the signal enhancing step could be successfully used to amplify the analytical sensitivity.
| BPA/ng mL−1 | S/E for antigen recognitiona | S/S of two sandwich reactions to competitive reactionb | |||
|---|---|---|---|---|---|
| Competitive reaction | MPC-PNP | Polystyrene | MPC-PNP | Polystyrene | |
| a S/E was calculated from frequency shift and experimental error bars in calibration curve tests of the competitive immunoreaction and two signal enhancing steps, respectively. b S/S was calculated from the comparison between the amplified signal of each concentration in signal enhancing steps and the corresponding signal in competitive immunoreaction. | |||||
| 1000 | 3.7 | 2.9 | 1.4 | 2.3 | 1.3 |
| 100 | 5.2 | 7.7 | 1.9 | 7.4 | 5.8 |
| 10 | 6.7 | 12.0 | 3.5 | 7.1 | 7.3 |
| 1 | 9.5 | 15.1 | 5.7 | 8.1 | 7.9 |
| 0.1 | 13.0 | 17.4 | 9.5 | 8.7 | 7.9 |
| 0.01 | 14.8 | 20.1 | 6.6 | 9.1 | 8.1 |
| 0.001 | 14.1 | 21.6 | 9.7 | 9.3 | 8.3 |
Normally, in conventional immunoassay methods, such as ELISA, the competitive immunoassay with analyte conjugated macromolecules has been regarded as the best way to detect extremely low molecular weight analytes which were often present in environmental pollutants. Also, the 2nd immunoreaction has only been regarded as a way to introduce enzyme or fluorescent molecules after the competitive immunoassay is completed because it uses UV or fluorescence spectroscopy to gain a quantitative signal. On the other hand, it has a very different meaning when these two immunoassay protocols were used in the immunosensor, especially the PZ immunosensor. As it was proven in this study, the PZ immunosensor could directly achieve a calibration curve for BPA, which had almost the same detection limits as the ELISA technique, with only the competitive immunoreaction between BPA and BPA-HRP. Furthermore, the PZ immunosensor could successfully amplify its analytical sensitivity and extend the detection limits by the introduction of the anti-BPA antibody conjugated MPC-PNP as a new material in the 2nd immunoreaction, i.e., the signal enhancing step.
Conclusion
We have investigated general considerations for integrating protocols with the competitive immunoreaction and the signal enhancing step to detect BPA in environmental pollutants using a PZ immunosensor. The anti-BPA monoclonal antibody was successfully immobilized via covalent binding between the amino residues of the antibody and aldehyde groups on the gold surface of the PZ crystal. The optimal immobilizing concentration of the anti-BPA antibody was then determined to be 3.77 µg mL−1, since its surface coverage was investigated using the mass change according to the antibody immobilization. Also, the cross reactivity of the anti-BPA monoclonal antibody by the PZ immunosensor and ELISA methods was considered using four BPA derivatives. BPA was successfully detected by the competitive immunoreaction when 30 ng mL−1 of BPA-HRP was used as the fixed concentration. Furthermore, its detect limitation was clearly extended from 0.1 ng mL−1 to 0.01 ng mL−1 by the signal enhancing step when the anti-BPA antibody conjugated MPC-PNP was used.Acknowledgements
This work was supported financially in part by a Grant-in-Aid for Scientific Research on Priority Areas (16040217) from MEXT, for “Application of permselective and biocompatible membranes for the improvement of quartz crystal microbalance biosensors” from the Daiwa Anglo-Japanese Foundation, and for “Study on monitoring of environmental risk compounds such as dioxins and endocrine disruptors using sensing systems” from the Ministry of the Environment, Japan.References
- Y. Hayashi, R. Matsuda, T. Maitani, W. Nishimura, K. Ito and M. Maeda, Anal. Chem., 2004, 76, 1295 CrossRef CAS.
- C. Kessler, Nonradioactive labeling and detection of biomolecules, Springer-Verlag, New York, 1992 Search PubMed.
- Y. Sugawara, S. J. Gee, J. R. Sanborn, S. D. Gilman and B. D. Hammock, Anal. Chem., 1998, 70, 1092 CrossRef CAS.
- P. Tijssen, Practice and theory of enzyme immunoassay, Elsevier, New York, 8th edn, 1985 Search PubMed.
- M. Dijksma, B. Kamp, J. C. Hoogvliet and W. P. Van Bennekom, Anal. Chem., 2001, 73, 901 CrossRef CAS.
- R. E. Ionescu, C. Gondran, L. A. Gheber, S. Cosnier and R. S. Marks, Anal. Chem., 2004, 76, 6808 CrossRef CAS.
- S. Laschi, M. Mascini, G. Scortichini, M. Franek and M. Mascini, J. Agric. Food Chem., 2003, 51, 1816 CrossRef CAS.
- G. D. Liu, Z. Y. Wu, S. P. Wang, G. L. Shen and R. Q. Yu, Anal. Chem., 2001, 73, 3219 CrossRef CAS.
- F. Yu and W. Knoll, Anal. Chem., 2004, 76, 1971 CrossRef CAS.
- W. Bundach, A. P. Abel, A. E. Bruno and D. Neuschafer, Anal. Chem., 1999, 71, 3347 CrossRef CAS.
- B. B. Haab, M. J. Dunham and P. O. Brown, Genome Biol., 2001, 2, 4.1 Search PubMed.
- T. Vo-Dinh, J. P. Alarie, N. Isola, D. Landis, A. L. Wintenberg and M. N. Ericson, Anal. Chem., 1999, 71, 358 CrossRef CAS.
- U. Narang, P. R. Gauger, A. W. Kusterbeck and F. S. Ligler, Anal. Biochem., 1998, 255, 13 CrossRef CAS.
- A. Brecht, A. Klotz, C. Barzen, G. Gauglitz, R. D. Harris, G. R. Quigley, J. S. Wilkinson, P. Sztajnbok, R. Abunknesha, J. Gascon, A. Oubina and D. Barcelo, Anal. Chim. Acta, 1998, 362, 69 CrossRef CAS.
- A. Klotz, A. Brecht, C. Barzen, G. Gauglitz, R. D. Harris, G. R. Quigley, J. S. Wilkinson and R. A. Abunknesha, Sens. Actuators, B, 1998, 51, 181 CrossRef.
- K. Schult, A. Katerkamp, D. Trau, F. Grawe, K. Cammann and M. Meusel, Anal. Chem., 1999, 71, 5430 CrossRef CAS.
- D. Trau, W. Yang, M. Seydack, F. Caruso, N. T. Yu and R. Renneberg, Anal. Chem., 2002, 74, 5480 CrossRef CAS.
- Y. Weizmann, F. Patolsky, E. Katz and I. Willner, J. Am. Chem. Soc., 2003, 125, 3452 CrossRef CAS.
- J. C. Richards and D. T. Bach, US Pat. 4847193, 1989 Search PubMed.
- M. D. Ward and R. C. Ebersole, US Pat. 5501986, 1996 Search PubMed.
- H. Aizawa, S. Kurosawa, M. Tozuka, J. Park, K. Kobayashi and H. Tanaka, Biosens. Bioelectron., 2003, 18, 765 CrossRef CAS.
- N. Y. Pan and J. S. Shin, Sens. Actuators, B, 2004, 98, 180 CrossRef.
- H. Sota, H. Yoshimine, R. F. Whittier, M. Gotoh, Y. Shinohara, Y. Hasegawa and Y. Okahata, Anal. Chem., 2002, 74, 3592 CrossRef CAS.
- S. Susmel, C. K. O'Sullivan and G. G. Guilbault, Enzyme Microb. Technol., 2000, 27, 639 CrossRef CAS.
- N. Tassew and M. Thompson, Anal. Chem., 2002, 74, 5313 CrossRef CAS.
- B. Zuo, S. Li, Z. Guo, J. Zhang and C. Chen, Anal. Chem., 2004, 76, 3536 CrossRef CAS.
- A. Shons and F. J. Dorman, J. Biomed. Mater. Res., 1972, 6, 565 CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 115, 206.
- S. R. Howe and L. Borodinsky, Food Addit. Contam., 1998, 15, 370 CAS.
- J. E. Biles, T. P. McNeal and T. H. Begley, J. Agric. Food Chem., 1997, 45, 4697 CrossRef CAS.
- J. A. Brontons, M. F. Olea-Serrano, M. Villalobos, V. Pedraza and N. Olea, Environ. Health Perspect., 1995, 103, 608 CrossRef.
- A. V. Krishnan, P. Stathis, S. F. Permuth, L. Tokes and D. Feldman, Endocrinology, 1993, 132, 2279 CrossRef CAS.
- P. Perez, R. Pulgar, F. Olea-Serrano, M. Villalobos, A. Rivas, M. Metzler, V. Pedraza and N. Olea, Environ. Health Perspect., 1998, 106, 167 CrossRef CAS.
- P. A. Hunt, K. E. Koehler, M. Susiarjo, C. A. Hodges, A. Hagan, R. C. Voigt, S. Thomas, B. F. Thomas and T. J. Hassold, Curr. Biol., 2003, 13, 546 CrossRef CAS.
- C. A. Staples, P. B. Dorn, G. M. Klecka, S. T. O'Block and L. R. Harris, Chemosphere, 1997, 36, 2149.
- M. Ema, S. Fujii, M. Furukawa, M. Kiguchi, T. Ikka and A. Harazono, Reprod. Toxicol., 2001, 15, 505 CrossRef CAS.
- J. Park, S. Kurosawa, J. Watanabe and K. Ishihara, Anal. Chem., 2004, 76, 2649 CrossRef CAS.
- S. Kurosawa, H. Aizawa and Y. Yoshimoto, IEEE Trans. Ultrason. Ferroelectrics Freq. Control, 2000, 47, 1256 Search PubMed.
- J. Park, S. Kurosawa, H. Aizawa, S. I. Wakida, S. Yamada and K. Ishihara, Sens. Actuators, B, 2003, 91, 158 CrossRef.
- K. Al-Malah, J. McGuire and R. Sproull, J. Colloid Interface Sci., 1995, 170, 261 CrossRef CAS.
- D. R. Jackson, S. Omanovic and S. G. Roscoe, Langmuir, 2000, 16, 5449 CrossRef CAS.
Footnote |
| † Presented at the Biosensors and Biomaterials Workshop, Tsukuba, Japan, March 7–9, 2005. |
| This journal is © The Royal Society of Chemistry 2006 |