The water/oil interface: the emerging horizon for self-assembly of nanoparticles
Dayang
Wang
*,
Hongwei
Duan
and
Helmuth
Möhwald
Max Planck Institute of Colloids and Interfaces, Research Campus Golm, D-14424, Potsdam, Germany. E-mail: dayang.wang@mpikg-golm.mpg.de; Fax: 49 331 567 9202
First published on 26th October 2005
Abstract
This article highlights our recent achievement on directing nanoparticles to self-assemble at the water/oil interface. We demonstrate that the contact angle of 90° is prerequisite for nanoparticles to localize at the interface, which is determined by the terminal groups of the capping ligands. With this peculiar surface wettability, nanoparticles of different sizes and chemical composition may self-assemble into homogeneous or composite thin films at the water/oil interface. The interfacially active nanoparticles may be employed to stabilize water-in-oil or oil-in-water droplets, creating the microcapsules whose shells are composed of randomly close packed nanoparticles. The permeability of the resulting microcapsules is defined by the sizes of the nanoparticles used.
 Dayang Wang | Dayang Wang studied chemistry at Jilin University, Changchun, China. He received a PhD degree in polymer chemistry and physics in 1998 there. In 1999, he moved to the Max Planck Institute of Colloidal and Interfaces (MPIKG), Potsdam, Germany, as a postdoctoral fellow. In 2000, he received an Alexander von Humboldt fellowship in MPIKG. Since 2003 he is a group leader in MPIKG, His main research interests are multi-functional colloidal particles, self-assembly, and patterning. |
 Hongwei Duan | Hongwei Duan received his BS and MS (with Prof. Ming Jiang) from Fudan University in 1999 and 2002, respectively. He completed his PhD under Prof. Helmuth Möhwald at Max Planck institute of Colloids and Interfaces in 2005. His current research interests include nanocrystals, polymer–nanocrystal hybrid colloids and polymer assemblies. |
 Helmuth Möhwald | Helmuth Möhwald received his Diploma in Physics in 1971 in University of Göttingen, Germany, and his PhD degree in Physics in 1974 in Max Planck Institute of Biophysical Chemistry, Göttingen. In 1978, he finished his habilitation in University of Ulm, Germany. In 1981–1987, he was C3 professor in the Technology University of München, Germany. In 1987–1993, he held the Chair of Physical Chemistry (C4 professor) in the University of Mainz, Germany. Since 1993, he is the director and scientific member of the Max Planck Institute of Colloids and Interfaces, Potsdam, Germany. His main research interests include biomimetic systems, chemistry and physics in confined spaces, dynamics at interfaces, and supramolecular interactions. |
1. Introduction
The advent of nanotechnology creates a renaissance in colloid science, shedding new light on the study of colloidal particles. Once colloidal particles are scaled down to the nanometre range, one can manipulate their properties by their size and shape. The last decade has seen tremendous progress in the wet-chemical fabrication of nanoparticles (NPs).1 Up to date one is able to tailor various materials into monodisperse nanoparticles, according to their size, shape, and surface chemistry, in a controlled manner.2 The driving force of nanotechnology is the miniature manufacture of devices, inspired by Feynman in 1960s.3 Nonetheless, nano-devices are currently recognized not as simply miniaturized micro-devices because the cooperative interaction between their components gives rise to novel collective properties, which can be manipulated by the distance of the neighboring components, for instance.2,4 Nanotechnology therefore demands the capability to control the way the components are integrated. Recently, accordingly, enormous effort is devoted to the exploitation of new methodologies to organize NPs.5The straightforward chemical way to organize NPs is to non- or covalently link them via interaction of their capping ligands.5a,c But the sophisticated design of the capping ligands is prerequisite for achieving ordered structures and defining the interaction between NPs. Self-assembly paves a simple and general way to organize NPs.5b,d Owing to their exceedingly narrow size distribution, lipophilic NPs may crystallize into micron-sized colloidal crystals by controlled evaporation of solvents. Besides, the Langmiur–Blodgett technique has been extended to organize lipophilic NPs into two-dimensional (2D) arrays at the water/air interface.2a This allows fine manipulation of the distance between NPs by means of mechanical compression, thus realizing the definition of the interaction between the particles.2a,4
Besides the above techniques, most recently, our group and others have successfully demonstrated the potential to recruit the water/oil interface to organize NPs.6–8 Although it is less familiar to the materials science community, the study of the interfacial attachment of colloidal particles, especially micron-sized ones, de facto has a century of history, pioneered by Pickering in 1907.9,10 The energy (ΔE) of attaching particles at the water–oil interface can easily be calculated by the following equation:
| ΔE = −π(d/2)2γw/o(1 ± cosθ)2 | (1) |
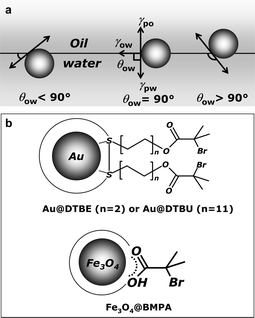 | ||
| Scheme 1 (a) Schematic representation of position of a particle at a water/oil interface for a contact angle with the interfaces less than 90° (left), equal to 90° (center), and larger than 90° (right). (b) Schematic illustration of the structures of Au@DTBE or Au@DTBU (upper) and γ-Fe2O3@BMPA NPs (lower). | ||
Most recently, Lin and coworkers observed that CdSe NPs, stabilized by tri-n-octylphosphine oxides, can self-assemble at the water/toluene interface and stabilize water droplets in toluene.7 Reincke and coworkers demonstrated that the addition of ethanol renders the contact angle of citrate-stabilized Au NPs close to 90°, thus pushing them to the water/hexane interface.8 We have established the generally-applicable protocol to render NPs interfacial active—contact angle of close to 90°—by an appropriate hydrophobic coating.6 With this success, we are capable of creating novel microcapsules, whose shells are composed of close packed NPs.6b Due to their large interface attachment energy, micron-sized colloidal particles have already been utilized to construct microcapsules.9 Dinsmore and coworkers name such capsule “colloidosomes” according to their analogy with liposomes.9c Herein we coin our NP-stabilized capsules as nanoparticlosomes—nanoparticle colloidosomes—with the intent of highlighting their nanoscale permeability defined by the sizes of NPs and their packing structures.
2. Directing nanoparticles to self-assemble at the water/oil interface
Our success of making NPs interface active stems from the current comprehensive understanding of surface wetting.11 The ligands used in our work have a 2-bromo-2-methyl-propionate terminal group, as shown in Scheme 1b. The carboxylic ester and bromide groups of this terminus are well-known to provide contact angles in the range of 80–110°.11 By means of ligand exchange, we succeeded in capping hydrophilic Au or Ag NPs with 2,2′-dithiobis[1-(2-bromo-2-methylpropionyloxy)ethane] (DTBE). After optimizing the ligand density, about 800 DTBE molecules per NP, the capping of DTBE does not deteriorate the colloidal stability of the resulting coated NPs in water. As observed in Fig. 1, once toluene was added into the aqueous solution of the resulting DTBE coated metal NPs, followed by a gentle shaking, all particles were trapped at the water/toluene interface, forming thin films with metallic luster. This bulk metallic reflection suggests the electronic coupling of the NPs, thus indicating close packing arrays of the NPs at the interfaces. After transfer onto solid substrates, e.g., glass slides, these thin films of metallic NPs exhibit a contract angle of 90° with the water/toluene interface. In contrast, the contact angle of the self-assembled films of original hydrophilic metallic NPs is about 60°. Transmission electron micrographs (TEM) demonstrate that metallic NPs self-assembled into a random close packed monolayer at the water/toluene interface (Fig. 2). Besides, nano-alloy thin films were also successfully formed by directing for instance both Au@DTBE and Ag@DTBE NPs to self-assemble at the water/toluene interface (Fig. 1c). The interfacial activity of NPs is demonstrated exclusively dependent on the 2-bromo-2-methyl-propionate terminus rather than the alkyl chain length of the ligands used; the alkyl chain can be as long as 11 carbons (Scheme 1b). Once using such a ligand, we have realized interfacial self-assembly of differently sized hydrophilic metallic NPs, ranging from 5 nm to 20 nm, suggesting a minor contribution of the particle sizes as compared to the contact angle. In contrast, the interfacial attachment of microparticles is mainly due to their giant dimension. | ||
| Fig. 1 Photographs of self-assemblies of 12 nm Au@DTBE NPs (a), 40 nm Ag@DTBE NPs (b) and their mixture with a molar ratio of 1∶1 (c) at the water/toluene interface in plastic eppendorf tubes. These tubes are tilted such that the colored area corresponds to the water/toluene interface. | ||
 | ||
| Fig. 2 TEM image of a monolayer of 12 nm Au@DTBE NPs, formed at the water/toluene interface. The inset is the high magnification TEM picture (the scale bar is 25 nm). | ||
In order to generalize the effect of the terminal group of 2-bromo-2-methyl-propionate, we extended our protocol to hydrophobic 4 nm, 5 nm and 8 nm Fe3O4 NPs, stabilized by oleic acid and oleylamine. After replacing their stabilizers via ligand exchange, 2-bromo-2-methylpropionic acid (BMPA) were capped on Fe3O4 NPs (Scheme 1b). As shown in Fig. 3, the terminal group of 2-bromo-2-methyl-propionate of BMPA renders Fe3O4 NPs interface-active; driving the capped NPs from the toluene bulk phase to the water/toluene interface and to self-assemble into a close-packed monolayer there. After transferring into glass slides, the BMPA capping gives Fe3O4 NPs a contact angle close to 90° with the water/toluene interface.
 | ||
| Fig. 3 Photograph (a) and TEM image (b) of a 8 nm Fe3O4@BMPA NP monolayer, formed at the water/toluene interface. | ||
3. Nanoparticlosomes: microcapsules with nanoscale defined permeability
The interface-active NPs can be expected to behave in a similar fashion as surfactants do. This inspired us to use the NPs obtained above as emulsion stabilizers. After adding water to the toluene suspension of Fe3O4@BMPA NPs, we observed that vigorous shaking led to formation of stable water droplets in toluene, whose shells were composed of close-packed NPs, determined by TEM. The resulting NP-stabilized droplets provide novel microcapsules, nanoparticlosomes. To improve the structural stability of nanoparticlosomes against the environmental change, we gelated the water cores by introducing agarose inside. Besides improvement of the structural stability, as shown in Fig. 4a, the gelated cores allow transfer and re-dispersion of the nanoparticlosomes into water. Due to close-packed Fe3O4@BMPA NPs within their shells, the nanoparticlcomes obtained are magnetic, allowing manipulation by external magnetic fields, which should be of great importance for microencapsulation applications.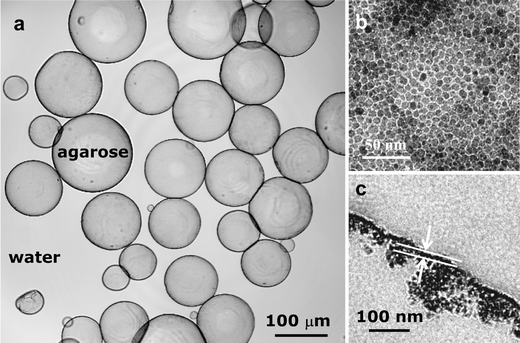 | ||
| Fig. 4 Confocal microscopy image of 8.0 nm Fe3O4 nanoparticlosomes containing agarose hydrogel cores (a) and TEM images of their shells (b) and their ultrathin sections (c). | ||
The permeability of nanoparticlosomes is obviously determined by the interstitial area between close-packed NPs in the shells. As observed by TEM, NPs are randomly close packed in the nanoparticlosome shells, so hexagonal, cubic and pentagonal packing arrays coexist. Based on simple geometry analysis of the interstitial sizes of these three packing arrays, as shown in Fig. 5a, the pentagonal arrangement gives the largest interstice, 0.7 of the NP size, which is expected as the cutoff for permeation of materials through the nanoparticlosome wall. In order to investigate the permeability of Fe3O4 nanoparticlosomes, monodisperse water-soluble CdTe NPs were recruited as size probes due to their well-defined size and geometry.12 As shown in Fig. 5, 5.0 nm Fe3O4 nanoparticlosomes selectively release 2.8 nm CdTe NPs (green emission at 540 nm) and keep 4.0 nm CdTe NPs inside (red emission at 650 nm) during storage in water because their cutoff is 3.5 nm, in between small and large CdTe NPs. The large cutoff of 8.0 nm Fe3O4 nanoparticlosomes (5.6 nm) allows complete release of 4.0 nm CdTe NPs. This demonstrates that the nanoscale permeability of Fe3O4 nanoparticlosomes is well defined by the sizes of the Fe3O4 NPs used. This also indicates that the NP shell has only few defects.
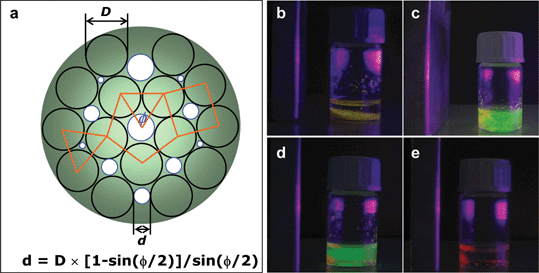 | ||
| Fig. 5 (a) Schematic illustration of close-packing arrays of NPs on nanoparticlosomes and the equation for calculating their interstitial dimension. (b) Fluorescence photograph of 5.0 nm Fe3O4 nanoparticlosomes with agarose gel cores formed in toluene. In their gel cores both 2.8 nm and 4.0 nm CdTe NPs are loaded. Fluorescence photographs of these nanoparticlosomes freshly transferred into water (c), stored in water for 10 min (d) and after washing away the released CdTe NPs by water (d). | ||
4. Conclusions and outlook
We have demonstrated the potential of recruiting the water/oil interface as a platform for self-assembly of NPs. By capping NPs with ligands with a 2-bromo-2-methyl-propionate terminus, we succeeded in directing self-assembly of hydrophilic and hydrophobic NPs of different sizes and chemical compositions at the water/oil interface, forming homogeneous and heterogeneous thin films. The interface activity of NPs is dominated by the surface wettability of NPs—the contact angle with the water/oil interface—rather than their sizes. The interfacial self-assembly of NPs can be realized only when their contact angles are close to 90°, which is determined by the terminal groups of their capping ligands. The interface-active NPs can be employed to stabilize water-in-oil droplets, nanoparticlosomes whose shells are composed of close packed NPs. The nanoparticlosome cutoff is defined by the interstitial area between close-packed NPs, which provides an immense flexibility to tune the permeability of microcapsules on the nanometer scale.The interfacial self-assembly of NPs of size less than 10 nm are starting to draw attention just since last year. There are plenty of important issues still required to be addressed. The contact angle of NPs at the water/oil interface obtained so far is macroscopic, which should embody a collective effect of NPs behind. To determine the contact angle of one individual NP should be a difficult but inevitable work in future. Another important task will be to extend the knowledge of interfacial attachment of sub- or microparticles to control the distance of neighboring NPs assembled at the interface, thus realizing manipulation of the NP cooperation interaction. Russell and coworkers demonstrated the potential to chemically cross-link the nanoparticles attached at the water/oil interface by use of cross-linkable ligands, obtaining free-standing thin films of the particles.7b This strategy should be extended to lock the particles of nanoparticlosomes to achieve a robust structural integrity. The incorporation of different functional nano-objects into nanoparticlosomes will create new micron-size compartments, in which electronic, optical, and magnetic cooperative properties may be expected. This will be a striking topic parallel to the microencapsulation applications.
Different from the solid substrate, most significantly, the water/oil interface is fluid, so the self-assembly of NPs at the interface may be expect dynamic. Russell and coworkers observed the spontaneous tendency to replace small CdSe NPs by big ones at the water/oil interface.7a This dynamic character is reminiscent of the self-assembly process occurring in nature, usually associated with self-healing.13 In this sense, we can not say that the self-assembly of NPs at the fluid water/oil interface is no more than an extension of that on solid surfaces. It should hold immense promise in generating biomimetic nanodevices.
References
- (a) H. Weller, Angew. Chem., Int. Ed., 1993, 32, 41 CrossRef; (b) A. P. Alivisatos, Science, 1996, 271, 933 CrossRef CAS.
- (a) C. Collier, T. Vossmeyer and J. Heath, Annu. Rev. Phys. Chem., 1998, 49, 371 CrossRef CAS; (b) C. Murray, C. Kagan and M. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545 Search PubMed; (c) H. Weller, Philos. Trans. R. Soc. London, Ser. A, 2003, 361, 229 CrossRef CAS.
- R. Feynman, Sci. Eng., 1960, 23, 22 Search PubMed.
- F. Remacle and R. Levine, ChemPhysChem, 2001, 2, 20 CrossRef CAS.
- (a) E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042 CrossRef CAS; (b) F. Redl, K. Cho, C. Murray and S. O'Brien, Nature, 2003, 423, 968 CrossRef CAS; (c) R. Shenhar and V. Rotello, Acc. Chem. Res., 2003, 36, 549 CrossRef CAS; (d) M. Brust and C. J. kiely, Colloids Surf., A, 2002, 202, 175 CrossRef CAS.
- (a) H. Duan, D. Wang, D. Kurth and H. Möhwald, Angew. Chem., Int. Ed., 2005, 43, 5639; (b) H. Duan, D. Wang, N. Sobal, M. Giersig, D. Kurth and H. Möhwald, Nano Lett., 2005, 5, 949 CrossRef CAS.
- (a) Y. Lin, H. Skaff, T. Emrick, A. Dinsmore and T. Russell, Science, 2003, 299, 226 CrossRef CAS; (b) Y. Lin, H. Skaff, A. Boker, A. Dinsmore, T. Emrick and T. Russell, J. Am. Chem. Soc., 2003, 125, 12690 CrossRef CAS.
- F. Reincke, S. Hickey, W. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458 CrossRef CAS.
- (a) S. Pickering, J. Chem. Soc., 1907, 91, 2001 RSC; (b) O. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374 CrossRef CAS; (c) A. Dinsmore, M. Hsu, M. Nikolaides, M. Marquez, A. Bausch and D. Weitz, Science, 2002, 298, 1006 CrossRef CAS.
- (a) B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21 Search PubMed; (b) R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100–102, 503 CrossRef CAS.
- Contact Angle, Wettability and Adhesion ed. K. L. Mittal, VSP BV, Utrecht, 1993 Search PubMed.
- N. Gaponik, D. Talapin, A. Rogach, K. Hoppe, E. Shevchenko, A. Komowski, A. Eychmüller and H. Weller, J. Phys. Chem. B, 2002, 106, 7177 CrossRef CAS.
- (a) G. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. USA, 2002, 99, 4769 CrossRef CAS; (b) M. Boncheva, D. Gracias, H. Jacobs and G. Whitesides, Proc. Natl. Acad. Sci. USA, 2002, 99, 4937 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |