Investigation of selenium-containing root exudates of Brassica juncea using HPLC-ICP-MS and ESI-qTOF-MS
Anne P.
Vonderheide
a,
Sandra
Mounicou
b,
Juris
Meija
b,
Heather F.
Henry
a,
Joseph A.
Caruso
*b and
Jodi R.
Shann
a
aDepartment of Biological Sciences, University of Cincinnati, Cincinnati, OH 45221-0006, USA
bDepartment of Chemistry, University of Cincinnati, Cincinnati, OH 45221-0172, USA. E-mail: Joseph.Caruso@uc.edu
First published on 22nd November 2005
Abstract
Selenium-containing root exudates were investigated in a known selenium accumulator model plant. Indian mustard (Brassica juncea) plants were grown hydroponically and supplemented with selenite (SeO32−) in a 25% Hoagland's nutrient solution. Additive concentrations were 0, 1, 5 and 20 µg mL−1 Se with five replicate plants per treatment level. Plants were exposed to the respective Se solutions for two weeks, then placed in deionized water for two more weeks. The hydroponic solutions were collected for analysis after the first two weeks of selenium supplementation (day 14) and twice during the deionized water period (days 21 and 28). Separation by ion-pairing high performance liquid chromatography was followed by inductively coupled plasma-mass spectrometry (ICP-MS) for selenium specific detection. Chromatographic peaks unable to be identified by retention-time matching were collected for analysis by electrospray ionization mass spectrometry (ESI-MS). Additional chemical experiments were performed for structural elucidation. Several selenium-containing compounds were identified in the exudate-containing solution and two were identified as selenocystine and the selenosulfate (SSeO32−) ion. The presence of dimethylselenide (CH3SeCH3) is also observed but cannot be attributed exclusively to plant exudation because plants were not grown in sterile conditions. Further, the incorporation of fortified selenoamino acids into peptide structures was found to occur under neutral pH conditions, suggesting that exuded enzymes might facilitate such a reaction. Finally, physiological differences resulting from selenium supplementations were noted and discussed.
Introduction
Selenium is an essential element that has a narrow window between necessity and toxicity to humans.1 It enters the food chain through the soil-to-plant pathway2 and hence its level in crops is dependent on the selenium content of the soil where the food is raised.3 Natural selenium levels in the soil are highly variable throughout the world. To avoid selenium toxicity in areas where selenium is a dominant contaminant of agricultural lands, phytoremediation has been explored as a low-cost removal remedy.4 Plant groups such as Astragalus, Arabidopsis, Neptunia and Brassica have members with the ability to extract selenium, either by accumulation or volatilization mechanisms.5 One species in particular, Brassica juncea, has been the focus of much research due to its relatively large biomass and its fast growth cycle. Many have used ICP-MS in an attempt to elucidate the selenium metabolic pathway, however, all focus on either the selenium species contained within various parts of the plant (i.e. roots, shoots, stems, leaves)6,7 or with selenium volatilization resulting from uptake.8 In order to gain a more comprehensive picture of this accumulator's treatment of selenium, the potential for selenium-containing root exudates of B. juncea grown in a selenium-rich environment was examined here.Root exudates are organic compounds released into the soil by plant roots9 where they may play several ecological roles. Exudates have been shown to provide nutrients to beneficial microorganisms, assist in acquisition of minerals by plants and act as toxins to inhibit competitive neighbors and pathogens.10,11 Hence, exudation tends to increase as a response to environmental stress such as nutrient deficiency or infection.12 Though the role of root exudates in phytoremediation has been well established in stimulating degradation of organic and/or halogenated environmental contaminants, less is known of their role in metal metabolism.13,14 There is growing evidence to support the role of root exudates as a plant defense for metal contamination. Investigations of other plant species have shown changes in root exudate composition specifically as a mechanism to prevent aluminium15−18 or cadmium19 phytotoxicity. In other works, Luongo and coworkers examined the role of root exudates on As mobilization and accumulation in both a hyperaccumulator and a sensitive species20 and Yang et al. studied the exudates of ryegrass and maize grown in the presence of nickel.21 In all cases, characterization of the exudate profile was undertaken under typical conditions and in the presence of the metal; concentrations of small organic acids capable of metal chelation were found to increase. These research findings support the idea that the metal-binding capabilities of these exudates may be an important mechanism for stabilizing metals in soil.
In plants, selenium, unlike other metal contaminants, can substitute for S in the sulfur metabolic pathways and subsequently be incorporated into amino acids or proteins. There is evidence this Se incorporation into amino acids is controlled and may be the mechanism of metal tolerance in hyperaccumulators. On the other hand, nonaccumulator species appear to non-specifically incorporate selenium into non-functional proteins, proteins that have no purpose. These lethal substitutions may be the cause of Se phytotoxicity.5 Because inorganic selenium can be metabolized by the plant to various organic compounds, identifying the molecular structure of selenium-containing compounds exuded by plant roots may yield further insight into the plant metabolism of selenium.
This work chemically identifies and follows the formation of selenium-containing root exudates in hydroponically grown B. juncea. After specified intervals, hydroponic solutions containing exudates were analyzed by high performance liquid chromatography-inductively coupled plasma-mass spectrometry (HPLC-ICP-MS) and selenium-containing compounds were detected by monitoring characteristic isotopes. A collision cell was employed to facilitate the detection of sulfur isotopes in further characterization experiments. Unknown compounds were collected and analyzed by electrospray ionization mass spectrometry (ESI-MS) for identification.
Experimental
Seeds of wild Indian mustard (Brassica juncea) were obtained from the USDA Agricultural Research Station (Ames, IA). After seed germination, plants were grown hydroponically in 25% Hoagland's solution (each plant was grown in its own individual container) and maintained in a controlled growth room (15/20 °C, 8 h dark/16 h light, 520 lx) for two months. Growth solutions were prepared to contain specific concentrations (0, 1, 5 and 20 µg mL−1) of inorganic selenium (as sodium selenite, Na2SeO3) corresponding to 0, 7.8, 39 and 156 µM SeO32−. Further, one vial (no replication) of non-planted nutrient solution was maintained for each of the supplementation levels alongside planted treatments throughout the duration of the experiment. After 14 d, the selenium-containing nutrient solution was replaced with deionized water. Plants remained in this nutrient-deficient state from day 15 to 28. At days 21 and 28, the deionized water was replaced and the collected sample was analyzed before and after concentration, as described in the following sections. Due to the role microorganisms play in stimulating root exudation, we chose to maintain plants in non-sterile conditions to maximize exudation.22 Also, a non-sterile set up is more realistic in terms of understanding plant metabolism of selenium in the environment.Reagents and standards
All the solutions were prepared in 18 MΩ cm distilled and deionized water (Sybron Barnstead, Boston, MA, USA). For reversed phase chromatography, the mobile phase contained 5% methanol (Tedia Company Inc., Fairfield, OH, USA).In further investigative experiments, 1 mL of 5 mg mL−1 dithiothreitol (Aldrich, Milwaukee, WI, USA) and 1 mL of 20 mg mL−1 of proteinase K from Tritirachium album (Sigma-Aldrich, St-Louis, MO, USA) were added individually to a 5 mL volume of the exudate solution. The samples were incubated overnight at 37 °C.
The selenium standards were prepared and diluted in distilled deionized water. Dimethyl selenide (CH3SeCH3) and sodium selenite (Na2SeO3) were purchased from Fluka (Fluka, Milwaukee, WI, USA) and ICN Biomedicals Inc. (ICN Biomedicals Inc., Aurora, OH, USA) respectively. Sodium selenide (Na2Se) and elemental selenium (Se, 300 mesh) were obtained from Alfa Aesar (Alfa Aesar, Ward Hill, MA, USA). Sodium selenate (Na2SeO4·10H2O, 99.999%), Se-methionine and Se-cystine were obtained from Sigma (Sigma-Aldrich, St-Louis, MO, USA). Anhydrous sodium sulfite (Na2SO3) was obtained from Fisher Scientific (Fairlawn, NJ, USA). Se-methyl–Se-methionine was prepared as follows: 2 mg selenomethionine was mixed with 20 µL methyl iodide. The mixture was kept at room temperature for 2 hours and then 50 µL of water was added. The mixture was left for four days and then evaporated. Specific details regarding characterization by electrospray mass spectrometry and chromatographic evaluation of purity are reported elsewhere.23 The Se-methyl–Se-cysteine standard was provided by the Department of Chemistry at the University of California, Berkeley. Sodium selenosulfate (Na2SSeO3) was prepared as follows:24 1.50 g Na2SO3 was dissolved in 10 mL of water and 0.30 g Se was slowly added at a temperature of 70 °C during a period of 2 h using magnetic stirring. After ∼5 h, the unreacted Se was removed by filtration and the solution was kept refrigerated under nitrogen. Identity of the product (in the mixture) was verified using direct infusion negative ionization electrospray mass spectrometry (Se patterns at m/z = 160, 161 and 183 corresponding to 80Se were observed).
Instrumentation
| Reversed phase chromatography | |
|---|---|
| Column | Alltima C8 with dimensions of 250 mm × 4.6 mm i.d. and a particle size of 5 µm |
| Mobile phase | 0.1% Heptafluorobutyric acid + 5% methanol, pH = 2.5 |
| Flow | 0.9 mL min−1 |
| Temperature | 25 °C |
| Injection volume | 100 µL |
| Anion-exchange chromatography | |
|---|---|
| Column | Mono Q 5/50 GL FPLC (5 mm × 50 mm) |
| Mobile phase | Start buffer (A): 30 mM Tris-HCl, pH = 7 |
| Elution buffer (B): 0.3 M CH3COONH4 + 0.25 M NH4Cl in 30 mM Tris-HCl, pH = 7 | |
| 0 min: 100% A | |
| 15 min: 75% B | |
| 15.5 min: 100% B | |
| 30 min to 40 min: 100% A | |
| Flow | 0.75 mL min−1 |
| Temperature | 25 °C |
| Injection volume | 100 µL |
| ICP-MS | |
|---|---|
| Forward power | 1500 W |
| Plasma gas flow rate | 15.0 L min−1 |
| Carrier gas flow rate | 1.08 L min−1 |
| Dwell time | 0.1 s per isotope |
| Isotopes monitored | 77Se, 78Se, 82Se, 33S, 34S |
| ESI-MS | |
|---|---|
| Capillary voltage | 3000 V |
| Cone voltage | 35 V |
| Nebulizing gas | N2 |
| CID gas | Ar |
| Mass range | 50–1000 m/z |
Results and discussion
Plant growth and exudate production
All plants survived throughout the duration and were healthy enough to flower and produce fruit. However, plants grown with the highest selenium treatment (20 µg mL−1) displayed noticeably different physical characteristics than the other treatment levels. Specifically, these plants experienced stunted growth and reduced flower size, though the overall flower production (in terms of number) appeared to be the same. Although variation in leaf trichomes was observed even in control plants, none of the 20 µg mL−1 selenium-containing plants had hairs on their leaf surfaces except occasionally in the leaf margins. Leaf veination was thick and white in the higher selenium treatments rather than thin and inconspicuous in the low concentrations or controls. Root tips and the axes of root hairs were blackened in the 20 µg mL−1 treatments, though without apparent loss of length or mass. It was also noted that the adult leaves of selenium-containing plants retained juvenile leaf shape (entire, lancelot), as opposed to the adult (ovate, toothed) leaves of the control plants.The hydroponic solutions were collected at day 14 (the selenium-containing nutrient solution) and days 21 and 28 (deionized water). Based on the area of chromatographic peaks, total exudates were higher after a week of nutrient deficiency (i.e. at days 21 and 28) than in the initial two week period growing in nutrient solution. Increases in the amount of root exudates during periods of nutrient deficiency have been noted by others.25,26 Further, a higher concentration of exudates was collected on day 28 than on day 21. This may be explained by the Prikryl and Vancura observation that the release of exudates was enhanced when exudate concentration in the vicinity of the roots was lowered by replacement of the hydroponic solution.27
HPLC-ICP-MS
The selenium-containing nutrient solution of each of the additive concentrations (1, 5 and 20 µg mL−1 SeO32−) was analyzed by HPLC-ICP-MS. As expected, with each of the treatments, one can observe the peak of the supplemented inorganic Se (20 µg mL−1 SeO32− treatment is shown in Fig. 1A) as well as the presence of two additional selenium-containing compounds. The elution of the first is just prior to that of SeO32−; the remainder of the chromatogram shows a less intense peak in the retention time region of 24 minutes. | ||
| Fig. 1 A. Chromatogram of hydroponic solution of plant supplemented with 20 µg mL−1 selenite (156 µM). B. Chromatogram of selenomethionine standard, 100 ppb (510 nM). C. Chromatogram of hydroponic solution of plant supplemented with 20 µg mL−1 selenite fortified with selenomethionine standard. | ||
After 14 days of supplementation, plants were placed in deionized water that was collected at days 21 and 28. The deionized growth water was analyzed by HPLC-ICP-MS for each of the three levels of previously dosed plants (1, 5, 20 µg mL−1). The two major selenium-containing species eluted just before SeO32− and at approximately 24 minutes. Incorporation of inorganic Se into sulfur-containing amino acids is a known pathway of this accumulator. Therefore, known available Se amino acids were chromatographed under the same conditions to determine possible matches. These included Se-methionine, Se-cystine, Se-methyl–Se-methionine and Se-methyl–Se-cysteine and the elution of Se-methionine was found to occur near the region of 24 minutes.
The selenium-containing nutrient solution of a plant supplemented with 20 µg mL−1 SeO32− was consequently fortified with Se-methionine and these results are given in Fig. 1. As stated earlier, Part A shows the chromatogram of the hydroponic solution of a plant supplemented with 20 µg mL−1 SeO32−. In Part B, one can view the chromatogram from the analysis of the Se-methionine standard. Finally, Part C shows the chromatographic results of the addition of Se-methionine to the hydroponic solution of Part A. This figure shows that, when added to this matrix, most of the Se-methionine rapidly forms a new selenium compound with an entity of the exudate solution (referred to as “SeMet-P” throughout the remainder of this paper). The same result occurred when Se-methionine was added to the control for samples collected during the time the plant was given nutrient solution as well as during the time of nutrient deficiency. This was done to establish that the entity with which the Se-methionine reacted was a root exudate and not a constituent of the nutrient solution. The peak resulting from the SeMet addition was collected for further characterization by electrospray mass spectrometry and results are discussed in the following section.
Additional experiments for further chemical characterization of unknown #1 and the peak eluting at 24 minutes were performed and initially the potential presence of sulfur was investigated. The detection of sulfur is less sensitive than that of selenium with ICP-MS as the molecular interference of 16O2+ on 32S+ precludes effectively monitoring the most abundant isotope. A collision cell was employed under specific conditions as discussed in the Experimental section so as to allow the effective monitoring of this element's isotopes at m/z 33 and 34. A peak at this m/z was observed to coelute with that of unknown 1, however, no sulfur signal was detected in the region of 24 minutes. Secondly, dithiothreitol (DTT) was added to the selenium-containing nutrient solution of a plant previously administered 20 µg mL−1 SeO32−. If the unknown compounds contained a Se–S or Se–Se bond, the addition of DTT would break such and the peaks would be expected to move, or at least diminish in intensity. When this reagent was added to the selenium-containing nutrient solution, unknown #1 disappeared while no change was observed in the intensity or retention time of the peak eluting at 24 minutes. This allowed the tentative conclusion regarding the presence of a Se–S or Se–Se bond in the unknown compound #1 as well as the absence of such in the peak eluting at 24 minutes. Finally, proteinase K was added to the 20 µg mL−1 selenium-containing nutrient solution in order to investigate a possible peptide structure. If the unidentified peaks represented peptides, the addition of this enzyme would serve to diminish the intensity of said peaks; however, no lessening in intensity was observed for either peak.
The sum of these chemical experiments led to speculation regarding the identity of unknown #1 as selenosulfate, SSeO32−. As a first step in the proof of this hypothesis, this ion was synthesized as previously documented by Ball and Milne24 and outlined in the Experimental section. Successful synthesis was confirmed through ESI spectra and purity was determined chromatographically. Chromatographic analysis also showed the elution of this ion at the same approximate time as unknown 1. Furthermore, both experiments performed above with the exudate solutions (monitoring of sulfur and addition of DTT) were effectively repeated with the standard material to ensure similar behavior. As a final step in confirming the presence of this ion, ion-exchange was chosen as an alternate form of chromatography. This selection was made in an effort to gain more retention of the ionic selenium species. Chromatographic conditions were used as established by Mounicou et al.28 and are described in the Experimental section. The analysis of the synthesized standard and the exudate solution showed the match in retention time of unknown 1 and the SSeO32− ion as can be viewed in Fig. 2.
 | ||
| Fig. 2 Chromatograms of SSeO32− standard and exudate sample analyzed by anion-exchange chromatography. | ||
As a further step to obtain a greater sensitivity in the search for selenium-containing exudates, all plant growth solutions were concentrated under a gentle stream of nitrogen. The exudate solutions were concentrated from ∼50 mL to ∼500 µL. The first significant observation was the disappearance of the peak at 24 minutes. As a consequence, several volatile selenium-containing compounds were investigated under the stated chromatographic conditions and dimethylselenide was found to elute at 24 minutes. The presence of this compound can not be exclusively attributed to root exudation as its presence has also been noted to be a result of microbial transformation.29 The presence of dimethylselenide, rather than diselenide, in the exudate medium is, however, consistent with the profile of selenium volatilization of this plant.8
Other peaks were observed in the concentrated root exudate solutions. Although no additional peaks were observed in the exudate solution of the plant supplemented with 20 µg mL−1, the solutions for plants supplemented with 1 µg mL−1 and 5 µg mL−1 SeO32− both exhibited earlier-eluting selenium-containing compounds. Fig. 3 shows the chromatographic results of the concentrated exudate solution obtained from the plant previously supplemented with 5 µg mL−1 and those for the plant supplemented with 1 µg mL−1 can be viewed in Fig. 4A. The solution of the plant supplemented with 1 µg mL−1 contains a peak eluting at approximately 8 minutes in the chromatogram of the concentrated exudate solution. In the chromatogram obtained from the analysis of the solution of the 5 µg mL−1 supplemented plant, an additional peak is evident, eluting at approximately 15 minutes (labeled as unknown #2).
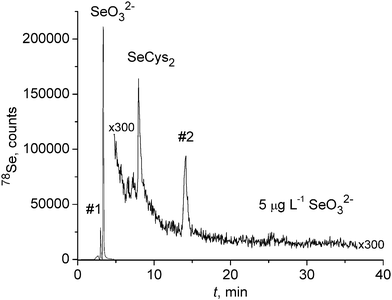 | ||
| Fig. 3 Chromatogram of hydroponic solution of plant previously supplemented with 5 µg mL−1 selenite. Solution was concentrated by evaporation with nitrogen. | ||
 | ||
| Fig. 4 A. Chromatogram of hydroponic solution of plant previously supplemented with 1 µg mL−1 selenite (7.8 µM). Solution was concentrated by evaporation with nitrogen. B. Chromatogram of selenocystine standard, 100 ppb (300 nM). C. Chromatogram of hydroponic solution of plant previously supplemented with 1 µg mL−1 selenite fortified with selenocystine standard. | ||
As mentioned earlier, Se-cystine eluted at approximately 8 minutes and a chromatogram of an analytical standard is shown in Fig. 4B. It is therefore apparent that the peak eluting at 8 minutes in both the plants supplemented with 1 µg mL−1 and 5 µg mL−1 concentrations matches the retention time of selenocystine. To further confirm, the respective exudate solutions were fortified with this selenium-containing amino acid. Interestingly, most of the fortified amino acid also seems to undergo species transformation, depicted graphically in Fig. 4C. The newly-formed species (labeled as (Se-Cys)2-P in the remainder of this paper) was collected for analysis by ESI-MS. Additionally, no commercial selenium standards were matched in retention time to the peak eluting at approximately 15 minutes and it too was collected for analysis by ESI-MS.
Electrospray mass spectrometry
Three unknowns were collected for analysis by ESI-MS and these included SeMet-P, (Se-Cys)2-P and the unknown #2. When analyzed by ESI-MS, (Se-Cys)2-P was found to contain a distinctive Se2 isotope pattern (Fig. 5). Also included in this figure is the theoretical Se isotope pattern for a molecule containing two selenium atoms. It is apparent that either (Se-Cys)2-P, or a fragment thereof, can be assigned a tentative protonated molecular weight of 865 u. Both the molecular ions at m/z 865 (80Se80Se and 78Se82Se) and m/z 863 (78Se80Se) were subjected to collision-induced dissociation (CID). Results showed very little fragmentation, with major ions at m/z 291 and 650 in both spectra, indicating that both Se atoms were lost from these fragments. An ion corresponding to 669 in the MS-MS spectrum of 865 and one at m/z 667 in the MS-MS spectrum of m/z 863 demonstrated that only these fragments were selenium-containing and represented a fragment lost containing just one of the selenium atoms. The fragment lost was calculated to have a molecular weight of 196 u and the proposed structure is given in Fig. 5. This structure represents one half of the Se-cystine amino acid plus a carboxylic acid group, leading to the hypothesis of (Se-Cys)2-P as a small peptide into which the fortified Se-cystine was incorporated upon addition. Further work with the species produced upon SeMet addition is discussed below and lends further support to the theory. It is also worth noting that this reaction is quite fast as exudate solutions were analyzed immediately after fortification. Furthermore, incorporation appears to be quite specific as results remained unchanged in numerous repetitive trials. Investigation of various additive concentrations showed that if the additive concentration was increased, an increase was observed in both the level of the complex and of the selenocystine. However, the increase in the level of the complex was found to be larger than the increase in the level of the selenocystine. | ||
| Fig. 5 Electrospray mass spectra of the (Se-Cys)2-P species formed upon (Se-Cys)2 addition. The inset shows the MS spectra matching the isotopic pattern of two selenium atoms. This mass was then subjected to CID, results also shown. (SeCys2-P peak in Fig. 4C). | ||
For the species produced upon SeMet addition, the highest-mass, most evident selenium isotopic pattern was found at m/z = 436 (80Se). Collision-induced dissociation of this ion yielded the mass spectrum as displayed in Fig. 6. It is first interesting to note the peaks at the higher masses of 454 and 472; each represents the addition of one water molecule to the base peak at m/z = 436 which is generally considered indicative of a peptide structure. Second is the fact that the majority of the primary peaks in the MS-MS spectrum were found to be selenium-containing (discernible Se isotope pattern) in the original MS data. Furthermore, one can see that the peaks at m/z = 242, 260 and 278 illustrate the same sequential addition of water molecules as well as, in the examination of the original MS spectrum, a pronounced selenium pattern. Finally worth noting is the peak at m/z = 109 as it is the major fragment of the selenomethionine molecule under the gentle ES conditions and represents CH2SeCH3+.
 | ||
| Fig. 6 Electrospray mass spectra of the SeMet-P species formed upon SeMet addition. The inset shows the MS spectra matching the isotopic pattern of one selenium atom. This mass was then subjected to CID, results also shown. (SeMet-P peak in Fig. 1C). | ||
As documented above, the peak at 242 was found to be selenium containing in the original spectrum. This represents an interesting phenomenon as the molecular weight of protonated Se-methionine is 198 u and therefore this fragment most likely represents the addition of a carbonyl and a water molecule and the proposed structure of this fragment is shown in Fig. 6. The attachment of a carbonyl to the selenomethionine yields further confidence to the theory of a peptide incorporation. The ion at m/z = 242 was subjected to collision-induced dissociation and two major peaks were found at m/z = 99 and 119. The structure of the m/z = 119 peak represents the loss of the CH2CH2SeCH3 chain and as such, this fragment ion strongly supports the structure assignment for the parent ion at m/z= 242. Finally, of further note is the fact that, although the characteristic fragment of selenomethionine was observed at m/z = 109 in the original spectrum, the fragment did not appear in the MS-MS spectrum for the fragment at m/z = 242.
Therefore, the characteristic addition of a water molecule to the parent ion as well as the structure of the fragment at m/z = 242 (Fig. 6) point to the fact that, when added to the exudates solution matrix, selenomethionine also undergoes a condensation reaction to form a small peptide. If the ion at m/z = 436 represents the parent ion of this peptide structure, there is only the possibility that this structure represents a small peptide containing selenomethionine, histidine and threonine. Summarily, there remains no doubt that the fortified amino acids (selenocystine and selenomethionine) undergo species transformation and that ESI data indicates peptide bond formation. The mechanism responsible for this condensation reaction, however, remains unclear.
Finally, the peak eluting at 15 minutes in the concentrated exudate solution obtained from the plant supplemented with 5 µg mL−1 was subjected to ESI-MS analysis. However, no clear Se isotope pattern could be discerned, most probably due to the low level present. Subsequent work performed in this group showed a large overall increase (∼1000 times) in exudate levels when plants were placed in a stressed state throughout the duration of supplementation. Specifically, in this work, plants were dosed with equivalent concentrations of Hg as well as Se. It is the authors hope that the large increase in exudate levels will yield a stronger signal in the ESI mass spectrum and hence a definitive characterization of this last unknown Se-containing exudate.
Conclusions
In conclusion, we observed three selenium-containing exudates released from plants of Brassica juncea grown in a selenium-rich environment. Two were identified as selenocystine and the selenosulfate (SSeO32−) ion. The presence of dimethylselenide (CH3SeCH3) is also observed but cannot be attributed exclusively to plant exudation because plants were not grown in sterile conditions. Additionally, the incorporation of fortified seleno-amino acids into peptide structures is observed in ESI spectra. As such, this study represents a preliminary investigation of the root exudates of this selenium-accumulating plant. Much future work lies ahead as it would be most interesting to study the exudates released from plants grown under sterile conditions and therefore gain a more comprehensive picture by comparison of the two results. Furthermore, a complete characterization of the exudate profile may lead to a better understanding of the selenium extracting abilities of this plant and the potential role of exudates in phytoremediation. Finally, the mechanism of the condensation reaction that takes place upon amino acid addition remains unclear and should be the object of future study.Acknowledgements
The authors would like to acknowledge NIEHS grant #ES04908 for partial funding of this research.References
- T. A. Brown and A. Shrift, Biol. Rev., 1982, 57, 59 CrossRef CAS.
- M. Stadlober, M. Sager and K. J. Irgolic, Food Chem., 2001, 73, 357 CrossRef CAS.
- M. P. Longnecker, P. R. Taylor, O. A. Levander, M. Howe, C. Weillon, P. A. McAdam, K. Y. Patterson, J. M. Holden, M. J. Stampfer, J. S. Morris and W. C. Willett, Am. J. Clin. Nutr., 1991, 53, 1288 CAS.
- G. S. Banuelos, S. Zambrzuski and B. Mackey, Plant Soil, 2000, 224, 251 Search PubMed.
- N. Terry, A. M. Zayed, M. P. d. Souza and A. S. Tarun, Annu. Rev. Plant Physiol. Plant Mol. Biol., 2000, 51, 401 CrossRef CAS.
- M. Montes-Bayon, E. G. Yanes, C. P. d. Leon, K. Jayasimhulu, A. Stalcup, J. Shann and J. A. Caruso, Anal. Chem., 2002, 74, 107 CrossRef CAS.
- M. Montes-Bayon, T. D. Grant, J. Meija and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 1015 RSC.
- J. Meija, M. Montes-Bayon, D. L. LeDuc, N. Terry and J. A. Caruso, Anal. Chem., 2002, 74, 5837 CrossRef.
- J. R. Shann and J. J. Boyle, Influence of plant species on in situ rhizosphere degradation, in Microbial Degradation of Organic Chemicals in the Rhizosphere: Implications for Bioremediation, ed. T Anderson and J. Coats, ACS Books, Washington, DC, 1994 Search PubMed.
- C. Bertin, X. Yang and L. A. Weston, Plant Soil, 2003, 256, 67 Search PubMed.
- F. D. Dakora and D. A. Phillips, Plant Soil, 2002, 245, 35 Search PubMed.
- Z. Rengel, Plant Soil, 2002, 245, 59 Search PubMed.
- S. P. McGrath, F. J. Zhao and E. Lombi, Plant Soil, 2001, 232, 207 Search PubMed.
- J. Kozdroj and J. D. v. Elsas, Soil Biol. Biochem., 2000, 32, 1405 CrossRef CAS.
- E. D. Mariano and W. G. Keltjens, Plant Soil, 2003, 256, 469 Search PubMed.
- Z. M. Yang, M. Sivaguru, W. J. HOrst and H. Matsumoto, Physiol. Plant., 2000, 110, 72 CrossRef CAS.
- U. Basu, A. G. Good, T. Aung, J. J. Slaski, A. Basu, K. G. Briggs and G. J. Taylor, Physiol. Plant., 1999, 106, 53 CrossRef CAS.
- A. Heim, I. Brunner, B. Frey, E. Frossard and J. Luster, J. Plant Nutr. Soil Sci., 2001, 164, 519 CrossRef CAS.
- K. A. Hill, L. W. Lion and B. A. Ahner, Environ. Sci. Technol., 2002, 36, 5363 CrossRef CAS.
- S. Tu, L. Ma and T. Luongo, Plant Soil, 2004, 258, 9 Search PubMed.
- X. E. Yang, V. C. Baligar, J. C. Foster and D. C. Martens, Plant Soil, 1997, 196, 271 Search PubMed.
- D. A. Barber and J. K. Martin, New Phytol., 1976, 76, 69 CAS.
- K. Wrobel, K. Wrobel, S. S. Kannamkumarath and J. A. Caruso, Anal. Bioanal. Chem., 2003, 377, 670 CrossRef CAS.
- S. Ball and J. Milne, Can. J. Chem., 1995, 73, 716 CAS.
- K. Tawaraya, K. Hashimoto and T. Wagatsuma, Mycorrhiza, 1998, 8, 67 CrossRef CAS.
- D. S. Lipton, R. W. Blanchar and D. G. Blevins, Plant Physiol., 1987, 85, 315 CrossRef CAS.
- S. Prikryl and V. Vancura, Plant Soil, 1980, 57, 69 Search PubMed.
- S. Mounicou, J. Meija and J. A. Caruso, Analyst, 2004, 129, 116 RSC.
- T. G. Chasteen and R. Bentley, Chem. Rev., 2003, 103, 1 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |