Three computational methods for studying permeation, selectivity and dynamics in biological ion channels
Shin-Ho
Chung
a and
Ben
Corry
b
aDepartment of Theoretical Physics, The Australian National University, Canberra, A.C.T. 0200, Australia. E-mail: shin-ho.chung@anu.edu.au.; Fax: +61 2 6247 2792; Tel: +61 2 6125 2024
bSchool of Biomedical, Biomolecular and Chemical Sciences, The University of Western Australia, Perth, Australia
First published on 9th November 2005
Abstract
The cell membrane, confining some ions and molecules on one side and exchanging others with the other side, is the ultimate unit of the physiology of life. The delicate task of regulating the transport of ions across the membrane is carried out by biological nanotubes called ‘ion channels’. Recently, there have been enormous strides in our understanding of the structure–function relationships of biological ion channels. The molecular structures of several ion channels have been determined from crystallographic analysis, including potassium channels, mechanosensitive channels, a chloride channel, as well as gramicidin channels and porins. It is expected that the X-ray structures of other ion channels will soon follow these discoveries, ushering in a new era of ion channel studies in which predicting the function of channels from their atomic structures will become the main quest. In parallel to these experimental findings, there have been important advances in computational biophysics. Here we summarize three theoretical approaches that have been utilized to understand the dynamics of ion permeation across bio-nanotubes, highlighting their advantages and shortcomings, and briefly describe some of the salient properties of ion channels uncovered through computational studies.
 Shin-Ho Chung | Shin-Ho Chung received B.Sc. degrees from Stanford University, Stanford, California and London University, London, England, and his Ph.D. degree from Harvard University, Cambridge, MA. He held a post-doctoral position at the Research Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA. Currently, he is the Head of the Biophysics Group in the Department of Theoretical Physics, Australian National University. His primary research effort is aimed at building theoretical models of biological ion channels. |
 Ben Corry | Ben Corry received a B.Sc., B.A. and Ph.D. from the Australian National University, Canberra, Australia. He held a post-doctoral position at the Department of Theoretical Physics, Australian National University. Subsequently, he was awarded an Australian Research Council Post-doctoral Fellowship. Currently, he is at the Department of Chemistry, the University of Western Australia, Perth, Australia. His research interests are centered on elucidating the permeation mechanisms of ions across bio-nanotubes, using computational and fluorescence methods. |
Introduction
Ionic channels in lipid membranes play a crucial role in the existence of living organisms. All electrical activities in the nervous system, including communication between cells and the influence of hormones and drugs on cell function, are regulated by the opening and closing of these membrane proteins. Because these channels are elementary building blocks of brain function, understanding their mechanisms at a molecular level is a fundamental problem in biophysics. Moreover, the elucidation of how single channels work will ultimately help us find the causes of, and potentially cures for, a number of neurological and muscular disorders.In the past few years, the field of ion channel research has entered into a rapid phase of development. The molecular structures of the Streptomyces lividans KcsA potassium channel as well as several other potassium channels, two mechanosensitive channels, a chloride channel and neurotransmitter receptor pores have been determined from crystallographic analysis.1–8 It is expected that crystal structures of other ion channels will soon follow these discoveries. Also, as new analytical methods have been developed and computational power increases, theoretical methods of studying the permeation of ions through ion channels and the structural dynamics of the protein have become increasingly advanced. Now it has become possible to relate the atomic structure of an ion channel to its function, through the fundamental laws of physics. The mechanisms underlying macroscopic observable properties of ion channels are being addressed by molecular and stochastic dynamics simulations. Intuitive and hand-waving explanations of the permeation and selectivity of ions are beginning to be replaced by quantitative statements based on rigorous physical laws.
Here we give an overview of recent advances in the biophysics of ion channels, placing a special emphasis on theoretical approaches that are currently under development. Computational methods of solving complex biological problems, such as permeation, selectivity and gating mechanisms in ion channels, will increasingly play prominent roles as the speed of computers increases. We give brief summaries of various methods that have been developed for treating the time-dependent, nonequilibrium processes that underlie the flow of currents through biological ion channels. This review is primarily devoted to three computational approaches—molecular dynamics, stochastic dynamics and continuum theories—used to unravel the inner workings of biomolecules. We give intuitive explanations of the physics underlying each of the methods, referring to more comprehensive publications for mathematical details. We discuss the merits and shortcomings of each computational approach and give examples of their application to ion channels. Detailed accounts of recent experimental findings on ion channels are not given here; the reader is referred to the latest edition of Hille9 which provides an excellent source of information in this regard. The reader is also referred to recent review articles10–14 for further details of recent advances in ion channel research.
Theoretical tools for studying ion channels
One important aim of theoretical biophysicists has been to provide a comprehensive physical description of biological ion channels. Such a theoretical model, once successfully formulated, will link channel structure to channel function through the fundamental processes operating in electrolyte solutions. It will also concisely summarize the experimental data, by interlacing all those seemingly unrelated and disparate observations into a connected whole. The theory will elucidate the detailed mechanisms of ion permeation: the location of binding sites in the channel, how fast an ion moves across the channel, how the protein discriminates between ion types and where the rate-limiting steps are in conduction. Finally, it will make predictions that can be confirmed or refuted experimentally.The tools of physics that are employed in this endeavor, from fundamental to phenomenological, are ab initio and classical molecular dynamics, stochastic dynamics and continuum theories. In ab initio molecular dynamics, the interactions between atoms are determined from first principles electronic structure calculations. Since there are no free parameters in these calculations, ab initio molecular dynamics represents the ultimate approach to the modelling of biomolecular systems. However, because of the extremely demanding nature of computations, its applications are generally limited to very small systems at present. As both computational power and the algorithms for implementing the quantum mechanical equations improve, we can expect such calculations to play an important role in ion channel analysis. In classical molecular dynamics, trajectories of all the atoms in a system are followed using Newton's equation of motion. Simulations are carried out using empirically determined pair-wise interaction potentials between the atoms. Although it is possible to model an entire ion channel with classical molecular dynamics, it is not feasible to simulate the system long enough to see the permeation of ions across a channel and to determine its conductance under physiological conditions, which is the most important channel property that can be measured experimentally. For that purpose, we utilize stochastic dynamics, the simplest form of which is Brownian dynamics, where water molecules that form the bulk of the system are integrated out and only the ions themselves are simulated explicitly. The continuum electro-diffusion theory of the Poisson–Nernst–Planck equations makes one further simplification known as the mean-field approximation. Here, ions are treated not as discrete entities but as continuous charge densities that represent the space-time average of the microscopic motion of the ions. In Poisson–Nernst–Planck theory, the flux of an ionic species is described by the Nernst–Planck equation that combines Ohm's law with Fick's law of diffusion, and the potential at each position is determined from the solution of Poisson's equation using the total charge density in the simulation assembly. Poisson–Nernst–Planck theory thus incorporates the channel structure and its solution yields the potential, concentration and flux of ions in the system in a self-consistent manner.
There is one other approach that has been fruitfully employed to model biological ion channels, namely, reaction rate theory.9 In this approach, an ion channel is represented by a series of ion binding sites separated by barriers, and ions are assumed to hop from one binding site to another, with the probability of each hop determined by the height of the energy barrier. Many useful insights have been gleaned about the mechanisms of ion permeation using this approach. The merits and demerits of this theory have been debated extensively in the literature,15–19 to which the interested reader is referred. We will not discuss the rate theories further in this review because the model parameters have no direct physical relation to the channel structure, whereas the current focus in ion channel research is on the structure–function relationships.
Poisson–Nernst–Planck theory
 | (1) |
To calculate the ionic current, a channel is placed in a simulation system by representing it as a rigid dielectric region and assigning partial charges to the locations of the atoms. Reservoirs with specified ion concentrations and potentials are attached at each end of the channel. The simulation system is then divided into small rectangular grids, and the Poisson–Nernst–Planck equations are solved at the grid points using a finite difference algorithm.20,21 The grid size has to be optimized for efficient running of the program. A smaller grid size improves the accuracy of the results but also takes a much longer computational time. The required inputs for the algorithm are: (i) the coordinates of all the atoms forming the channel, (ii) the magnitude of electronic charge carried by each atom, (iii) the dimensions of the reservoirs and ionic concentrations in each reservoir, (iv) the dielectric constant of the protein and the solution, (v) the membrane potential, and (vi) the diffusion coefficients of cations and anions. Once these parameters are specified, the solutions of the Poisson–Nernst–Planck equations give the concentration and potential throughout the system as well as the steady state ionic currents through the channel.
There is one severe shortcoming of PNP theory when it is applied to study a mesoscopic system. In this theory, ions are not treated as discrete entities but as an average concentration. Now, let us consider a cylindrical channel with a radius of 3 Å, spanning a 30 Å thick membrane, whose volume is ∼103 Å3. If ions were treated as discrete spherical entities, then at physiological concentrations, on average either a cation or anion can be expected in the pore only 10% of the time. To carry out the PNP calculation, the simulation system needs to be divided into small cubic cells with a volume of, for example, 1 Å3 and the concentration of anions and cations is computed in each cell. In the example given, a cubic cell contains ∼10−4 cations and an equal number of anions. In reality there will be either a whole cation, a whole anion or no ion at any point in the channel. The presence of both cationic and anionic fractional charges in each cell introduces errors in the results obtained with the PNP equations, with the magnitude of the errors increasing steadily as the radius of the pore decreases and the chance of having both cations and anions in the channel diminishes.
The systematic errors inherent in the theory when it is applied to mesoscopic systems arise predominantly from the fact that the effects of “induced surface charges” are underestimated in PNP theory. When a charged particle in a high dielectric medium (electrolyte solution) approaches a region of a low dielectric medium (the protein wall), it induces surface charges of the same polarity at the protein–water interface. Water molecules near a cation in an electrolyte solution will align themselves such that the oxygen atoms, with their partial negative charges, are positioned nearest to the ion. Because polar or carbonyl groups on the protein wall cannot rotate as freely as water molecules, there will be excesses of hydrogen atoms at the water–protein interface. Viewed from the ion, these excess hydrogen atoms at the boundary appear as surface charges, exerting a repulsive force on it. Macroscopically, we say that a charge q located at a distance d from a slab of protein induces charge at the surface of the dielectric boundary. For an idealized infinite plane, the magnitude of the repulsive force this ion experiences is nearly the same as when we place another charge q′, on the other side, at a distance d from the surface, and remove the boundary. This effect is greatly magnified in a narrow, cylindrical water-filled pore created by a low dielectric material. Thus, in reality, an ion attempting to navigate across a narrow conduit formed by the protein wall encounters an insurmountable energy barrier owing to induced surface charges. This energy barrier can usually only be overcome by ions with the aid of charged residues or dipoles near the pore. The interaction between the two forces on a permeating ion—the repulsive force resulting induced surface charges and attractive force induced by fixed charges—play a crucial role in the permeation dynamics in ionic channels.
In PNP theory, in contrast, the presence of both positive and negative fractional charges on any grid point means that the total charge is zero and no surface charges are induced. Even if partial charges in the protein create a net charge at any point, the surface charge induced by this is always smaller than if the entire ionic charge of an ion was localized there. These fractional charges diffuse across the pore, one cubic grid to another, under the influence of the membrane potential, unencumbered by any induced surface charges on the protein boundary. In short, one aspect of the ion–protein interaction, which is the dominant force in the process of ion permeation across a narrow pore, is not accounted for in PNP theory. There is a further problem in applying PNP theory to biological ion channels, where two or more ions are resident. In such situations, the ion–ion interactions play an important role in conduction processes. These Coulombic interactions taking place inside of the pore cannot be satisfactorily dealt with in PNP theory. PNP theory also suffers the problems of using a rigid protein structure and assigning dielectric constants that are discussed in more detail with reference to Brownian dynamics simulations.
Im and Roux36 have examined the properties of ion permeation across the channel formed by OmpF porin from Escherichia coli, using both PNP theory and Brownian dynamics. This wide pore passively transports small molecules down their concentration gradient. It is weakly cation selective, and its selectivity depends on the ionic strength of the solution. The theory is able to capture some of the salient features of the permeation dynamics, including the electrostatic interactions between ions and the charge distribution of the channel, the number of ions in the pore, and the current–voltage–concentration profile. PNP theory overestimates the current by about 50%. A similar study has been carried out on the pore formed by staphylococcal α-hemolysin, a toxin protein that causes urinary tract infections. The channel conductance calculated from PNP theory is consistently larger (by 30 to 50%), compared to the experimental value.37 From these studies, we conclude that PNP theory can be used to study ion permeation through wide molecular pores, provided one is prepared to accept a large error in calculated conductances.
Nonner and Eisenberg38 made an attempt to relate the observed properties of the calcium channel to its structure with PNP theory. They modelled the pore as a cylinder connected to a tapered atrium at each end. To mimic the important cluster of four glutamate residues found in the real calcium channel, the central cylinder is assigned a low dielectric material with a uniform volume density of fixed charge. Applying PNP theory to this model the calculated currents successfully match the current–voltage relations, anomalous mole fraction effects and saturation effects of varied Ca2+ and Na+ concentrations. Despite the success of this method the proposed mechanism of selectivity relies on an excess chemical potential of unspecified physical origin, and some of the parameters used to fit the channel data appear physically unrealistic. For example, the calcium diffusion coefficient used in this study to fit the data is about four orders of magnitude smaller than the microscopic estimates obtained from molecular dynamics simulations.39
Brownian dynamics
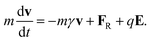 | (2) |
The first two terms on the right-hand side of eqn. (2) describe the effects of collisions with the surrounding water molecules. The first term corresponds to an average frictional force with a friction coefficient given by mγ. The second term, FR, represents the random part of the collisions and rapidly fluctuates around a zero mean. Finally, E in eqn. (2) denotes the total electric field at the position of the ion arising from other ions, fixed charges in the protein, membrane potential and induced surface charges.
To carry out Brownian dynamics simulations of ion channels, one needs to specify the boundaries of the system. This is a relatively simple problem for 1-dimensional Brownian dynamics simulations,40–42 but requires the addition of reservoirs to the channel system in the more realistic case of 3-dimensional Brownian dynamics simulations. Here we describe a simple stochastic boundary that has been used successfully in applications of Brownian dynamics simulations to a number of ion channels.43–47
Fig. 1 shows a schematic illustration of a Brownian dynamics simulation assembly. An ion channel representing the potassium channel is placed at the center of the system. The positions of all the atoms forming the channel are given by its X-ray structure, and the charge on each atom is assigned. Then, a large reservoir with a fixed number of K+ (or Na+) and Cl− ions is attached at each end of the channel (Fig. 1A). The membrane potential is imposed by applying a uniform electric field across the channel (Fig. 1B). This is equivalent to placing a pair of large plates far away from the channel and applying a potential difference between them. Since the space between the electrodes is filled with electrolyte solution, each reservoir is in iso-potential. That is, the average potential anywhere in the reservoir is identical to the applied potential at the voltage plate on that side, and the potential drop occurs almost entirely across the channel. When an ion strikes the reservoir boundary during simulations, it is elastically scattered back into the reservoir, equivalent to letting an ion enter the reservoir whenever one leaves the simulation system. Thus the concentrations of ions in the reservoirs are maintained at the desired values at all times. During simulations of current measurements, the chosen concentration values in the reservoirs are maintained by recycling ions from one side to the other whenever there is an imbalance due to a conduction event, mimicking the current flow through a closed circuit.
 | ||
| Fig. 1 Simulation assembly for Brownian dynamics. (A) The shape of the KcsA channel is modified such that the minimal radius of the intracellular gate (on the left hand side) is 3 Å. The ribbons represent the outer helices, pore helices and inner helices. A reservoir containing a fixed number of anions (red) and cations (blue) is attached at each end of the KcsA potassium channel. The charge on each atom forming the channel is assigned. (B) A uniform electric field is applied across the channel to mimic the membrane potential. This is equivalent to placing two voltage plates at a distance and applying a potential (±V) on each plate. | ||
In Brownian dynamics simulations, the Langevin equation is solved repeatedly to trace the trajectory of every ion in the assembly. Snapshots of the simulation system are taken at short time intervals for millions of time-steps. At each time-step, usually 2 fs, the forces acting on each ion are calculated and the Langevin equation is used to determine where it will move in the next time-step. By repeating this process for a sufficiently long period of time, usually many microseconds, one can determine how many ions move across the channel in a fixed period of simulation time and deduce the single channel current.
The forces acting on the ions at each step can be calculated in many ways. Examples exist in which these are fed into the simulation from the results of phenomenological46 or molecular dynamics simulations,47 an approach that can potentially avoid the difficulties of assuming a rigid protein. However, since the water and protein are already represented as continuous media, the forces are most often calculated by solving Poisson's equation. A crucial issue is whether such a continuum approximation can be justified in a narrow, biological nanotube. In bulk water, molecules polarize so as to shield electrostatic interactions by a factor of approximately 1/80. However, given the likely preferential alignment of water in narrow pores and regions of high charge, this shielding is likely to be far less effective in an ion channel. Thus, one should use a lower value of the dielectric constant for the water in the channel when solving Poisson's equation. But exactly what value of the dielectric constant should be used is unknown. Determining the appropriate values using molecular dynamics simulations or otherwise would be a useful project. The validity of treating the channel protein as a static structure in Brownian dynamics also deserves further investigation. It should be noted that thermal fluctuations of proteins occur in the time-scale of femtoseconds, whereas a conduction event across a typical ionic channel takes place once in 100 ns—approximately six to seven orders of magnitude slower time-scale. Thus, it is likely that rapid thermal fluctuations of the atoms forming the channel are not important for channel selectivity and conduction. Alterations in the average positions of the protein atoms caused by the presence of permeating ions may play a role, and their effects should be examined both experimentally and by using molecular dynamics simulations. If found to be important, some of the motions of the protein, such as the bending of carbonyl groups, can readily be incorporated in Brownian dynamics modelling of ion channels. Finally, size-dependent selectivity among ions with the same valence cannot be easily understood within the Brownian dynamics framework, and one has to appeal to molecular dynamics or semi-microscopic Monte Carlo simulations48 for that purpose.
We summarize two examples of how Brownian dynamics has been applied recently in elucidating the dynamics of ion permeation in biological channels. Using the experimentally determined potassium channel structure as a template, as shown in Fig. 2A, Chung et al.49 proposed a plausible explanation for the diversity of potassium channels seen in nature. There are many different types of potassium channels, which differ widely in their conductances and gating characteristics while having a similar primary structure. Conductance levels of various types of potassium channels range from 4 to 270 pS (1 pS equals 0.1 pA of current across the channel with the driving force of 100 mV). Despite this diversity, they all share the common feature of being highly selective to potassium ions and display broadly similar selectivity sequences for monovalent cations. To understand this feature, Chung et al.49 investigated the possible structural differences that could give rise to different potassium channels. They systematically change the radius of the intracellular pore entrance, leaving the dimensions of the selectivity filter and cavity unaltered. As the intrapore radius is increased from 2 to 5 Å, the channel conductance changes from 0.7 to 197 pS (0.17 to 48 pA). In Fig. 2B, the simulated current across the model ion channel determined from Brownian dynamics is plotted against the radius of the intrapore gate. By examining the energy profiles and the probabilities of ion occupancies in various segments of the channel, they deduce the rate-limiting step for conduction in the potassium channels. Ion distributions revealed that the selectivity filter was occupied by two K+ ions most of the time. A conduction event was triggered when a third ion climbed over the energy barrier located between the cavity and the intracellular mouth and moved toward the selectivity filter. Potential energy profiles encountered by an ion traversing along the central axis of the channel when there are two ions in or near the selectivity filter are shown for the channels with radii 2 Å (solid line in Fig. 2C), 3 Å (long-dashed line) and 4 Å (dashed line). Ions need to climb over the energy barrier, whose height is denoted as ΔU, to move across the channel. This barrier is the rate-limiting step in the permeation process: as its height increases with a decreasing intrapore radius, the channel conductance drops exponentially.
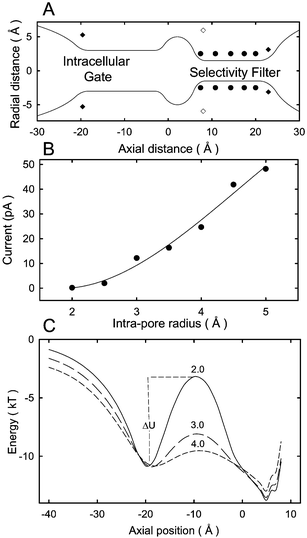 | ||
| Fig. 2 Brownian dynamics simulations of model potassium channels. (A) Solid line shows the outline of a simplified model channel. A three dimensional channel shape is obtained by rotating the curves by 180°. The positions of dipoles on the channel wall are indicated with filled circles (carbonyl and hydroxyl oxygen atoms), filled square (mouth dipoles) and open square (helix dipoles). A set of such channels with varying radii of the intracellular gate is constructed for Brownian dynamics simulations. (B) The outward current is plotted against the radius of the intracellular aspect of the channel entrance. The applied field to obtain the current is 2 × 107 V/m. (C) The energy profiles obtained from the channels with the radii of 2 (solid line), 3 (long-dashed line) and 4 Å (dashed line) are superimposed. The curves show the barrier ΔU encountered by an ion attempting to move towards the selectivity filter. The height of the barrier ΔU an ion needs to surmount to traverse the channel decreases progressively as the radius of the intrapore gate is widened. | ||
Brownian dynamics simulations were similarly applied to elucidate the dynamics of ion permeation across ClC-type channels.50,51 The ClC family of Cl− channels, present in the cell membranes of every living organism, perform diverse roles, such as the control of cellular excitability, acidification of intracellular vesicles, and cell volume regulation.52–54 The X-ray structures of two prokaryotic ClC proteins were determined by Dutzler et al.3,4 Corry et al.51 created an open-state homology model of a eukaryotic ClC channel, ClC-0, using the crystal structure of the prokaryotic protein as a basis, as shown in Fig. 3A. This channel, from Torpedo electroplax, was first discovered by Miller55 and is one of the most thoroughly studied channels of this family. As illustrated in Fig. 3A, the ionic pathway of ClC-0 takes a tortuous course through the protein, unlike that of the potassium channel, which is straight and perpendicular to the membrane surface. The channel is quite narrow, having a minimum radius of 2.5 Å near the center, but opens up quite rapidly at each end. The distance from one end of the pore to the other is 55 Å and it is lined with many charged and polar amino acid residues. The pore is lined with four acidic residues and ten basic residues. Their locations along the ion conducting pathway are shown in Fig. 3B.
 | ||
| Fig. 3 Brownian dynamics simulations of an open-state ClC channel, ClC-0. (A) The open state of the channel is based on the crystal structure of the bacterial ClC protein. The front half of the atoms is removed to reveal the ion-conducting path across the protein. (B) The water-filled pore through which Cl− ions move is lined with both acidic and basic residues. The channel is normally occupied by two Cl− ions, shown here in green. (C) The current–voltage relationship obtained with symmetrical solutions of 150 mM, using Brownian dynamics simulations (filled circles) is compared with the experimental measurements obtained by Miller,55 shown in open circles. (D) The current–concentration relationship obtained with symmetrical solutions of varying concentrations of NaCl in the reservoirs under an applied potential of −80 mV (filled circles) is fitted with the Michaelis–Menten equation. The experimental measurements obtained by Tsung-Yu Chen (personal communication) are shown in open circles. The half-saturation points determined from the fitted curves are 163 ± 51 mM for the simulated data and 136 ± 8 mM for the experimental data. | ||
The conduction properties of the model shown in Fig. 3B were examined with Brownian dynamics simulations. A current–voltage relationship obtained with symmetrical solutions of 150 mM in both reservoirs is shown in Fig. 3C. The relationship is linear, with a conductance of 11.3 ± 0.5 pS that agrees well with experimental measurements reported by Miller55 (superimposed open circles). The current–concentration relationship obtained from the homology model using Brownian dynamics (filled circles) is also accord with the experimental observations (open circles) as shown in Fig. 3D.
Molecular dynamics
 | (3) |
At every time step, the potential function is recalculated using the new positions of the particles to determine their positions a short time later, and this process is iterated for a large number of steps until a statistically satisfactory data set is generated. The trajectory data thus generated is stored at certain intervals, which can later be analyzed to determine the structural and dynamical properties of the simulation assembly. Quantities such as free energy, mean square displacement, radial distribution and other correlation functions can be calculated from an ensemble average over several simulations or parts of a long simulation. Ion channel simulations will typically include tens to hundreds of thousands of atoms and the trajectories of all atoms can be followed as they move in real time typically for up to ∼100 ns, although longer simulations have also been reported. Because all the atoms in the system (including water molecules) are represented explicitly in molecular dynamics, there are no frictional or random forces to deal with as in stochastic dynamics. Also, no assumptions are required about the nature of the solvent, and there is no need to choose dielectric values or boundaries as all atoms are present, and in principle all interactions (water–ions, water–protein, water–lipid, lipid–protein) are incorporated. This method automatically includes the dynamics of the ion channel protein itself as well as any dynamic effects of the lipids or solvent.
The success of molecular dynamics simulations in capturing the dynamics of the real system hinges critically on how accurately the potential functions or force fields are selected. In the past two decades, numerous studies have been carried out to develop force fields for biomolecular applications, and these are incorporated into several user-friendly computer programs for simulation of biomolecular systems.56–59 In these programs, the non-bonded interactions between atoms are represented by Coulomb and Lennard-Jones potentials. Pairwise interactions are used for all the atoms in the system, and the potential parameters are determined empirically from spectroscopic data and fits to bulk properties. Most atoms in the system are also covalently bonded to other atoms, and these bonds are represented in molecular dynamics by a set of force parameters that describe the stretching of a bond between two atoms, the bending of a bond angle formed by three atoms, and the torsion of a dihedral angle between the 123 and 234 planes in four atoms.
The second limitation of this technique is the inadequacy of the empirical force fields employed in the molecular dynamics simulation packages currently available for describing ion permeation. The electrons around atoms are not inert but move according to quantum mechanical laws. When an atom is placed in an electric field, the position of the electron cloud shifts with respect to the nucleus. This induces a dipole, which in turn creates an electric field of its own and further polarizes the surrounding atoms. It is computationally expensive to compute all these polarization effects, including dispersion forces (which arises from quantum fluctuations that induce correlations between the electrons of two atoms). Thus, polarization effects implemented in most programs are incorporated implicitly by invoking a mean field approximation. That is, an average induced dipole term is added to the monomer value such that the results of simulations reproduce the bulk property of a system. For example, the dipole moment of water is taken as 2.3 Debye in most molecular dynamics programs, which is larger than the experimental value of 1.85 Debye. These crude approximations of the polarization effects may introduce large errors in the results of simulations when the technique is applied to a mesoscopic system, such as ion channels, for which the parameters have not been empirically fitted. Thus, it is desirable to develop polarizable force fields that could provide more realistic pictures of quantities such as the free-energy profiles of an ion in a channel.
Ideally, the parameters of force fields to be used in molecular dynamics should be derived from electronic structure calculations using ab initio molecular dynamics methods, rather than determining them empirically from fits to data. Ab initio molecular dynamics has already been applied in condensed matter physics and in studies of water, electrolytes and bio-molecules. Density functional theory has been adapted to calculate the potentials between atoms on the fly.65,66 Examples of the use of static ab initio methods in examining ion binding in potassium channels67 have been reported and the development of linear scaling techniques mean that these methods will undoubtedly be applied for modelling of ion channels in the near future.
The availability of an X-ray structure of the KscA potassium channel prompted many groups to investigate ion permeation through it using molecular dynamics. Largely because it was the first biological ion channel to have its tertiary structure determined, it has been a prime target for simulation and modelling studies and is the only example we will discuss here. These studies have examined the mechanisms underlying the permeation of ions across the channel, the basis of ion selectivity, and the conformational changes that occur in the KcsA protein when the channel opens. Such simulations reveal that the potassium channel is usually occupied by three K+ ions, two in the selectivity filter and one in the cavity, as observed in X-ray diffraction experiments.1,74,75 There is also some evidence that the K+ ion in the cavity approaching the selectivity filter triggers a conduction event, and permeation across the filter occurs through the recycling of ions as 2 K+ → 3 K+ → 2 K+.76,77 Potential of mean force calculations with multiple K+ ions in the selectivity filter indicate that the free energy barriers between the bindings sites are about 3 to 4 kT.77 Thus, Coulomb repulsion exerted by a third K+ ion enables the resident ion to climb over this energy barrier and conduction to take place.
The crystal radius of sodium is 0.99 Å, whereas that of potassium is 1.33 Å. Yet, all potassium channels effectively exclude Na+ ions passing through the pore. The key differences between potassium and sodium appear to be only a small difference in radius and in polarizability. On the basis of the X-ray structure of the KcsA potassium channel, it has been suggested that a ‘rigid’ selectivity filter provides stronger cation–oxygen interactions for K+ ions than for Na+ ions. Thus, the energetic cost of dehydrating K+ ions is repaid by ion–protein interactions, while ion–protein interactions are too weak to balance the cost of dehydrating Na+ ions. Several simulations have tried to address this question through free-energy perturbation calculations, where a K+ ion is alchemically transformed into a Na+ ion. The barrier Na+ encounters in crossing the selectivity filter is estimated to be 11 kT,78 8 kT79 or 5 kT,80 in rough agreement with the experimental value of 9 kT extracted from the K+/Na+ selectivity ratio of 104.
The conventional view that the carbonyl oxygen atoms lining the pore are held rigidly in place has been questioned by several molecular dynamics studies. Domene and Sansom81 placed different cations (Na+, K+, Rb+, Cs+) in the selectivity filter and observed its shape. Significant flexibility of the filter is observed, as well as concerted motions of ions and water. In particular, pronounced distortions of the filter occur when no ions are present, which is also found from crystallographic studies at low salt concentration.82 The two most readily permeant ions, K+ and Rb+, are similar in their interactions with the selectivity filter, while Na+ ions tend to distort the filter by binding to a ring of four carbonyl oxygens. The presence of a larger Cs+ ion results in a small degree of expansion of the filter relative to the X-ray structure and these ions show some tendency to bind within the gate region of the channel, near the cavity. Although these results are very interesting, it should be kept in mind that ion parameters are difficult to obtain with high accuracy and such differences may result from the choice of force field parameters. Further studies in this area would certainly be worthwhile. More recently, Noskov et al.83 suggested that selectivity may result from the electrostatic interactions between various atoms and the permeating ion. Noting that the protein forming the potassium channel undergoes structural fluctuations of about 0.75 Å, far greater than the difference of the atomic radii of Na+ and K+ ions, they suggest that the tight coordination of a K+ ion by 8 carbonyl oxygens lining the filter cannot be the underlying mechanism for selectivity. In this context, it should be emphasized once again that that the characteristic oscillation periods of atomic motions are of the order of 10 fs (bond stretch) to 30 fs (dihedral motion),84 whereas a conduction event across the potassium channel takes place on average once every 100 ns. Thus, these atomic motions take place at the rate of six to seven orders of magnitude faster than a conduction event. A permeating ion is therefore likely to see the average position of the atoms forming the conduit. If so, root-mean-square atomic fluctuations may have no relevance to the selectivity or conductivity of the channel, but structural flexibility that alters the average positions of the protein atoms may be important.
Concluding remarks
We have briefly summarized three computational tools—Poisson–Nernst–Planck theory, Brownian dynamics and molecular dynamics—that will play increasingly prominent roles in understanding how biological ion channel work. Each of these approaches has its strengths and limitations, and involves a degree of approximation. The main defects of PNP theory are errors stemming from the mean-field assumption. In particular, it ignores the effects of induced surface charges created as a charged particle in electrolyte solutions approaches the protein boundary. The magnitude of the errors introduced by the mean-field approximation become large when the theory is applied to narrow ionic channels. By incorporating a term in the PNP equations to account for the barrier created by induced surface charges, the magnitude of the errors can be reduced somewhat.34 However, doing this removes much of the simplicity of the PNP theory, one of its main advantages over the other approaches, and also it is still hard to know the accuracy of the results without comparison to a more detailed model. One of the main caveats to the application of Brownian dynamics to biological ion channels is the use of Poisson's equation to estimate the forces encountered by permeant ions. The issue here is whether one can legitimately employ macroscopic electrostatics in regions that are not much larger than the diameters of the water molecules and ions. In the narrow constricted region of the channel, such as in the selectivity filter of the potassium channel, the representation of the channel contents as a continuous medium is a poor approximation. Nevertheless, the results derived from Brownian dynamics simulations accurately replicate a wide variety of experimental observations when a relatively high effective dielectric constant (usually 60) is assigned to the water-filled pore. The greatest limitations of molecular dynamics is the computational power required that limits the possible simulation times. While the calculation of free energy profiles provides useful information on ion permeation, it is not a substitute for a direct estimation of conductance from simulations. Thus, virtually no predictions derived from molecular dynamics simulations can be directly compared with experimental data. If no such comparisons can be made, there can only be a limited interaction between experimenters and theoreticians. With the current doubling of computer speeds every two years, this computational limitation will eventually be overcome. Then, the force fields employed in molecular dynamics simulations may need to be improved to include polarization effects, perhaps using ab initio molecular dynamics as a guide.All three theoretical approaches are useful in elucidating the mechanisms underlying selectivity and permeation of ions across biological nanotubes. For ion channels with large pore radii, such as mechano-sensitive channels, PNP theory can be fruitfully utilized. Also, if one is interested in simply obtaining order-of-magnitude estimates of conductances of various model channels, this simple theory will provide the answers with little computational cost. To study the mechanisms underlying the selectivity sequences of monovalent ions or to determine the precise conformational changes of the protein when a channel undergoes the transition from the closed to the open state, one has to rely on molecular dynamics simulations. Thus far, a combined use of molecular and Brownian dynamics has proved to be well suited for studying structure-function relationships in ion channels. Brownian dynamics enables the calculation of conductance properties while molecular dynamics can provide input and justification for the parameters of the stochastic simulations as well as explaining finer details such as size-based selectivity. Now and in the near future, as we attempt to understand membrane channels in terms of rigorous molecular physics, there will be an increasing interplay between experiment and theory, the former providing hints and clues for building and refining models and the latter making testable predictions.
References
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohne, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef CAS.
- G. Chang, R. H. Spencer, A. T. Lee, M. T. Barclay and D. C. Rees, Science, 1998, 282, 2220–2226 CrossRef CAS.
- R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS.
- R. Dutzler, E. B. Campbell and R. MacKinnon, Science, 2003, 300, 108–112 CrossRef CAS.
- R. B. Bass, P. Stropo, M. Baraclay and D. C. Reece, Science, 2002, 298, 1582–1587 CrossRef CAS.
- S. B. Long, E. B. Campbell and R. MacKinnon, Science, 2004, 309, 897–903.
- S. B. Long, E. B. Campbell and R. MacKinnon, Science, 2004, 309, 903–908.
- N. Unwin, J. Mol. Biol., 2005, 346, 967–989 CrossRef CAS.
- B. Hille, Ionic Channels of Excitable Membranes, Sinauer Associates, Sunderland, MA, 3rd edn., 2001 Search PubMed.
- M. B. Partenskii and P. C. Jordan, Q. Rev. Biophys., 1992, 25, 477–510 CrossRef CAS.
- R. S. Eisenberg, J. Membr. Biol., 1999, 171, 1–24 Search PubMed.
- B. Roux, S. Bernéche and W. Im, Biochemistry, 2000, 39, 13295–13306 CrossRef CAS.
- D. P. Tieleman, P. C. Biggin, G. R. Smith and M. S. P. Sansom, Q. Rev. Biophys., 2001, 34, 473–561 CAS.
- S. H. Chung and S. Kuyucak, Biochim. Biophys. Acta, 2002, 1565, 267–286 CAS.
- R. R. Eisenberg, J. Membr. Biol., 1996, 150, 1–25 Search PubMed.
- E. W. McCleskey, J. Gen. Physiol., 1999, 113, 765–772 CrossRef CAS.
- W. Nonner, D. P. Chen and B. Eisenberg, J. Gen. Physiol., 1999, 113, 773–782 CrossRef CAS.
- C. Miller, J. Gen. Physiol., 1999, 113, 783–787 CrossRef CAS.
- D. G. Levitt, J. Gen. Physiol., 1999, 113, 789–794 CrossRef CAS.
- M. G. Kurnikova, R. D. Coalson, P. Graf and A. Nitzan, Biophys. J., 1999, 76, 642–656 CrossRef CAS.
- B. Corry, S. Kuyucak and S. H. Chung, Biophys. J., 2000, 78, 2364–2381 CrossRef CAS.
- J. Bockris and A. K. N. Reddy, Modern Electrochemistry, 1970, Plenum, New York, vol. 1 Search PubMed.
- B. Honig and A. Nicholls, Science, 1995, 268, 1144–1149 CrossRef CAS.
- D. G. Levitt, Biophys. J., 1985, 48, 19–31 CrossRef CAS.
- P. C. Jordan, R. J. Bacquet, J. A. McCammon and P. Tran, Biophys. J., 1989, 55, 1041–1052 CrossRef CAS.
- M. Cai and P. C. Jordan, Biophys. J., 1990, 57, 883–891 CrossRef CAS.
- E. Mason and E. McDaniel, Transport Properties of Ions in Gases, John Wiley, New York, 1988 Search PubMed.
- T. F. Weiss, Cellular Biophysics, MIT Press, Cambridge, MA, 1996, vols. 1– 2 Search PubMed.
- D. P. Chen, J. Lear and R. S. Eisenberg, Biophys. J., 1997, 75, 97–116 Search PubMed.
- D. P. Chen, L. Xu, A. Tripathy, G. Meissner and R. S. Eisenberg, Biophys. J., 1999, 76, 1346–1366 CrossRef CAS.
- P. Graf, A. Nitzan, M. G. Kurnikova and R. D. Coalson, J. Chem. Phys. B, 2000, 104, 12324–12338 Search PubMed.
- Z. Schuss, B. Nadler and R. S. Eisenberg, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2001, 64, 0361161–14.
- R. D. Coalson, M. G. Kurnikova, P. Graf and A. Nizan, Biophys. J., 2002, 82, 340a.
- B. Corry, S. Kuyucak and S. H. Chung, Biophys. J., 2003, 84, 3594–3606 CrossRef CAS.
- B. Nadler, U. Hollerbach and R. S. Eisenberg, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2003, 68, 0219051–9.
- W. Im and B. Roux, J. Mol. Biol., 2002, 322, 851–869 CrossRef CAS.
- S. Yu. Noskov, W. Im and B. Roux, Biophys. J., 2004, 87, 2299–2309 CrossRef CAS.
- W. Nonner and B. Eisenberg, Biophys. J., 1998, 75, 1287–1305 CrossRef CAS.
- T. W. Allen, S. Kuyucak and S. H. Chung, J. Chem. Phys., 1999, 111, 7985–7999 CrossRef CAS.
- K. E. Cooper, E. Jakobsson and P. Wolynes, Prog. Biophys. Mol. Biol., 1985, 46, 51–96 CrossRef CAS.
- E. Jakobsson and W. W. Chiu, Biophys. J., 1987, 52, 33–45 CrossRef CAS.
- S. Bek and E. Jakobsson, Biophys. J., 1994, 66, 1028–1038 CrossRef CAS.
- S. H. Chung, M. Hoyles, T. Wallen and S. Kuyucak, Biophys. J., 1998, 75, 793–809 CrossRef CAS.
- S. H. Chung, T. W. Allen, M. Hoyles and S. Kuyucak, Biophys. J., 1999, 77, 2517–2533 CrossRef CAS.
- B. Corry, T. W. Allen, S. Kuyucak and S. H. Chung, Biophys. J., 2001, 80, 195–214 CrossRef CAS.
- S. Edwards, B. Corry, S. Kuyucak and S. H. Chung, Biophys. J., 2002, 83, 1348–1360 CrossRef CAS.
- S. Bernéche and B. Roux, Proc. Natl. Acad. Sci. USA, 2003, 100, 8644–8648 CrossRef CAS.
- S. Garofoli and P. C. Jordan, Biophys. J., 2003, 84, 2814–2830 CrossRef CAS.
- S. H. Chung, T. W. Allen and S. Kuyucak, Biophys. J., 2002, 83, 263–277 CrossRef CAS.
- B. Corry, M. O'Mara and S. H. Chung, Chem. Phys. Lett., 2004, 386, 233–238 CrossRef CAS.
- B. Corry, M. O'Mara and S. H. Chung, Biophys. J., 2004, 86, 846–860 CrossRef CAS.
- T. J. Jentsch, T. Friedrich, A. Schriever and H. Yamada, Pflügers Arch., 1999, 437, 783–795 Search PubMed.
- M. Maduke, C. Miller and J. A. Mindell, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 411–438 CrossRef CAS.
- C. Fahlke, Am. J. Renal Physiol., 2001, 280, F748–F758 Search PubMed.
- C. Miller, Philos. Trans. R. Soc. London, Ser. B, 1982, 299, 401–411 CrossRef CAS.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765–784 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 187–217 CrossRef CAS.
- J. Hermans, H. J. C. Berendsen, W. F. van Gunsteren and J. P. M. Postma, Biopolymers, 1984, 23, 1513–1518 CAS.
- L. Kalé, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, J. Comput. Phys., 1999, 151, 283–312 CrossRef.
- H. M. Chun, C. E. Padilla, D. N. Chin, M. Watanabe, V. I. Karlov, H. E. Alper, K. Soosaar, K. B. Blair, O. M. Becker, L. S. D. Caves, R. Nagle, D. N. Haney and B. L. Farmer, J. Comput. Chem., 2000, 21, 159–184 CrossRef CAS.
- P. S. Crozier, R. L. Rowley, N. B. Holladay, D. Hendersen and D. D. Busath, Phys. Rev. Lett., 2001, 86, 2467–2470 CrossRef CAS.
- M. Jensen, S. Park, E. Tajkhorshid and K. Schulten, Proc. Natl. Acad. Sci. USA, 2002, 99, 6731–6736 CrossRef CAS.
- V. L. Dorman and P. C. Jordan, Biophys. J., 2004, 86, 3529–3541 CrossRef CAS.
- B. Corry and S. H. Chung, Eur. Biophys. J., 2005, 34, 208–216 Search PubMed.
- W. Kohn, Rev. Mod. Phys., 1999, 71, 1253–1266 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS.
- L. Guidoni and P. Carloni, Biochim. Biophys. Acta, 2002, 1563, 1–7 CAS.
- D. P. Tieleman and H. J. C. Berendsen, Biophys. J., 1998, 74, 2786–2801 CrossRef CAS.
- M. A. Eriksson and B. Roux, Biophys. J., 2002, 83, 2595–2609 CrossRef CAS.
- D. P. Tieleman, L. R. Forrest, M. S. Sansom and J. J. Berendsen, Biochemistry, 1998, 37, 17554–17561 CrossRef CAS.
- H. I. Petrache, D. M. Zuckerman, J. N. Sachs, J. A. Killian, R. E. Koeppe and T. B. Woolf, Langmuir, 2002, 18, 1340–1351 CrossRef CAS.
- B. Roux and M. Karplus, J. Am. Chem. Soc., 1993, 115, 3250–3262 CrossRef CAS.
- T. W. Allen, T. Bastug, S. Kuyucak and S. H. Chung, Biophys. J., 2003, 84, 2159–2168 CrossRef CAS.
- J. H. Morais-Cabral, Y. Zhou and R. MacKinnon, Nature, 2001, 414, 37–42 CrossRef CAS.
- Y. Zhou, J. H. Morais-Cabral, A. Kaufman and R. MacKinnon, Nature, 2001, 414, 43–48 CrossRef CAS.
- J. Åqvist and V. B. Luzkov, Nature, 2000, 404, 881–884 CrossRef CAS.
- S. Bernéche and B. Roux, Biophys. J., 2000, 78, 2900–2917 CrossRef CAS.
- S. Bernéche and B. Roux, Nature, 2001, 414, 73–77 CrossRef CAS.
- T. W. Allen, A. Bliznyuk, A. P. Rendell, S. Kuyucak and S. H. Chung, J. Chem. Phys., 2000, 112, 8191–8204 CrossRef CAS.
- V. B. Luzhkov and J. Åqvist, Biochim. Biophys. Acta, 2000, 1481, 360–370 CAS.
- C. Domene and M. S. Sansom, Biophys. J., 2003, 85, 2787–2800 CrossRef CAS.
- Y. Zhou and R. MacKinnon, J. Mol. Biol., 2003, 333, 965–975 CrossRef CAS.
- S. Noskov, S. Bernéche and B. Roux, Nature, 2004, 431, 830–834 CrossRef CAS.
- A. A. Feenstra, B. Hess and J. J. C. Berendsen, J. Comput. Chem., 1999, 20, 786–798 CrossRef.
| This journal is © The Royal Society of Chemistry 2005 |