DOI:
10.1039/B510100J
(Paper)
Org. Biomol. Chem., 2005,
3, 3514-3523
Heterocyclic ring scaffolds as small-molecule cholesterol absorption inhibitors
Received 15th July 2005, Accepted 1st August 2005
First published on 24th August 2005
Abstract
Enantio- and diastereoselective syntheses of a substituted oxazolidinone, isoxazoline and pyrazoline as β-lactam surrogates are described. The substituted heterocycles were designed to incorporate side chains closely resembling those found in the β-lactam cholesterol absorption inhibitor ezetimibe (1). Additionally, the in vitro inhibitory efficacy of the novel compounds as cholesterol absorption inhibitors is reported using a brush border membrane vesicle assay.
Introduction
Enantio- and diastereoselective methods for the synthesis of substituted non-aromatic heterocycles are of prime importance. When incorporating multiple derivatization sites such methods facilitate diversity oriented synthesis. A recent example is the drug ezetimibe (1, Fig. 1),1 which inhibits cholesterol absorption and contains a non-aromatic heterocycle in the form of a β-lactam ring. In the development of ezetimibe, the β-lactam was proposed to be essential for inhibitory activity,2,3 with the corresponding ring-opened β-amino acid derivative being completely inactive.3 In the course of an ongoing project aimed at the characterization and further study of intestinal cholesterol uptake, we became interested in the design of structurally well-defined, non-aromatic heterocycles which can mimic the β-lactam scaffold. The β-lactam ring is a rigid, almost planar heterocycle that defines out of plane vectors from a central core. Given our objectives of identifying structural congeners of β-lactams, we focused on generating structures in which the geometrical alignment of the three exit vectors of the substituents in the β-lactam are conserved (Fig. 1). Importantly, we additionally wished to identify β-lactam mimics that would not be prone to undergo hydrolysis as seen for β-lactams in general. In this report, we document the enantio- and diastereoselective syntheses of three β-lactam surrogates, namely an oxazolidinone, an isoxazoline, and a pyrazoline, which do not suffer from hydrolytic instability and display a set of exit vectors closely resembling those found in the β-lactam scaffold. We furthermore report their activities as cholesterol absorption inhibitors using our recently developed brush border membrane vesicle assay.4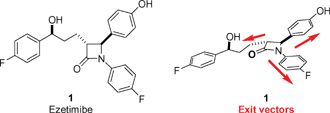 |
| | Fig. 1 Ezetimibe and the exit vectors of the β-lactam core. | |
Results and discussion
The oxazolidinone scaffold 2 has previously been suggested to serve as a structural mimic of the β-lactam of ezetimibe (1).2 Our ab initio geometry optimizations5
(Fig. 2) additionally suggested that the isoxazoline 3 and the pyrazoline 4 position three out of plane exit vectors in a manner that corresponds well to the β-lactam ring of ezetimibe. The excellent overlaps of the substituents are illustrated by superposition of each of the three heterocycles 2–4 with the β-lactam core found in ezetimibe. In order to focus on the exit vectors from the heterocyclic core, the flexible hydroxypropyl side chain in ezetimibe, which is not expected to strongly favor any single conformation, was replaced by a methyl group in the calculations.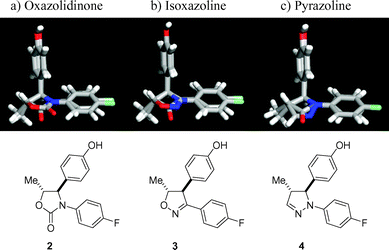 |
| | Fig. 2 Geometric overlap of oxazolidinone 2
(a), isoxazoline 3
(b), and pyrazoline 4
(c) with the β-lactam core structure of ezetimibe (1). | |
The synthesis of the desired oxazolidinone 16 commenced with a Staudinger cycloaddition of imine 66 and the ketene derived from acid 57
(Scheme 1). The reaction proceeded in 75% yield (cis∶trans
= 95∶5) to give a mixture of cis-diastereomers 7 and 8
(dr = 2∶3 as determined by 1H NMR spectroscopy).8 These were separable by silica gel chromatography and afforded 8 as a single isomer. Acid-mediated cleavage of the ketal furnished α-hydroxy-β-lactam 9 in enantiomerically pure form and 51% yield. Although yield and diastereoselectivity were modest, the ready availability of the inexpensive starting materials as well as the straightforward and scalable reaction protocol were decisive in our synthetic plan. Cleavage of the β-lactam under alkaline conditions delivered amino alcohol 10 in 92% yield, which was converted to oxazolidinone 11 in 89% yield using triphosgene.
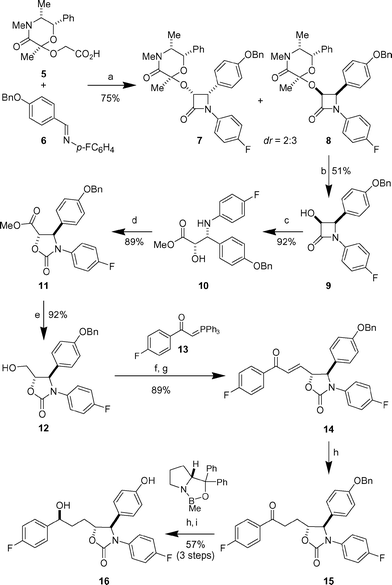 |
| | Scheme 1 Reagents and conditions: a) triphosgene, Et3N, CH2Cl2, 0 °C to 23 °C. b) CSA, THF–H2O, reflux. c) NaOMe, MeOH. d) Triphosgene, iPr2NEt, DMAP, CH2Cl2, −78 °C to 23 °C. e) NaBH4, EtOH. f)
(COCl)2, DMSO, Et3N, CH2Cl2, −78 °C. g)
13. h) H2, Pd/C, EtOH. i)
(R)-CBS catalyst, BH3·SMe2, CH2Cl2, −20 °C to 0 °C. | |
The methyl ester in 11 served as an appropriate handle to attach the 3′-aryl-3′-hydroxypropyl side chain found in ezetimibe (1). Initially, we envisaged reduction of the ester to the aldehyde and subsequent introduction of the side chain by an aldol condensation reaction. However, all attempts to isolate the aldehyde derived from 11 failed. Reduction of ester 11 or the corresponding Weinreb amide9 with DIBAL-H resulted only in decomposition products, attributed to the presumed instability of the product aldehyde. In 1985 Ireland documented the manipulation of unstable aldehydes through in situ Swern oxidation of the corresponding alcohols and subsequent Wittig reaction.10 Consequently, ester 11 was reduced to the corresponding alcohol 12 by treatment with NaBH4 in ethanol at 23 °C. Subsequent Swern oxidation11 at −78 °C for 5 min furnished the intermediate aldehyde, which was subjected to in situ reaction with stabilized phosphorous ylide 13.12 The unusual low reaction temperature for this Wittig reaction (< −40 °C)13 underscores the high electrophilicity of the intermediate aldehyde. Through this procedure trans-enone 14 could be conveniently prepared in 89% yield. Hydrogenation of the conjugated double bond afforded ketone 15, which was diastereoselectively reduced using the (R)-CBS catalyst14
(dr > 99∶1 according to 19F-NMR). Finally hydrogenolysis of the benzyl ether furnished the targeted oxazolidinone 16 in 57% yield over three steps. The above route thus furnished a rapid and straightforward access to the oxazolidinone scaffold with the desired side chains.
The stereoselective synthesis of the desired isoxazoline 26 was then pursued (Scheme 2) through a diastereoselective dipolar cycloaddition reaction of nitrile oxides and optically active allylic alcohols, which provides access to chiral optically active isoxazolines.15 However, at the outset of our synthesis it was far from clear whether allylic alcohols wherein the olefin is conjugated to a functionalized aromatic ring could be used as dipolarophiles in this cycloaddition, since the vast majority of the described magnesium-mediated cycloadditions have been conducted with non-conjugated allylic alcohols. In order to test the strategy, the cinnamyl aldehyde 18 was prepared from commercially available 4-hydroxycinnamic acid (17) in 76% yield over 4 steps. Subsequent Brown allylation using (+)-β-allyldiisopinocampheyl borane (19)16 afforded homoallylic alcohol 20 in 77% yield and 93% ee as determined by chiral HPLC. Cycloaddition of this allylic alcohol with the nitrile oxide derived from 22 delivered the product isoxazoline largely derived from cycloaddition to the terminal double bond. We speculated that this undesired regioselectivity could be circumvented by conversion of the terminal double bond to a corresponding disubstituted olefin, thereby reducing the rate of the cycloaddition reaction at this site. In this regard, 20 was subjected to Heck arylation17 to give 21 in 51% yield. In initial investigations of the cycloaddition reaction we noted a major by-product resulting from dimerization of the nitrile oxide. In order to minimize the formation of this by-product, the reaction was conducted at low concentration of the nitrile oxide in the reaction mixture by slow addition of the hydroximinoyl chloride (generated from oxime oxidation with tert-butyl hypochloride) to the dipolarophile over a period of 30 h. Thus, cycloaddition of allylic alcohol 21 with the nitrile oxide derived from 22 proceeded completely regio- and diastereoselectively (dr > 99∶1 as determined by NMR) to give isoxazoline 23 in 36% yield with 46% recovered starting material. Installation of the desired substituents commenced by conversion of 23 to aldehyde 24. In analogy to the synthesis of the oxazolidinone 16 described earlier, aldehyde 24 was allowed to react with phosphorous ylide 1312 to afford enone 25
(30% over 4 steps). Hydrogenation of the double bond followed by diastereoselective ketone reduction (dr > 99∶1 as determined by 1H NMR) using the (R)-CBS catalyst14 afforded the desired isoxazoline 26 in 75% yield over two steps.
 |
| | Scheme 2 Reagents and conditions: a) SOCl2, MeOH. b) MsCl, Et3N, THF. c) DIBAL-H, CH2Cl2, 0 °C. d) MnO2, CH2Cl2. e)
19, Et2O, −78 °C. f) C6H5I, Pd(OAc)2, PPh3, Et3N, MeCN. g)
22, tBuOCl, iPrOH, EtMgBr, CH2Cl2. h) MsCl, pyr, CH2Cl2. i) DBU, CH2Cl2, reflux. j) K2OsO4·2H2O, NaIO4, THF–H2O. k)
13. l) H2, Pd/C, MeOH. m)
(R)-CBS catalyst, BH3·SMe2, CH2Cl2, −20 °C to 0 °C. | |
In the approach to the desired substituted pyrazolines, a diastereoselective 1,3-dipolar cycloaddition of TMS-diazomethane18 was utilized to construct the heterocyclic core (Scheme 3). The synthesis commenced with a Zn-mediated enantioselective alkyne addition19 to p-fluorobenzaldehyde 27 to give propargylic alcohol 28 in 75% yield (96% ee as determined by chiral HPLC). The yields were higher when the reaction was conducted slightly below room temperature (8–13 °C). Subsequent silylation was immediately followed by sequential reduction of the triple bond and removal of the benzyl group to give alcohol 29 in 80% overall yield. This was necessary because the intermediary propargylic silyl ether proved unstable and difficult to isolate. The propensity of the benzylic and propargylic C–OSi bond to undergo hydrogenolytic cleavage necessitated stepwise hydrogenation of the alkyne prior to removal of the benzyl group. Subsequent Dess–Martin oxidation20
(80% yield) and Horner–Wadsworth–Emmons olefination using the camphorsultam derived phosphonate 3021,22 and LiCl–DBU23 afforded the (E)-olefin 31 in 71% yield. The pyrazoline heterocyclic core was generated using a diastereoselective 1,3-dipolar cycloaddition of TMS-diazomethane,18 which furnished the desired pyrazoline 32 in 94% combined yield (89∶11 dr based on the yields of the isolated diastereomers). The diastereomeric products were readily separated by chromatography on silica gel to afford diastereomerically pure 32.
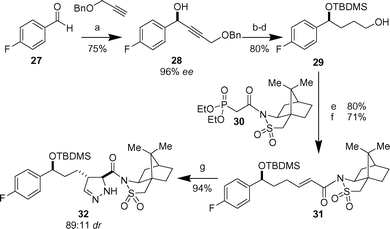 |
| | Scheme 3 Reagents and conditions: a) Zn(OTf)2, (+)-N-methylephedrine, Et3N, toluene, 8–13 °C. b) TBDMSCl, imidazole, DMF. c) H2, Pd/C, Na2CO3, EtOH. d) H2, Pd/C, EtOH. e) Dess–Martin periodinane, CH2Cl2. f)
30, DBU, LiCl, MeCN. g) TMSCHN2, toluene–hexane; then TFA, CH2Cl2. | |
Typical conditions employed in Pd-mediated N-arylations24 proved incompatible with the camphorsultam imide. However, a Cu-mediated N-arylation proceeded successfully employing either a boronic acid25 or a triarylbismuth derivative26
(Scheme 4). Optimal yields (63%) were obtained using the organobismuth reagent, (p-FC6H4)3Bi, which was readily obtained by reaction of p-fluorophenylmagnesium bromide with BiCl3. With this intermediate 33 in hand, we envisioned a rapid synthesis of various analogues by conversion of the carboxylic acid derivative into an oxazole as a substitute for the phenol substituent of ezetimibe. In this regard, substitution of the camphorsultam auxiliary with glycine catalyzed by KCN27
(75%) and dehydration using the water-soluble DCC analogue 3528 afforded the desired, but rather unstable, oxazolone 36 in 39% isolated yield. Generation of the oxazole 37 was effected by benzene sulfonate ester formation and desilylation in 40% and 39% yields, respectively.
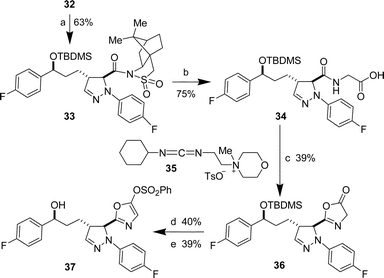 |
| | Scheme 4 Reagents and conditions: a)
(p-FC6H4)3Bi, Cu(OAc)2, Et3N, CH2Cl2. b) Glycine, KCN, MeOH, 50 °C. c)
35, CH2Cl2, reflux. d) PhSO2Cl, Et3N, CH2Cl2. e) HF·pyr, pyr, THF. | |
As an alternative pyrazoline derivatization, the chiral camphorsultam auxiliary of 33 was reductively removed (LiAlH4, 76% yield) to give a primary alcohol which, following tosylation (83% yield), was subjected to nucleophilic displacement of the sulfonate by hydroquinone in 86% yield (Scheme 5). Subsequent desilylation afforded the pyrazoline 38 in 99% yield featuring an oxymethylene linker between the pyrazoline and the aromatic ring substituent.
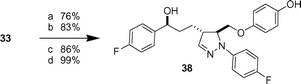 |
| | Scheme 5 Reagents and conditions: a) LiAlH4, THF, −78 °C. b) TsCl, DMAP. Et3N, CH2Cl2. c) Hydroquinone, Cs2CO3, DMF, 80 °C. d) HF·pyr, pyr, THF. | |
The heterocyclic compounds 16, 26, and 37–38 were subsequently evaluated for inhibition of intestinal cholesterol uptake using our recent brush border membrane vesicle in vitro assay (Fig. 3).4 We were pleased to observe that oxazolidinone 16 showed a similar in vitro activity (19% inhibition) to ezetimibe (1)
(16% inhibition). Despite previous promising in vitro results for a wide range of sulfonate ester phenolic derivatives of ezetimibe,4b the sulfonate ester substituted isoxazoline (26) and pyrazoline (37) did not show any activity as cholesterol absorption inhibitors. The remaining pyrazoline 38 was likewise void of inhibitory activity. This attests that small changes of the heterocyclic core can result in marked differences as cholesterol absorption inhibitors even though the geometric deviations of the exit vectors are only subtle (Fig. 2).
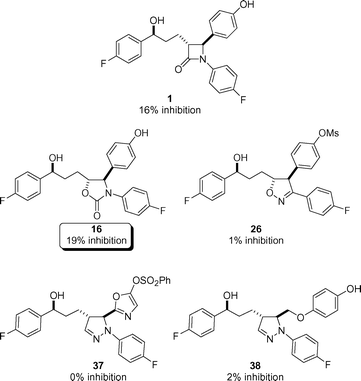 |
| | Fig. 3 Percentage inhibition in the brush border membrane vesicle assay using rabbit small intestine at nominal concentrations of 6 µM.4 The average standard deviations were ±3% inhibition. | |
Conclusions
We have documented the enantio- and diastereoselective syntheses of three β-lactam surrogates with side chains resembling those found in the cholesterol absorption inhibitor ezetimibe (1). In the course of these investigations we expanded the substrate scope of the highly diastereoselective hydroxyl-directed nitrile oxide cycloadditions. The pyrazoline synthesis featured a diastereoselective dipolar cycloaddition of TMS-diazomethane and a copper-mediated N-arylation using an organobismuth reagent as the key steps. When evaluated in the brush border membrane vesicle assay, the oxazolidinone 16 showed similar activity as ezetimibe (1) as a cholesterol absorption inhibitor. This promising result suggests that an oxazolidinone ring scaffold could effectively replace the β-lactam of ezetimibe. Synthesis of additional analogues and their biological evaluation are underway and will be reported in due course.Experimental
General experimental details
Reactions in anhydrous solvents were all performed using oven dried glassware under an atmosphere of argon. Reagent grade solvents were all purchased from chemical companies and used without prior purification. Anhydrous THF, ether, toluene, CH3CN and CH2Cl2 were dried and purified through activated alumina columns as described.29 Diisopropylamine, triethylamine and pyridine were distilled from KOH. For chromatographic purification, technical grade solvents were distilled prior to use. TLC was performed using Machery-Nagel Alugram Sil G/UV254 or Merck 0.25 mm silica gel 60 F254 TLC glass plates. Visualization of the developed chromatogram was performed by UV fluorescence at 254 nm and oxidative stain by either ceric ammonium molybdate solution, KMnO4–NaHCO3 water solution, phosphomolybdic acid or H2SO4–MeOH. Chromatographic purification of products was accomplished by dry column vacuum chromatography30 on either Merck silica gel 60 (15–40 µm) or Brunschwig silica 18–32, 60 Å
(18–32 µM) or by flash chromatography on silica gel 60 (230–400 mesh, 0.04–0.063 mm) from Merck at rt and 0.3–0.5 mbar air pressure. Concentration under reduced pressure was performed by rotary evaporation at 40 °C and the purified compounds were subsequently dried under high vacuum (<0.5 Torr). NMR spectra were recorded on a Varian Mercury 300 MHz apparatus operating at 300 MHz, 75 MHz and 282 MHz for 1H, 13C/DEPT and 19F, respectively, and chemical shifts (δ) were referenced to the internal solvent signals for 1H and 13C. Multiplicities are reported as follows: 1H: s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet; 13C: C, CH, CH2, CH3
(determined by DEPT); coupling constants are reported in Hz. Melting points were measured on a Büchi 510 apparatus in open capillaries and all melting points are uncorrected. IR-Spectra were recorded in CHCl3 on a Perkin Elmer Spectrum RX I FT-IR apparatus (thin films on NaCl plates) and are reported as absorption maxima in cm−1. Optical rotations are reported in 10−1deg cm2 g−1. Elemental analysis was performed by the Mikroelementaranalytisches Laboratorium at the ETH, Zürich. High resolution matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) and electrospray ionization (ESI-MS) were performed by the mass spectrometry service of the LOC at the ETH, Zürich.(2S,5R,6S)-2-[(1S,2R)-2-(4-Benzyloxyphenyl)-3-(4-fluorophenyl)-4-oxocyclobutoxy]-2,4,5-trimethyl-6-phenylmorpholin-3-one (8). To a solution of acid 57
(30.0 g, 102 mmol, 1.11 eq.) in CH2Cl2
(600 ml) was added triethylamine (64.0 ml, 461 mmol, 5.00 eq.) followed by imine 66
(28.1 g, 92.1 mmol, 1.00 eq.). The solution was cooled to −20 °C and triphosgene (16.4 g, 55.8 mmol, 0.600 eq.) was added in 50 ml CH2Cl2 over a period of 20 min. The solution was warmed to 23 °C over a period of 8 h and stirred for an additional 10 h at this temperature. The solution was poured onto 600 ml ice water and 200 ml CH2Cl2. The aqueous phase was extracted with CH2Cl2
(3 × 100 ml). The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel eluting with hexane–EtOAc (3∶2 to 1∶2 gradient) and then chromatography on silica gel eluting with EtOAc–CH2Cl2
(7∶1 to 3∶1 gradient) to afford β-lactam 8 as a colorless solid in 45% yield along with 30% yield of the undesired diastereomer 7. Mp: 132 °C. Rf
= 0.38 [hexane–EtOAc 1∶1 (v/v)]. α30.5D
=
+77°, (c 1.075 in CHCl3). 1H-NMR (300 MHz, CDCl3): δ 7.46–7.07 (16 H, m), 6.92–6.84 (2 H, m), 5.34 (1 H, d, J
= 5.3 Hz), 5.06 (2 H, s), 4.95 (1 H, d, J
= 5.3 Hz), 4.60 (1 H, d, J
= 2.5 Hz), 3.23–3.14 (1 H, m), 2.90 (3 H, s), 1.70 (3 H, s), 0.83 (3 H, d, J
= 6.2 Hz). 13C-NMR (75 MHz, CDCl3): δ 165.4, 165.0, 159.3 (d, J
= 244 Hz), 159.1, 137.1 (d, J
= 5 Hz), 133.7, 129.9, 128.9, 128.6, 128.3, 128.0, 127.7, 125.7, 119.0 (d, J
= 8 Hz), 116.0 (d, J
= 23 Hz), 115.1, 100.1, 76.9, 71.2, 70.1, 62.2, 59.0, 33.8, 23.6, 12.4. IR (thin film): 2938, 1756, 1667, 1612, 1511, 1382, 1223, 1177, 1112, 1092, 834, 734. HRMS (EI): found, 580.2369. C35H33FN2O5+ requires 580.2374. (3S,4R)-4-(4-Benzyloxyphenyl)-1-(4-fluorophenyl)-3-hydroxyazetidin-2-one (9). To a solution of ketal 8
(17.0 g, 29.0 mmol, 1.00 eq.) in THF (242 ml) and water (48 ml) was added p-toluenesulfonic acid monohydrate (55.7 g, 293 mmol, 10.0 eq.). The solution was heated to reflux for 5 h. The solution was concentrated to an approximate volume of 60 ml and then poured onto EtOAc (150 ml) and water (250 ml). The aqueous phase was extracted with EtOAc (4 × 100 ml). The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (3∶2 to 2∶3 gradient), to afford β-lactam 9 as a colorless solid in 51% yield. Mp: 168 °C. Rf
= 0.26 [hexane–EtOAc 3∶2 (v/v)]. α29.5D
=
−129°, (c 1.22 in acetone). 1H-NMR (300 MHz, acetone-d6): δ 7.50–7.47 (2 H, m), 7.42–7.29 (5 H, m), 7.10–7.01 (4 H, m), 5.33 (1 H, d, J
= 5.3 Hz), 5.27 (1 H, dd, J
= 7.2 Hz, 5.3 Hz), 5.11 (2 H, s), 5.07 (1 H, d, J
= 7.2 Hz). 13C-NMR (75 MHz, acetone-d6): δ 166.5, 159.2, 159.0 (d, J
= 241 Hz), 137.7, 134.7, 129.6, 128.6, 128.0, 127.8, 118.9 (d, J
= 8 Hz), 115.8 (d, J
= 23 Hz), 114.8, 78.0, 69.8, 62.3. IR (thin film): 3120, 1756, 1667, 1612, 1511, 1382, 1223, 1177, 1112, 1092, 834, 734. HRMS (EI): found, 363.1268. C22H18FNO3+ requires 363.1271. Anal.: found, C, 77.73; H, 5.20; N, 3.91. C22H18FNO3 requires C, 72.72; H, 4.99; N, 3.85%.
(2S,3R)-3-(4-Benzyloxyphenyl)-3-(4-fluorophenylamino)-2-hydroxypropionic acid methyl ester (10). To a suspension of β-lactam 9
(2.00 g, 5.50 mmol, 1.00 eq.) in methanol (55.0 ml) was added sodium methoxide (1.49 g, 27.5 mmol, 5.00 eq.). The suspension was stirred at 23 °C for 2 h. To the forming solution was added NH4Cl(s) and the suspension was concentrated in vacuo. To the solid was added EtOAc (50 ml) and water (50 ml). The aqueous phase was extracted with EtOAc (3 × 20 ml). The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (3∶2 to 1∶1 gradient), to afford amino alcohol 10 as a colorless solid in 89% yield. Mp: 103 °C. Rf
= 0.45 [hexane–EtOAc 3∶2 (v/v)]. α25.3D
=
+13.9°, (c 1.10 in CH2Cl2). 1H-NMR (300 MHz, CDCl3): δ 7.44–7.24 (4 H, m), 6.97–6.91 (2 H, m), 6.84–6.76 (2 H, m), 6.53–6.46 (2 H, m), 5.02 (2 H, s), 4.81 (1 H, s), 4.60 (1 H, s), 4.46 (1 H, m), 3.79 (3 H, s), 3.07 (1 H, d, J
= 3.7 Hz). 13C-NMR (75 MHz, CDCl3): δ 158.2, 155.8 (d, J
= 233 Hz), 142.5, 136.8, 131.0, 128.5, 127.9, 127.9, 127.4, 155.5 (d, J
= 22 Hz), 114.9, 114.8, 74.6, 70.0, 59.1, 53.1, 114.8, 78.0, 69.8, 62.3. IR (thin film): 3390, 1737, 1610, 1510, 1221, 1113, 823. MS (EI): 306.1748 (2.54%), 186.2356 (18.8%), 91.0908 (100%). Anal.: found, C, 69.88; H, 5.78; N, 3.54. C23H22FNO4 requires C, 69.86; H, 5.61; N, 3.54%.
(4R,5S)-4-(4-Benzyloxyphenyl)-3-(4-fluorophenyl)-2-oxooxazolidine-5-carboxylic acid methyl ester (11). To a solution of amino alcohol 10
(1.92 g, 4.86 mmol, 1.00 eq.) in CH2Cl2
(24.0 ml) was added diisopropylethylamine (2.54 ml, 14.6 mmol, 3.00 eq.) and 4-N,N-dimethylaminopyridine (59.0 mg, 0.486 mmol, 0.10 eq.). The solution was cooled to −78 °C and triphosgene (1.44 g, 4.86 mmol, 1.00 eq.) in CH2Cl2
(4.0 ml) was added over a period of 5 min. The solution was warmed to 23 °C over 8 h and stirred at this temperature for an additional 5 h. To this solution was added water (50 ml) and concentrated aqueous ammonium hydroxide solution (3 ml). The aqueous phase was extracted with CH2Cl2
(3 × 20 ml). The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (2∶1 to 1∶1 gradient), to afford methyl ester 11 as a colourless solid in 82% yield. Mp: 118 °C. Rf
= 0.54 [hexane–EtOAc 3∶2 (v/v)]. α29.3D
=
+18°, (c 1.10 in CHCl3). 1H-NMR (300 MHz, CDCl3): δ 7.40–7.32 (7 H, m), 7.29–7.22 (2 H, m), 6.98–6.93 (4 H, m), 5.33 (1 H, d, J
= 4.4 Hz), 5.03 (2 H, s), 4.73 (1 H, d, J
= 4.4 Hz), 3.89 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 168.9, 160.1 (d, J
= 244 Hz), 159.7, 154.3, 136.7, 132.7, 129.5, 128.9, 128.4, 127.8, 127.7, 123.2 (d, J
= 8 Hz), 116.1 (d, J
= 22 Hz), 116.0, 77.9, 70.3, 36.6, 53.5. IR (thin film): 1769, 1552, 1388, 1227, 1099, 834. HRMS (MALDI): found, 444.1224. C24H20FNO5Na+ requires 444.1224. Anal.: found, C, 68.18; H, 4.91; N, 3.38. C24H20FNO5 requires C, 68.40; H, 4.78; N, 3.32%.
(4R,5S)-4-(4-Benzyloxyphenyl)-3-(4-fluorophenyl)-5-hydroxymethyloxazolidin-2-one (12). To a suspension of methyl ester 11
(1.68 g, 3.99 mmol, 1.00 eq.) in ethanol (27.0 ml) was added, at 23 °C, sodium borohydride (226 mg, 5.98 mmol, 1.50 eq.). The suspension was stirred for 2 h at this temperature after which point all solids had dissolved. To this solution was added NH4Cl(s) and the volume was concentrated to 5 ml in vacuo. To this suspension was added water (50 ml) and EtOAc (50 ml). The aqueous phase was extracted with EtOAc. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (1∶1 to 2∶3 gradient), to afford alcohol 12 as a colorless solid in 92% yield. Mp: 143 °C. Rf
= 0.40 [hexane–EtOAc 2∶3 (v/v)]. α32.4D
=
−16°, (c 1.54 in CHCl3). 1H-NMR (300 MHz, CDCl3): δ 7.42–7.19 (9 H, m), 6.97–6.90 (4 H, m), 5.26 (1 H, d, J
= 6.5 Hz), 5.02 (2 H, s), 4.39 (1 H, m), 3.99 (1 H, d, J
= 12.5 Hz), 3.74 (1 H, d, J
= 12.5 Hz), 2.77 (1 H, s). 13C-NMR (75 MHz, CDCl3): δ 159.7 (d, J
= 245 Hz), 159.0, 136.4, 132.7, 129.4, 128.5, 128.0, 127.9, 127.4, 123.6 (d, J
= 8 Hz), 115.6 (d, J
= 22 Hz), 115.6, 82.0, 70.1, 61.6, 61.2. IR (thin film): 3418, 2930, 2871, 1748, 1611, 1512, 1394, 1234. HRMS (EI): found, 393.1389. C23H20FNO4+ requires 393.1376. Anal.: found, C, 70.26; H, 5.21; N, 3.61. C23H20FNO4 requires C, 70.22; H, 5.12; N, 3.56%.
(4R,5R)-4-(4-Benzyloxyphenyl)-3-(4-fluorophenyl)-5-[(E)-3-(4-fluorophenyl)-3-oxopropenyl]oxazolidin-2-one (14). To a solution of oxalyl chloride (508 mg, 4.00 mmol, 2.00 eq.) in CH2Cl2
(15.0 ml) was added, at −78 °C, dimethyl sulfoxide (0.355 ml, 5.00 mmol, 2.50 eq.). After 10 min at −78 °C, alcohol 12
(787 mg, 2.00 mmol, 1.00 eq.) in CH2Cl2
(15.0 ml) was added over a period of 5 min. After an additional 5 min at this temperature, triethylamine (1.14 ml, 8.00 mmol, 8.00 eq.) was added. After 5 min, 1-(4-fluorophenyl)-2-(triphenyl-λ5-phosphanylidene)ethanone 1312 was added and the resulting suspension was warmed to 20 °C and stirred for an additional 30 min. To the solution was added saturated aqueous Na2HCO3 solution. The aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (2∶1 to 1∶1 gradient), to afford enone 14 as a colorless solid in 89% yield. Mp: 152 °C. Rf
= 0.56 [hexane–EtOAc 3∶2 (v/v)]. α25.6D
=
+100°, (c 0.60 in CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.06–7.99 (2 H, m), 7.42–7.06 (14 H, m), 7.00–6.92 (4 H, m), 5.05–5.00 (4 H, m). 13C-NMR (75 MHz, CDCl3): δ 187.1, 165.9 (d, J
= 254 Hz), 159.8 (d, J
= 243 Hz), 159.4, 154.8, 140.0, 136.2, 133.2, 132.3, 131.4 (d, J
= 9 Hz), 128.6, 128.1, 128.1, 127.9, 127.4, 125.8, 123.5 (d, J
= 9 Hz), 115.9 (d, J
= 24 Hz), 115.8 (d, J
= 24 Hz), 115.8, 80.5, 70.2, 66.0. IR (thin film): 1760, 1675, 1597, 1511, 1385, 1227. HRMS (MALDI): found, 534.1482. C31H23F2NO4Na+ requires 534.1493. Anal.: found, C, 72.51; H, 4.78; N, 2.73. C31H23F2NO4 requires C, 72.79; H, 4.53; N, 2.74%. (4R,5R)-3-(4-Fluorophenyl)-5-[(S)-3-(4-fluorophenyl)-3-hydroxypropyl]-4-(4-hydroxyphenyl)oxazolidin-2-one (16). To enone 14
(910 mg, 1.78 mmol, 1.00 eq.) in ethanol (15.0 ml) was added, at 23 °C, Pd on carbon (10%)
(100 mg). The suspension was vigorously stirred under 1 atm of hydrogen gas for 3 h. The suspension was filtered through a pad of celite, eluting with EtOAc, concentrated and the residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (2∶1 to 1∶1 gradient). A portion of the resulting benzyl ether 15
(310 mg, 0.604 mmol, 1.00 eq.) was dissolved in CH2Cl2 and cooled to −20 °C. (R)-3,3-Diphenyl-1-methyltetrahydro-3H-pyrrolo-oxazaborole-2-methyl oxazaborolidine [solution in toluene (0.5 M) 0.600 ml, 0.302 mmol, 0.50 eq.] was added, followed by borane–dimethylsulfide complex (0.080 ml, 0.905 mmol, 1.50 eq.). The solution was stirred at −20 °C for 2 h, then warmed to 0 °C and quenched with methanol. To the solution was added saturated aqueous Na2HCO3 solution and CH2Cl2. The aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (3∶2 to 1∶1 gradient). A portion of the resulting alcohol (53 mg, 0.10 mmol, 1.0 eq.) was dissolved in ethanol and Pd on carbon (10 mg) was added. The suspension was vigorously stirred under an atmosphere of hydrogen for 2.5 h. The suspension was filtered through a plug of celite eluting with EtOAc. The residue was purified by chromatography on silica gel eluting with hexane–EtOAc (1∶1 to 1∶2 gradient) to afford oxazolidinone 16 as a colorless solid in 57% yield from enone 14. Mp: 98 °C. Rf
= 0.41 [hexane–EtOAc 2∶3 (v/v)]. α27.6D
=
−1°, (c 0.84 in CHCl3). 1H-NMR (300 MHz, acetone-d6): δ 7.47–7.35 (4 H, m), 7.29–7.24 (2 H, m), 7.09–6.97 (4 H, m), 6.85–6.79 (2 H, m), 5.15 (1 H, d, J
= 6.7 Hz), 4.76–4.68 (1 H, m), 4.43–4.34 (2 H, m), 2.02–1.76 (4 H, m). 13C-NMR (75 MHz, acetone-d6): δ 162.0 (d, J
= 243 Hz), 159.5 (d, J
= 242 Hz), 157.9, 155.3, 142.2 (d, J
= 3 Hz), 134.3 (d, J
= 2 Hz), 129.1, 128.7, 127.8 (d, J
= 8 Hz), 123.8 (d, J
= 9 Hz), 116.1, 115.2 (d, J
= 23 Hz), 114.9 (d, J
= 21 Hz), 82.4, 72.3, 65.6, 35.0, 30.3. IR (thin film): 3316, 2925, 1726, 1603, 1511, 1224, 835. HRMS (MALDI): found, 448.1326. C24H21F2NO4Na+ requires 448.1337. The diastereoselectivity of the CBS reduction was established by integration of the fluorine signals in the 19F-NMR spectrum by comparison to a mixture of 16 and (4R,5R)-3-(4-fluorophenyl)-5-[(R)-3-(4-fluorophenyl)-3-hydroxypropyl]-4-(4-hydroxyphenyl)oxazolidin-2-one, obtained by NaBH4 reduction of the corresponding ketone.
Methanesulfonic acid 4-[(E)-3-oxopropenyl]phenyl ester (18). To a suspension of 4-hydroxycinnamic acid 17
(8.85 g, 53.5 mmol, 1.00 eq.) in methanol (70 ml) at 0 °C was added dropwise thionyl chloride (6.40 g, 53.5 mmol, 1.00 eq.). The ice bath was removed and the solution was stirred at 23 °C for 16 h. A stream of air was bubbled through the solution for 2 h and the solution was concentrated in vacuo to afford an off-white solid. This solid was dissolved in THF (75 ml) and triethylamine (6.05 g, 60.0 mmol, 1.20 eq.) was added and the solution was cooled to 0 °C. To this solution was added methanesulfonyl chloride (6.30 g, 55.0 mmol, 1.10 eq.). The ice bath was removed and the suspension was stirred at 23 °C for 2 h. This suspension was poured onto saturated, aqueous NH4Cl (25 ml), water (50 ml), and EtOAc (150 ml). The phases were separated and the aqueous phase was extracted with EtOAc (3 × 30 ml). The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was recrystallized from hexane–EtOAc (1∶3, 200 ml) to afford a colorless solid. The solid was suspended in CH2Cl2
(100 ml) and the suspension was cooled to 0 °C. To this suspension was added, over a period of 15 min, DIBAl-H (15.3 g, 108 mmol, 2.15 eq.) and the solution was stirred at 0 °C for 15 min. To this solution was added saturated, aqueous NaK-tartrate solution (100 ml) followed by Et2O (100 ml). This emulsion was vigorously stirred for 12 h. The phases were separated and the aqueous phase was extracted with Et2O. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo to afford a yellow solid. This solid was dissolved in CH2Cl2
(200 ml) and MnO2
(34.7 g, 400 mmol, 8.00 eq.) was added and the suspension was stirred for 6 h. The suspension was filtered over a plug of celite, eluting with CH2Cl2. The filtrate was concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (1∶1 to 2∶3 gradient), to afford the target compound as a bright yellow solid in 76% yield (four steps). Rf
= 0.35 [hexane–EtOAc 1∶1 (v/v)]. Mp: 78 °C. 1H NMR (300 MHz, CDCl3): δ 9.69 (1 H, d, J
= 7.5 Hz), 7.62–7.60 (2 H, m), 7.45 (1 H, d, J
= 15.9 Hz), 7.36–7.33 (2 H, m), 6.67 (1 H, dd, J
= 15.6 Hz, 7.8 Hz), 3.18 (3 H, s). 13C NMR (75 MHz, CDCl3): δ 193.6, 151.0, 150.8, 133.5, 130.4, 129.7, 123.0, 38.0. IR (thin film) 3035, 2939, 2826, 2744, 1678, 1627, 1600, 1504, 1367, 1177, 1155, 1126, 974, 873, 775, 693, 526 (cm−1). HRMS-EI (m/z): found, 226.0301. C10H10O4S requires 226.0300. Anal.: found, C, 53.14; H, 4.56. C10H10O4S requires C, 53.09; H, 4.45%.
Methanesulfonic acid 4-[(E)-(S)-3-hydroxyhexa-1,5-dienyl]phenyl ester (20). To (+)-β-chloro diisopinocampheyl borane (8.24 g, 25.6 mmol, 1.25 eq.) in Et2O (50 ml) at −78 °C was added allylmagnesium bromide (1.0 M in Et2O, 24.7 ml, 24.7 mmol, 1.20 eq.). The emulsion was warmed to 23 °C and stirred at this temperature for 2 h to afford a grey slurry. This slurry was cooled to −78 °C and aldehyde 18 was added portionwise over a period of 15 min. The yellow slurry was stirred at −78 °C for 2 h. The reaction was quenched with methanol (1.0 ml), 10% aqueous NaOH (25 ml), and H2O2
(30%)
(25 ml). The emulsion was vigorously stirred for 20 h. The phases were separated and the aqueous phase was extracted with Et2O. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo to afford a yellow oil. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (2∶1 to 1∶2 gradient), to afford the target compound as a colorless oil (77% yield, 93% ee). Rf
= 0.35 [hexane–EtOAc 1∶1 (v/v)]. 1H NMR (300 MHz, CDCl3): δ 7.42–7.38 (2 H, m), 7.24–7.20 (2 H, m), 6.59 (1 H, d, J
= 16.2 Hz), 6.22 (1 H, dd, J
= 15.9 Hz, 5.9 Hz), 5.91–5.77 (1 H, m), 5.22–5.15 (2 H, m), 4.39–4.32 (1 H, m), 3.13 (3 H, s), 2.44–2.36 (2 H, m). 13C NMR (75 MHz, CDCl3): δ 148.3, 136.0, 133.7, 132.9, 128.5, 127.8, 122.1, 118.7, 71.3, 42.0, 37.4. IR (thin film) 3370, 3029, 1501, 1375, 1356, 1178, 1151, 969, 872, 695 (cm−1). HRMS-EI (m/z): found, 268.0760. C13H16O4S requires 268.0769. Chiral HPLC (Daicel Chiralpak AD-H, hexane–i-PrOH = 9∶1, flow rate = 1.00 ml min−1)
tR
= 21.0 min (major), tR
= 18.9 min (minor).
Methanesulfonic acid 4-[(1E,5E)-(S)-3-hydroxy-6-phenylhexa-1,5-dienyl]phenyl ester (21). To Pd(OAc)2
(82.0 mg, 0.365 mmol, 0.10 eq.) in acetonitrile (14.0 ml) at 25 °C was added triphenylphosphine (192 mg, 0.730 mmol, 0.20 eq.), triethylamine 140 (7 ml), iodobenzene (820 mg, 4.02 mmol, 1.10 eq.), and allyl alcohol 20
(980 mg, 3.65 mmol, 1.00 eq.). The suspension was heated at reflux for 4 h. The solution was concentrated in vacuo and the residue was dissolved in EtOAc (20 ml) and filtered over a plug of silica gel, eluting with EtOAc. The solution was concentrated in vacuo and the residue was recrystallized from toluene (4 ml). The crystals were filtered off, washed with toluene (3 ml), and dried in vacuo to afford the target compound as a beige solid (51% yield). Rf
= 0.35 [hexane–EtOAc 1∶1 (v/v)]. Mp (toluene) 134 °C. α29.0D
−4°
(c 0.85 in CH2Cl2). 1H NMR (300 MHz, acetone-d6): δ 7.55–7.51 (2 H, m), 7.40–7.37 (2 H, m), 7.30–7.16 (5 H, m), 6.68 (1 H, dd, J
= 15.9 Hz, 1.0 Hz), 6.53–6.33 (3 H, m), 4.46–4.38 (1 H, m), 4.10 (1 H, d, J
= 4.7 Hz), 3.26 (3 H, s), 2.55–2.51 (2 H, m). 13C NMR (75 MHz, acetone-d6): δ 149.4, 138.4, 137.3, 135.3, 132.7, 129.1, 128.4, 128.2, 127.6, 127.5, 126.7, 123.0, 72.3, 42.3, 37.5. IR (thin film) 3370, 3029, 2935, 1501, 1375, 1356, 1197, 1178, 1151, 969, 872, 777, 748, 695 (cm−1). HRMS-EI (m/z): 157.0344 (84.80%), 114.0275 (62.10%), 113.0204 (54.88%), 17.9497 (100%). Anal.: found, C, 66.41; H, 5.84. C19H20O4S requires C, 66.26; H, 5.85%.
Methanesulfonic acid 4-{(4S,5S)-3-(4-fluorophenyl)-5-[(E)-(S)-1-hydroxy-4-phenylbut-3-enyl]-4,5-dihydroisoxazol-4-yl}phenyl ester (23). To allyl alcohol 21
(600 mg, 1.74 mmol, 1.00 eq.) in CH2Cl2
(8.5 ml) at 0 °C was added isopropanol (345 mg, 5.75 mmol, 3.30 eq.) followed by ethylmagnesium bromide (3.0 M)
(1.74 ml, 5.23 mmol, 3.00 eq.). The solution was allowed to warm to 23 °C. Separately, 4-fluorobenzaldehyde oxime 22
(303 mg, 2.18 mmol, 1.25 eq.) was dissolved in CH2Cl2
(8.5 ml). This solution was cooled to −78 °C and tert-butyl hypochloride (239 mg, 2.18 mmol, 1.25 eq.) was added. The ice bath was removed and the solution allowed to reach 23 °C. This solution was added over a period of 30 h by syringe pump to the solution containing allyl alcohol 23. To the solution was added 10% aqueous HCl (10 ml). The phases were separated and the aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo to afford a yellow oil. The residue was purified by chromatography on silica gel, eluting with toluene–EtOAc (4∶1 to 2∶1 gradient), to afford the target compound as a colorless solid (36% yield, dr > 99∶1) and starting material 21
(46%). Rf
= 0.37 [hexane–EtOAc 1∶1 (v/v)]. Mp 78 °C. α29.0D
+147°
(c 0.65 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 7.56–7.50 (2 H, m), 7.36–7.18 (9 H, m), 7.00–6.93 (2 H, m), 6.51 (1 H, d, J
= 15.9 Hz), 6.24 (1 H, dd, J
= 15.9 Hz, 6.2 Hz), 4.79 (1 H, d, J
= 5.6 Hz), 4.51 (1 H, dd, J
= 5.6 Hz, 3.4 Hz), 3.90–3.82 (1 H, m), 3.13 (3 H, s), 2.62–2.58 (2 H, m), 2.14 (1 H, d, J
= 6.8 Hz). 13C NMR (75 MHz, CDCl3): δ 161.2 (d, J
= 242 Hz), 157.4, 148.4, 138.0, 136.8, 133.6, 129.2, 129.1, 128.5, 127.4, 126.1, 124.7, 123.0, 115.8 (d, J
= 24 Hz), 91.9, 72.1, 55.9, 37.7, 37.3. IR (thin film) 3387, 3027, 2936, 1603, 1512, 1502, 1368, 1235, 1177, 1151, 969, 913, 871, 838, 772, 749 (cm−1). HRMS-EI (m/z): found, 481.1355. C26H24FNO5 requires 481.1359.
Methanesulfonic acid 4-{(4S,5R)-3-(4-fluorophenyl)-5-[(E)-3-(4-fluorophenyl)-3-oxopropenyl]-4,5-dihydroisoxazol-4-yl}phenyl ester (25). To homoallyl alcohol 23
(80.0 mg, 0.155 mmol, 1.00 eq.) in CH2Cl2
(1.5 ml) at 25 °C was added pyridine (24.6 mg, 0.310 mmol, 2.00 eq.) followed by methanesulfonyl chloride (26.7 mg, 0.233 mmol, 1.50 eq.). The solution was stirred at 23 °C for 16 h, then diluted with CH2Cl2 and water. The phases were separated and the aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was dissolved in EtOAc and passed over a small plug of silica gel, eluting with EtOAc. The solution was concentrated in vacuo. The residue was dissolved in CH2Cl2
(1.0 ml) and DBU (0.10 ml) was added. The solution was heated at reflux for 12 h, then cooled and diluted with saturated aqueous NH4Cl and CH2Cl2. The phases were separated and the aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was dissolved in EtOAc and passed over a small plug of silica gel, eluting with EtOAc. The solution was concentrated in vacuo. The residue was dissolved in THF (2.6 ml) and water (2.6 ml) and NaIO4
(222 mg, 1.04 mmol, 8.00 eq.) was added followed by K2OsO4·2H2O (9.5 mg, 26 µmol, 0.20 eq.). The suspension was stirred for 14 h, then diluted with EtOAc and saturated aqueous Na2SO3. The phases were separated and the aqueous phase was extracted with EtOAc. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was immediately dissolved in CH2Cl2
(2.0 ml) and 1-(4-fluorophenyl)-2-(triphenyl-λ5-phosphanylidene)ethanone 1312
(200 mg, 0.516 mmol, 3.30 eq.) was added. The solution was stirred at 23 °C for 30 min and then concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (3∶2 to 1∶1 gradient), to afford the target compound as a colorless solid (30% yield, four steps). Rf
= 0.51 [hexane–EtOAc 1∶1 (v/v)]. Mp (MeOH) 64 °C. α27.8D
+255°
(c 1.00 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 8.03–7.99 (2 H, m), 7.58–7.54 (2 H, m), 7.35–6.96 (8 H, m), 5.20 (1 H, ddd, J
= 4.7 Hz, 4.7 Hz, 1.6 Hz), 4.62 (1 H, d, J
= 5.0 Hz), 3.16 (3 H, s). 13C NMR (75 MHz, CDCl3): δ 187.7, 165.7 (d, J
= 255 Hz), 163.6 (d, J
= 251 Hz), 156.6, 148.6, 142.6, 136.9, 133.4, 131.3 (d, J
= 9 Hz), 129.2 (d, J
= 9 Hz), 129.0, 125.2, 123.9, 123.2, 116.0 (d, J
= 22 Hz), 115.8 (d, J
= 22 Hz), 88.4, 59.4, 37.8. IR (thin film) 3028, 2938, 1672, 1626, 1598, 1511, 1369, 1235, 1153, 972, 872, 838 (cm−1). HRMS-EI (m/z): found, 483.0948. C25H19F2NO5S requires 483.0952. Methanesulfonic acid 4-{(4S,5R)-3-(4-fluorophenyl)-5-[(S)-3-(4-fluorophenyl)-3-hydroxypropyl]-4,5-dihydroisoxazol-4-yl}phenyl ester (26). To alkene 25
(6.7 mg, 14 µmol) in methanol (1.3 ml) was added Pd on carbon (10%)
(2 mg). The atmosphere was changed to hydrogen (1 atm) and the suspension was stirred for 10 min at 23 °C. The suspension was diluted with EtOAc (15 ml) and filtered through a plug of silica gel, eluting with EtOAc. The filtrate was concentrated in vacuo and redissolved in CH2Cl2
(0.50 ml). The solution was cooled to −20 °C and (R)-3,3-diphenyl-1-methyltetrahydro-3H-pyrrolo-oxazaborole-2-methyl-oxazaborolidine (solution in toluene, 0.5 M)
(10 µl, 5.0 µmol, 0.50 eq.) was added, followed by borane–dimethylsulfide complex (1.5 mg, 20 µmol, 2.0 eq.). The solution was stirred at −20 °C for 2 h, then warmed to 0 °C and stirred for an additional 1 h. To this solution was added methanol (50 µl) followed by saturated aqueous NaHCO3 solution and CH2Cl2. The phases were separated and the aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel, eluting with hexane–EtOAc (1∶1 to 1∶2 gradient), to afford the target compound as a colorless solid (75% yield, two steps). Rf
= 0.44 [hexane–EtOAc 1∶2 (v/v)]. Mp 68 °C. α26.2D
+135°
(c 0.25 in CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 7.55–7.51 (2 H, m), 7.33–7.24 (6 H, m), 7.06–6.95 (4 H, m), 4.75–4.71 (1 H, m), 4.52–4.46 (1 H, m), 4.36 (1 H, d, J
= 5.0 Hz), 3.14 (3 H, s), 1.99–1.82 (5 H, m). 13C NMR (75 MHz, CDCl3): δ 163.8 (d, J
= 250 Hz), 162.5 (d, J
= 245 Hz), 157.1, 148.7, 140.2, 138.4, 129.3 (d, J
= 9 Hz), 129.2, 127.6 (d, J
= 8 Hz), 125.0, 123.3, 116.2 (d, J
= 22 Hz), 115.7 (d, J
= 22 Hz), 90.7, 73.7, 59.3, 37.8, 35.1, 31.6. IR (thin film) 3388, 2930, 1604, 1151, 1368, 1222, 1151, 871, 837 (cm−1). HRMS-EI (m/z): found, 487.1256. C25H23F2NO5S requires 487.1265.
4-Benzyloxy-1-(4-fluorophenyl)but-2-yn-1-ol (28). A 50 ml Schlenk flask was charged with Zn(OTf)2
(12.647 g, 34.79 mmol) and heated to 120 °C under high-vacuum (0.2 Torr) for 3.5 h. After cooling, (+)-N-methylephedrine (6.595 g, 36.79 mmol) was added and the flask was purged with Ar for 15 min. Anhydrous toluene (14 ml) followed by Et3N (3.874 g, 38.3 mmol) were added and, after 3 h stirring, benzyl propargyl ether31
(5.556 g, 38.00 mmol) was added in one portion. After 20 min stirring, the mixture was transferred to a pre-cooled acetone bath (8 °C), stirred for 5 min and p-FC6H4CHO (3.632 g, 29.26 mmol) was added in one portion. After 15 h stirring at 9–12 °C, the suspension was diluted with EtOAc (125 ml) and washed with sat. aq. NH4Cl (2 × 30 ml) and brine (30 ml). The organic layer was evaporated on celite and purified by dry column vacuum chromatography (5.4 × 5.5 cm) on silica gel, eluting with a gradient of 0–50% EtOAc in hexane (v/v), to give alcohol 28
(5.896 g, 75%) as a light yellow oil. Enantiomeric excess as determined by HPLC analysis: 96% ee; tR 20 min (R-28), 28 min (S-28)
(Chiracel OD-H 25 cm, 6%
iPrOH in hexane, flow 1.0 ml min−1, 254 nm). Rf [EtOAc–hexane 1∶3 (v/v)] 0.28. 1H-NMR (300 MHz, CDCl3): δ 7.50 (2 H, dd, J
= 5.6, 8.7 Hz), 7.38–7.32 (5 H, m), 7.06 (2 H, t, J
= 8.7 Hz), 5.48 (1 H, s), 4.60 (2 H, s), 4.26 (2 H, s), 2.84 (1 H, s). 13C-NMR (75 MHz, CDCl3): δ 164.01, 160.75, 136.95, 136.04 (C), 128.30, 128.21, 127.92, 127.81, 115.43, 115.13 (CH), 86.13, 82.62 (C), 71.74 (CH2), 63.74 (CH), 57.35 (CH2). 19F-NMR (282 MHz, CDCl3): δ
−113.28 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 3390, 3066, 3032, 2859, 1604, 1508, 1455, 1413, 1386, 1355, 1224, 1158, 1121, 1096, 1072, 1028, 1014, 842, 772, 744, 699, 592, 561, 498. MALDI-MS: found, 293.0947 [MNa]+. C17H15FO2Na requires 293.0954. Anal.: found, C, 75.39; H, 5.62. C17H15FO2 requires C, 75.54; H, 5.59%. 4-(tert-Butyldimethylsilanyloxy)-4-(4-fluorophenyl)butan-1-ol (29). Alcohol 28
(4.108 g, 15.20 mmol) was dissolved in anhydrous DMF (50 ml). Imidazole (2.123 g, 31.1 mmol) and TBDMSCl (3.590 mg, 23.8 mmol) were added sequentially and the solution was stirred for 3.5 h, followed by addition of 50% sat. aq. NaHCO3
(150 ml). After extraction with ether (4 × 50 ml), the combined organic phase was washed successively with sat. aq. NaHCO3
(50 ml) and H2O (50 ml), evaporated and dried shortly under high vacuum. The residue was dissolved in EtOH (40 ml). Na2CO3
(3.229 g, 30.5 mmol) and Pd/C [10%
(w/w), 223 mg] were added and the suspension was evacuated 4 times with H2 and stirred under an H2 atmosphere for 19 h. The suspension was diluted with 10% EtOAc–hexane [250 ml (v/v)] and filtered through a short plug of silica gel [2 × 25 ml 20% EtOAc–hexane washings (v/v)], evaporated and dried shortly under high vacuum. The residue was dissolved in EtOH (40 ml). Pd/C [10%
(w/w), 142 mg] was added and the suspension was evacuated 4 times with H2 and stirred under an H2 atmosphere for 1 h. Additional Pd/C [10%
(w/w), 190 mg] was added and the suspension was evacuated 4 times with H2 and stirred under an H2 atmosphere for 1.25 h. The suspension was evaporated on celite and purified by dry column vacuum chromatography (5.2 × 5.5 cm) on silica gel, eluting with a gradient of 0–25% EtOAc in hexane (v/v), to give alcohol 29
(3.643 g, 80%) as a light yellow oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.37. 1H-NMR (300 MHz, CDCl3): δ 7.24 (2 H, dd, J
= 5.6, 8.7 Hz), 6.97 (2 H, t, J
= 8.7 Hz), 4.69 (1 H, dt, J
= 1.2, 5.0 Hz), 3.59 (2 H, dt, J
= 1.2, 6.2 Hz), 2.18 (1 H, bs), 1.77–1.45 (4 H, m), 0.87 (9 H, s), 0.02 (3 H, s), −0.15 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 163.37, 160.13, 140.96, 140.91 (C), 127.32, 127.23, 114.94, 114.64, 74.16 (CH), 62.76, 37.19, 28.47 (CH2), 25.76 (CH3), 18.15 (C), −4.71, −5.05 (CH3). IR (cm−1): 3339, 2954, 2930, 2885, 2858, 1606, 1510, 1472, 1463, 1362, 1252, 1223, 1156, 1092, 1060, 984, 890, 836, 776, 668, 560. MALDI-MS: found, 321.1643 [MNa]+. C16H27FO2SiNa requires 321.1662. Anal.: found, C, 64.36; H, 9.15. C16H27FO2Si requires C, 64.39; H, 9.12%.
Olefin (31). Alcohol 29 was dissolved in CH2Cl2
(50 ml). Dess–Martin periodinane (5.658 g, 13.3 mmol) was added and the milky solution was stirred at room temperature for 1.5 h. Sat. aq. Na2SO3
(100 ml) was added and the layers were swirled until the solid had dissolved. The layers were separated and the aqueous phase was extracted with CH2Cl2
(2 × 40 ml). The combined organic phase was evaporated on celite and purified by dry column vacuum chromatography (5.1 × 5.5 cm) on silica gel, eluting with a gradient of 0–10% EtOAc in hexane (v/v), to give the intermediary aldehyde (2.093 g, 80%) as a light yellow oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.63. 1H-NMR (300 MHz, CDCl3): δ 9.73 (1 H, d, J
= 1.5 Hz), 7.25 (2 H, dd, J
= 5.6, 8.7 Hz), 6.99 (2 H, t, J
= 9.0 Hz), 4.74 (1 H, dt, J
= 5.0, 6.8 Hz), 2.52–2.35 (2 H, m), 2.06–1.88 (2 H, m), 0.88 (9 H, s), 0.02 (3 H, s), −0.16 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 201.91 (CH), 163.35, 160.10, 140.13 (C), 127.20, 127.10, 115.04, 114.75, 73.03 (CH), 39.69, 33.11 (CH2), 25.85 (CH3), 18.21 (C), −4.61, −4.95 (CH3). IR (cm−1): 2955, 2938, 2888, 2858, 2720, 1727, 1606, 1509, 1472, 1464, 1412, 1390, 1362, 1254, 1223, 1156, 1090, 1014, 837, 776, 670, 540. Anal.: found, C, 64.95; H, 8.36. C16H25FO2Si requires C, 64.82; H, 8.50%. LiCl (140.8 mg, 3.32 mmol) was heated shortly with a heat gun under high-vacuum and, after cooling, anhydrous CH3CN (5 ml), phosphonate 3021
(660 mg, 1.68 mmol) and DBU (221 mg, 1.45 mmol) were added sequentially. After 3 min stirring, the aldehyde (407.3 mg, 1.37 mmol) was added and the suspension was stirred at room temperature for 2.5 h, followed by addition of 50% sat. aq. NaHCO3
(60 ml). After extraction with ether–hexane [1∶1 (v/v), 4 × 25 ml], the combined organic phase was washed with brine (25 ml), evaporated on celite and purified by dry column vacuum chromatography (4.6 × 3.3 cm) on silica gel, eluting with a gradient of 0–20% EtOAc in hexane (v/v), to give olefin 31
(520.7 mg, 71%) as a colourless oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.43. 1H-NMR (300 MHz, CDCl3): δ 7.25 (2 H, dd, J
= 5.6, 8.7 Hz), 7.10–6.94 (3 H, m), 6.53 (1 H, d, J
= 14.9 Hz), 4.65 (1 H, dd, J
= 5.0, 7.5 Hz), 3.91 (1 H, dd, J
= 5.6, 6.8 Hz), 3.50 (1 H, d, J
= 13.7 Hz), 3.42 (1 H, d, J
= 13.7 Hz), 2.30–2.23 (2 H, m), 2.09–2.02 (2 H, m), 1.90–1.70 (5 H, m), 1.43–1.30 (2 H, m), 1.15 (3 H, s), 0.95 (3 H, s), 0.85 (9 H, s), 0.01 (3 H, s), −0.20 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 163.88, 163.39, 160.14, 150.06, 140.63 (C), 127.35, 127.26, 120.91, 114.98, 114.69, 73.24, 64.99 (CH), 53.04 (CH2), 48.33, 47.67 (C), 44.58 (CH), 38.61, 38.39, 32.71, 28.32, 26.40 (CH2), 25.72, 20.72, 19.78 (CH3), 18.04 (C), −4.74, −5.10 (CH3). IR (cm−1): 2956, 2885, 2859, 1684, 1640, 1605, 1509, 1472, 1414, 1374, 1332, 1295, 1250, 1220, 1165, 1134, 1083, 1049, 995, 970, 860, 836, 774, 544. MALDI-MS: found, 558.2479 [MNa]+. C28H42FNO4SSiNa requires 558.2486. Anal.: found, C, 62.84; H, 7.78; N, 2.58. C28H42FNO4SSi requires C, 62.77; H, 7.90; N, 2.61%. Pyrazoline (32). Olefin 31 was dissolved in anhydrous toluene (2.0 ml). TMSCHN2
(2 M in hexanes, 1.50 ml, 3.0 mmol) was added and the solution was stirred at room temperature for 64 h. After evaporation, the residue was dissolved in CH2Cl2
(10 ml). TFA (202 mg, 1.77 mmol) was added and the solution was stirred for 20 min. Sat. aq. NaHCO3
(1.5 ml) was added and the mixture was evaporated on celite and purified by dry column vacuum chromatography (4.5 × 3.3 cm) on silica gel, eluting with a gradient of 0–40% EtOAc in hexane (v/v), to give diastereomeric pyrazolines 32
(468 mg, 84%) and 32A
(54.3 mg, 10%) as light yellow foams. 32: Rf [EtOAc–hexane 1∶3 (v/v)] 0.25. 1H-NMR (300 MHz, CDCl3): δ 7.21 (2 H, dd, J
= 5.6, 8.7 Hz), 6.95 (2 H, t, J
= 8.7 Hz), 6.60 (1 H, s), 6.16 (1 H, d, J
= 5.6 Hz), 4.65 (1 H, t, J
= 5.0 Hz), 4.33 (1 H, dd, J
= 5.9, 9.7 Hz), 3.87 (1 H, dd, J
= 5.0, 7.5 Hz), 3.67–3.62 (1 H, bs), 3.53 (1 H, d, J
= 13.7 Hz), 3.44 (1 H, d, J
= 13.7 Hz), 2.15–1.99 (2 H, m), 1.91–1.86 (3 H, m), 1.66–1.51 (3 H, m), 1.47–1.21 (3 H, m), 1.14 (3 H, s), 0.95 (3 H, s), 0.86 (9 H, s), 0.01 (3 H, s), −0.17 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 167.96, 163.12, 159.89 (C), 146.91 (CH), 140.52, 140.49 (C), 127.15, 127.05, 114.83, 114.54, 73.37, 66.44, 65.09 (CH), 52.81 (CH2), 48.91 (C), 48.04 (CH), 47.79 (C), 44.33 (CH), 37.98, 37.79, 32.55, 26.76, 26.45 (CH2), 25.82, 20.68, 19.84 (CH3), 18.16 (C), −4.64, −4.90 (CH3). IR (cm−1): 3360, 2955, 2857, 1700, 1604, 1509, 1472, 1390, 1329, 1273, 1250, 1236, 1221, 1166, 1134, 1086, 1066, 994, 939, 836, 775, 694, 542. MALDI-MS: found, 600.2691 [MNa]+. C29H44FN3O4SSiNa requires 600.2704. Anal.: found, C, 60.25; H, 7.83; N, 7.16. C29H44FN3O4SSi requires C, 60.28; H, 7.67; N, 7.27%. 32A: Rf [EtOAc–hexane 1∶3 (v/v)] 0.11. 1H-NMR (300 MHz, CDCl3): δ 7.21 (2 H, dd, J
= 5.3, 8.4 Hz), 6.96 (2 H, t, J
= 8.7 Hz), 6.62 (1 H, s), 6.14 (1 H, d, J
= 3.1 Hz), 4.59 (1 H, dd, J
= 5.0, 6.8 Hz), 4.39 (1 H, dd, J
= 3.1, 7.5 Hz), 3.90 (1 H, dd, J
= 5.0, 7.5 Hz), 3.52 (1 H, d, J
= 13.7 Hz), 3.45 (1 H, d, J
= 13.7 Hz), 3.37 (1 H, dd, J
= 6.2, 13.7 Hz), 2.08–1.13 (2 H, m), 1.00 (3 H, s), 0.96 (3 H, s), 0.85 (9 H, s), 0.00 (3 H, s), −0.19 (3 H, s). MALDI-MS: found, 600.2691 [MNa]+. C29H44FN3O4SSiNa requires 600.2704.
(p-FC6H4)3Bi. p-FC6H4Br (5.446 g, 31.1 mmol) dissolved in anhydrous ether (100 ml) was added to Mg turnings (844 mg, 34.7 mmol) and I2
(28 mg, 0.11 mmol) and the suspension was refluxed for 1 h 20 min and cooled to 0 °C. BiCl3
(3.931 g, 12.5 mmol) was added and, after 15 min stirring at 0 °C, the suspension was refluxed for 4 h. The suspension was cooled, H2O (3 ml) was added and the suspension was evaporated on celite and purified by dry column vacuum chromatography (4.8 × 5.5 cm) on silica gel, eluting with a gradient of 0–14% EtOAc in hexane (v/v), to give (p-FC6H4)3Bi (2.862 g, 56%) as a light yellow solid. Rf [EtOAc–hexane 1∶9 (v/v)] 0.42. 1H-NMR (300 MHz, CDCl3): δ 7.72 (2 H, dd, J
= 6.2, 8.1 Hz), 7.13 (2 H, t, J
= 9.0 Hz). 13C-NMR (75 MHz, CDCl3): δ 164.19, 160.92, 149.36 (C), 139.02, 138.92, 117.96, 117.70 (CH). 19F (282 MHz, CDCl3): δ
−111.97 (1 F, m).
N-Aryl pyrazoline (33). Pyrazoline 32
(409.8 mg, 0.709 mmol), Cu(OAc)2
(296 mg, 1.63 mmol) and (p-FC6H4)3Bi (950 mg, 1.92 mmol) were dissolved in anhydrous CH2Cl2
(5 ml). Anhydrous Et3N (165 mg, 1.63 mmol) was added and the dark green suspension was stirred at room temperature for 12.5 h. After evaporation on celite the residue was purified by dry column vacuum chromatography (4.5 × 3.3 cm) on silica gel, eluting with a gradient of 0–30% EtOAc in hexane (v/v), to give pyrazoline 33
(320.8 mg, 63%) as a light yellow foam. Rf [EtOAc–hexane 1∶3 (v/v)] 0.33. 1H-NMR (300 MHz, CDCl3): δ 7.24 (2 H, dd, J
= 5.3, 8.4 Hz), 7.01–6.94 (4 H, m), 6.89 (2 H, t, J
= 8.7 Hz), 6.68 (1 H, d, J
= 1.9 Hz), 5.05 (1 H, d, J
= 3.7 Hz), 4.62 (1 H, t, J
= 5.3 Hz), 3.85 (1 H, dd, J
= 4.4, 7.5 Hz), 3.59 (1 H, d, J
= 14.3 Hz), 3.58 (1 H, d, J
= 14.3 Hz), 3.41–3.35 (1 H, m), 1.98–1.78 (5 H, m), 1.72–1.60 (3 H, m), 1.41–1.23 (3 H, m), 1.21 (3 H, s), 0.98 (3 H, s), 0.88 (9 H, s), 0.04 (3 H, s), −0.17 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 169.54, 163.38, 160.14, 158.46, 155.30 (C), 142.10 (CH), 140.75 (C), 127.32, 127.22, 115.71, 115.40, 114.96, 114.67, 114.22, 114.12, 73.99, 65.48, 64.93 (CH), 53.02 (CH, CH2), 49.05, 47.77 (C), 44.31 (CH), 37.98, 36.95, 32.76, 27.79, 26.25 (CH2), 25.75, 20.37, 19.77 (CH3), 18.07 (C), −4.77, −5.01 (CH3). 19F (282 MHz, CDCl3): δ
−116.27 (1 F, m), −125.73 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 2957, 2857, 1699, 1606, 1510, 1471, 1413, 1362, 1334, 1268, 1250, 1221, 1166, 1136, 1113, 1088, 1063, 987, 836, 776, 759, 538. MALDI-MS: found, 540.2127 [MH − TBDMSOH]+. C29H32F2N3O3S requires 540.2132; found, 694.2909 [MNa]+. C35H47F2N3O4SSiNa requires 694.2922. Anal.: found, C, 62.37; H, 7.05; N, 6.03. C35H47F2N3O4SSi requires C, 62.56; H, 7.05; N, 6.25%.
Carboxylic acid (34). Pyrazoline 33
(228.5 mg, 0.340 mmol) was dissolved in anhydrous MeOH (5 ml), glycine (226 mg, 3.01 mmol) and KCN (305 mg, 4.68 mmol) were added and the suspension was stirred at 50 °C in a sealed flask for 19 h. After cooling, the suspension was evaporated on celite and purified twice by dry column vacuum chromatography (4.8 × 2.0 cm) on silica gel, eluting first with a gradient of 0–60% MeOH in EtOAc and second with 0–20% MeOH in CH2Cl2
(v/v), to give carboxylic acid 34
(135.6 mg, 75%) as a light yellow oil. Rf [MeOH–EtOAc 1∶3 (v/v)] 0.38. 1H-NMR (300 MHz, CDCl3): δ 7.33 (2 H, dd, J
= 5.6, 8.7 Hz), 7.01–6.93 (6 H, m), 6.80 (1 H, s), 4.78–4.73 (1 H, m), 3.95–3.78 (3 H, m), 3.40–3.32 (1 H, m), 1.88–1.65 (4 H, m), 0.86 (9 H, s), 0.04 (3 H, s), −0.17 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 174.55, 165.01, 161.79, 160.58 (C), 145.83 (CH), 144.14, 144.11, 142.57, 142.53 (C), 128.94, 128.82, 116.58, 116.34, 116.24, 115.95, 115.66, 74.91, 70.88, 55.50 (CH), 42.25, 38.40, 30.07 (CH2), 26.38 (CH3), 19.06 (C), −4.40, −4.71 (CH3). 19F (282 MHz, CDCl3): δ
−116.75 (1 F, m), −125.36 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 3325, 2954, 2930, 2858, 1737, 1671, 1606, 1508, 1472, 1410, 1361, 1252, 1224, 1157, 1088, 1006, 984, 835, 776, 760, 668, 608, 554. MALDI-MS: 576.2 [M − H + 2Na]+.
2-{2-(4-Fluorophenyl)-4-[3-(4-fluorophenyl)-3-hydroxypropyl]-3,4-dihydro-2H-pyrazol-3-yl}-4H-oxazol-5-one (36). Carboxylic acid 34
(22.0 mg, 0.041 mmol) was dissolved in anhydrous CH2Cl2
(5 ml), N-cyclohexyl-N′-2-(N-methylmorpholinio)ethylcarbodiimide p-toluenesulfonate 35
(19.0 mg, 0.045 mmol) was added and the mixture was stirred at reflux for 2 h. The solution was cooled, diluted with CH2Cl2
(10 ml), washed with sat. aq. NaHCO3
(10 ml) and H2O (10 ml), dried (Na2SO4), filtered through a short plug of silica gel [15 ml EtOAc–hexane washings, 1∶1 (v/v)] and evaporated to give the oxazolone 36
(8.3 mg, 39%) as a colourless oil. Rf [EtOAc–hexane 1∶1 (v/v)] 0.58. 1H-NMR (300 MHz, CDCl3): δ 7.23 (2 H, dd, J
= 5.6, 8.7 Hz), 7.06–6.93 (6 H, m), 6.73 (1 H, s), 4.68–4.64 (1 H, m), 4.39 (1 H, d, J
= 6.2 Hz), 4.21 (2 H, s), 3.47–3.39 (1 H, m), 1.80–1.57 (4 H, m), 0.86 (9 H, s), 0.00 (3 H, s), −0.17 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 174.69, 165.28, 163.58, 160.32, 159.11, 155.94, 142.46, 141.30, 140.28, 127.29, 127.18, 115.89, 115.60, 115.26, 114.97, 114.87, 114.76, 73.61, 63.26, 54.23, 52.64, 37.50, 29.70, 27.96, 25.78, 18.14, −4.63, −5.0. 19F (282 MHz, CDCl3): δ
−115.81 (1 F, m), −124.45 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 2930, 2858, 1835, 1674, 1606, 1509, 1472, 1362, 1252, 1224, 1157, 1088, 1021, 905, 836, 777, 736, 608, 553. MALDI-MS: 429.2 [M − C3H2NO2]+.
Benzenesulfonic acid 2-{2-(4-fluorophenyl)-4-[3-(4-fluorophenyl)-3-hydroxypropyl]-3,4-dihydro-2H-pyrazol-3-yl}oxazol-5-yl ester (37). Oxazolone 36
(24 mg, 0.047 mmol) was dissolved in anhydrous CH2Cl2
(5 ml), Et3N (0.2 ml, 1.4 mmol) followed by PhSO2Cl (0.1 ml, 0.78 mmol) were added and the mixture was stirred at room temperature for 22 h. Sat. aq. NaHCO3
(1 ml) was added and the mixture was evaporated on celite and purified by dry column vacuum chromatography (4.5 × 2.0 cm) on silica gel, eluting with a gradient of 0–60% EtOAc in hexane (v/v), to give the intermediary oxazole (12.2 mg, 40%) as a yellow oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.29. 1H-NMR (300 MHz, CDCl3): δ 7.76 (2 H, d, J
= 8.1 Hz), 7.63 (1 H, tt, J
= 1.2, 7.5 Hz), 7.42 (2 H, dd, J
= 7.5, 8.7 Hz), 7.21 (2 H, dd, J
= 5.3, 8.4 Hz), 6.99 (2 H, t, J
= 8.7 Hz), 6.91 (4 H, d, J
= 6.2 Hz), 6.66 (1 H, d, J
= 1.2 Hz), 6.52 (1 H, s), 4.64–4.60 (1 H, m), 4.51 (1 H, d, J
= 7.5 Hz), 3.35–3.30 (1 H, m), 1.74–1.53 (4 H, m), 0.85 (9 H, s), 0.01 (3 H, s), −0.18 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 157.31, 142.38, 135.27, 129.41, 128.62, 127.29, 127.18, 115.73, 115.44, 115.21, 114.90, 114.81, 112.25, 73.56, 63.05, 53.78, 27.75, 25.78, 18.14, −4.65, −5.02. 19F (282 MHz, CD3OD): δ
−115.91 (1 F, m), −124.69 (1 F, p, J
= 6.4 Hz). IR (cm−1): 2930, 2857, 1606, 1509, 1451, 1398, 1224, 1193, 1157, 1090, 999, 914, 829, 777, 752, 685, 618, 578, 554. This oxazole (12.0 mg, 0.018 mmol) was dissolved in anhydrous THF (1.0 ml, teflon bottle), anhydrous pyridine (0.20 ml) followed by HF·pyridine complex (0.20 ml) were added and the solution was stirred at room temperature for 10 h, diluted with ether (10 ml) and washed with sat. aq. NaHCO3
(2 × 5 ml). The organic layer was evaporated on celite and purified by dry column vacuum chromatography (3.2 × 2.0 cm) on silica gel, eluting with a gradient of 0–100% EtOAc in hexane (v/v), to give oxazole 37
(3.9 mg, 39%) as a light brown oil. Rf [EtOAc–hexane 1∶1 (v/v)] 0.19. 1H-NMR (300 MHz, CDCl3): δ 7.76 (2 H, d, J
= 7.5 Hz), 7.64 (1 H, t, J
= 7.5 Hz), 7.44 (2 H, t, J
= 7.8 Hz), 7.34–7.25 (2 H, m), 7.03 (2 H, t, J
= 8.4 Hz), 6.92 (4 H, d, J
= 6.2 Hz), 6.69 (1 H, d, J
= 1.9 Hz), 6.52 (1 H, s), 4.66–4.62 (1 H, m), 4.57 (1 H, d, J
= 7.5 Hz), 3.42–3.36 (1 H, m), 1.90–1.53 (4 H, m). 13C-NMR (75 MHz, CDCl3): δ 142.30, 139.76, 135.32, 129.44, 128.62, 127.39, 115.76, 115.65, 115.45, 115.37, 114.92, 114.82, 112.28, 73.17, 63.09, 53.68, 35.69, 28.19. 19F (282 MHz, CD3OD): δ
−114.03 (1 F, m), −123.73 (1 F, p, J
= 6.4 Hz). IR (cm−1): 3300, 2926, 1606, 1509, 1450, 1396, 1224, 1192, 1090, 998, 828, 736, 685, 618, 578.
4-{2-(4-Fluorophenyl)-4-[3-(4-fluorophenyl)-3-hydroxypropyl]-3,4-dihydro-2H-pyrazol-3-ylmethoxy}phenol (38). Pyrazoline 33
(101.5 mg, 0.151 mmol) was dissolved in anhydrous THF (5 ml)
−78 °C, LiAlH4
(33 mg, 0.87 mmol) was added and the suspension was stirred at −78 °C for 4.5 h. Sat. aq. NaHCO3
(1 ml) was added and the mixture was evaporated on celite and purified twice by dry column vacuum chromatography (4.6 × 2.0 cm) on silica gel, eluting with a gradient of 0–30% EtOAc in hexane (v/v), to give the intermediary alcohol (52.7 mg, 76%) as a light yellow oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.23. 1H-NMR (300 MHz, CDCl3): δ 7.23 (2 H, dd, J
= 5.6, 8.7 Hz), 7.04–6.92 (6 H, m), 6.67 (1 H, d, J
= 1.2 Hz), 4.64 (1 H, t, J
= 5.9 Hz), 3.81 (1 H, dd, J
= 4.0, 11.5 Hz), 3.68–3.58 (2 H, m), 3.12 (1 H, dd, J
= 6.2, 6.8 Hz), 1.86 (1 H, bs), 1.77–1.67 (2 H, m), 1.58–1.48 (2 H, m), 0.86 (9 H, s), 0.00 (3 H, s), −0.17 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 163.47, 160.22, 158.83, 155.68 (C), 144.84 (CH), 142.35, 140.67, 140.62 (C), 127.26, 127.16, 115.75, 115.46, 115.11, 115.06, 114.96, 114.83, 73.76, 66.81 (CH), 62.37 (CH2), 50.05 (CH), 37.72, 28.28 (CH2), 25.75 (CH3), 18.12 (C), −4.67, −5.01 (CH3). 19F (282 MHz, CDCl3): δ
−115.25 (1 F, septet, J
= 4.3 Hz), −124.25 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 3401, 2953, 2930, 2885, 2858, 1672, 1605, 1509, 1472, 1463, 1416, 1362, 1296, 1252, 1223, 1156, 1086, 1006, 979, 938, 861, 835, 776, 666, 608, 554. MALDI-MS: found, 429.2175 [M − CH2OH]. C24H31F2N2O2Si requires 429.2174. Found, 459.2279 [M − H]+. C25H33F2N2O2Si requires 459.2279. This alcohol (70.8 mg, 0.154 mmol) was dissolved in anhydrous CH2Cl2
(5 ml), anhydrous Et3N (0.50 ml, 3.9 mmol), DMAP (6.8 mg, 0.056 mmol) and TsCl (69 mg, 0.36 mmol) were added and the solution was stirred at room temperature for 12.5 h, evaporated on celite and purified by dry column vacuum chromatography (4.4 × 2.0 cm) on silica gel, eluting with a gradient of 0–20% EtOAc in hexane (v/v), to give the intermediary tosylate (78.4 mg, 83%) as a colourless oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.44. 1H-NMR (300 MHz, CDCl3): δ 7.68 (2 H, d, J
= 8.7 Hz), 7.25 (4 H, t, J
= 8.1 Hz), 6.99 (2 H, t, J
= 8.7 Hz), 6.92–6.80 (4 H, m), 6.64 (1 H, d, J
= 1.2 Hz), 4.65 (1 H, dd, J
= 4.4, 6.8 Hz), 4.12 (1 H, dd, J
= 2.5, 9.3 Hz), 3.92–3.81 (2 H, m), 3.08–3.01 (1 H, m), 2.42 (3 H, s), 1.80–1.43 (4 H, m), 0.87 (9 H, s), 0.01 (3 H, s), −0.17 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 163.49, 160.24, 158.59, 155.43, 145.16 (C), 143.40 (CH), 140.54, 132.21 (C), 129.84, 127.82, 127.32, 127.21, 115.79, 115.48, 115.12, 114.83, 114.31, 114.22, 73.50 (CH), 67.45 (CH2), 62.42, 50.74 (CH), 37.35, 27.87 (CH2), 25.77, 21.59 (CH3), 18.10 (C), −4.67, −5.01 (CH3). 19F (282 MHz, CDCl3): δ
−116.01 (1 F, m), −125.40 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 3055, 3034, 2953, 2930, 2886, 2857, 1603, 1509, 1472, 1463, 1365, 1307, 1294, 1252, 1223, 1190, 1177, 1156, 1096, 979, 862, 835, 775, 666, 608, 555. MALDI-MS: found, 483.1559 [MH − TBDMSOH]+. C26H25F2N2O3S requires 483.1554. Found, 637.2330 [MNa]+. C32H40F2N2O4SSiNa requires 637.2344. This tosylate was dissolved in anhydrous DMF (2.5 ml), hydroquinone (263 mg, 2.39 mmol) and Cs2CO3
(102.1 mg, 0.313 mmol) were added and the suspension was stirred at 80 °C for 12 h. EtOAc (30 ml) was added and the organic phase was washed with sat. aq. NaHCO3
(10 ml) and H2O (10 ml), evaporated on celite and purified by dry column vacuum chromatography (4.5 × 2.0 cm) on silica gel, eluting with a gradient of 0–30% EtOAc in hexane (v/v), to give the intermediary phenol (70.9 mg, 86%) as a colourless oil. Rf [EtOAc–hexane 1∶3 (v/v)] 0.33. 1H-NMR (300 MHz, CDCl3): δ 7.24 (2 H, dd, J
= 5.3, 8.4 Hz), 7.06–6.93 (6 H, m), 6.75–6.68 (5 H, m), 4.67 (1 H, dd, J
= 4.4, 6.8 Hz), 4.10–3.98 (2 H, m), 3.74 (1 H, dd, J
= 1.2, 7.5 Hz), 3.17–3.11 (1 H, m), 1.86–1.54 (4 H, m), 0.88 (9 H, s), 0.02 (3 H, s), −0.15 (3 H, s). 13C-NMR (75 MHz, CDCl3): δ 163.32, 160.08, 158.48, 155.35, 152.31, 149.86 (C), 143.85 (CH), 141.52, 141.49, 140.63 (C), 127.23, 127.12, 115.99, 115.73, 115.51, 115.44, 115.04, 114.75, 114.67, 114.57, 73.78 (CH), 67.79 (CH2), 63.88, 51.51 (CH), 37.77, 28.38 (CH2), 25.89 (CH3), 18.25 (C), −4.46, −4.80 (CH3). 19F (282 MHz, CDCl3): δ
−115.31 (1 F, m), −124.71 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 3350, 3056, 2953, 2930, 2885, 2858, 1605, 1509, 1472, 1462, 1362, 1297, 1226, 1156, 1100, 1086, 1050, 1006, 939, 828, 776, 667, 609, 553, 518. MALDI-MS: found, 421.1720 [MH − TBDMSOH]+. C25H23F2N2O2 requires 421.1728. Found, 553.2677 [MH]+. C31H39F2N2O3Si requires 553.2698. Found, 575.2505 [MNa]+. C31H38F2N2O3SiNa requires 575.2517. This phenol (18.4 mg, 0.0333 mmol) was dissolved in anhydrous THF (1.0 ml, teflon bottle) at 0 °C, anhydrous pyridine (0.20 ml) followed by HF·pyridine complex (0.20 ml) were added and the solution was allowed to warm to room temperature over several hours and stirred at room temperature for 22 h. Ether (20 ml) was added and the solution was washed with sat. aq. NaHCO3
(2 × 5 ml), evaporated on celite and purified by dry column vacuum chromatography (4.5 × 2.0 cm) on silica gel, eluting with a gradient of 0–60% EtOAc in hexane (v/v), to give diol 38
(14.4 mg, 99%) as a colourless oil. Rf [EtOAc–hexane 1∶1 (v/v)] 0.27. 1H-NMR (300 MHz, CDCl3): δ 7.29 (2 H, dd, J
= 5.3, 8.4 Hz), 7.06–6.93 (6 H, m), 6.75–6.67 (5 H, m), 4.70 (1 H, t, J
= 6.5 Hz), 4.09–4.03 (2 H, m), 3.72 (1 H, t, J
= 10.0 Hz), 3.18 (1 H, dd, J
= 4.4, 6.2 Hz), 1.99–1.50 (4 H, m). 13C-NMR (75 MHz, CDCl3): δ 163.72, 160.47, 155.31, 152.26, 149.95 (C), 143.53 (CH), 141.41, 139.78 (C), 127.41, 127.29, 116.01, 115.77, 115.51, 115.23, 114.54, 114.42, 73.49 (CH), 67.60 (CH2), 63.67, 51.35 (CH), 35.89, 28.70 (CH2). 19F (282 MHz, CDCl3): δ
−114.89 (1 F, septet, J
= 4.3 Hz), −124.64 (1 F, septet, J
= 4.3 Hz). IR (cm−1): 3320, 2927, 1604, 1508, 1453, 1366, 1225, 1157, 1102, 1044, 910, 826, 733, 609. MALDI-MS: found, 421.1717 [MH − H2O]+. C25H23F2N2O2 requires 421.1728. Found, 438.1755 [M]+.
C25H24F2N2O3 requires 438.1755. Found, 439.1825 [MH]+. C25H25F2N2O3 requires 439.1833. Found, 461.1650 [MNa]+. C25H24F2N2O3Na requires 461.1653.
Acknowledgements
This research was supported by a KTI grant (6813.2 BTS-LS) and Lipideon AG. L. K. was supported by a fellowship from The Technical University of Denmark. T. R. thanks the Fonds der Chemischen Industrie for a Kekulé Fellowship.References
- S. B. Rosenblum, T. Huynh, A. Afonso, H. R. Davis, N. Yumibe, J. W. Clader and D. A. Burnett, J. Med. Chem., 1998, 41, 973–980 CrossRef CAS; J. W. Clader, J. Med. Chem., 2004, 47, 1–9 CrossRef CAS ; FDA Drug Approvals List [online]
http://www.fda.gov/cder/foi/label/2002/21445lbl.pdf.
- S. Dugar, M. P. Kirkup, J. W. Clader, S. I. Lin, R. Rizvi, M. E. Snow, H. R. Davis and S. W. McCombie, Bioorg. Med. Chem. Lett., 1995, 5, 2947–2952 CrossRef CAS.
- J. W. Clader, D. A. Burnett, M. A. Caplen, M. S. Domalski, S. Dugar, W. Vaccaro, R. Sher, M. E. Browne, H. R. Zhao, R. E. Burrier, B. Salisbury and H. R. Davis, J. Med. Chem., 1996, 39, 3684–3693 CrossRef CAS.
- For a detailed description of the development and use of the brush border membrane vesicle assay, see:
(a) L. Kværnø, T. Ritter, M. Werder, H. Hauser and E. M. Carreira, Angew. Chem. Int. Ed., 2004, 43, 4653–4656 CrossRef CAS;
(b) L. Kværnø, M. Werder, H. Hauser and E. M. Carreira, J. Med. Chem., 2005, 48 Search PubMed , in press.
- Ab initio calculations: initial energy-minimization by molecular mechanics using MacroModel v. 7.0 [pseudosystematical Monte Carlo conformational search (100 steps, limit 30 kJ mol−1) with CHCl3 as solvent, MMFFs force field] was followed by a quantum-mechanical geometry optimization using Jaguar v. 4.2 (B3LYP/6-31G*) in Maestro v. 4.1.
- T. Kambara and K. Tomioka, J. Org. Chem., 1999, 64, 9282–9285 CrossRef CAS.
- B. A. Shinkre, V. G. Puranik, B. M. Bhawal and A. Deshmukh, Tetrahedron: Asymmetry, 2003, 14, 453–459 CrossRef CAS.
- The 3∶2 diastereoselectivity induced by the chiral ephedrine-based auxiliary corresponds to those previously reported for similar substrates, see: S. V. Pansare, B. A. Shinkre and A. Bhattacharyya, Tetrahedron, 2002, 58, 8985–8991 Search PubMed; S. V. Pansare, R. G. Ravi and R. P. Jain, J. Org. Chem., 1998, 63, 4120–4124 CrossRef CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- R. E. Ireland and D. W. Norbeck, J. Org. Chem., 1985, 50, 2198–2200 CrossRef CAS.
- K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef CAS.
- R. K. Bansal, S. Mathur, J. K. Jain and D. Sharma, J. Indian Chem. Soc., 1988, 65, 134–136 CAS.
- Wittig reactions of aldehydes with similar stabilized ylides are normally carried out 130–150 °C warmer, in refluxing benzene or toluene. For examples, see: A. F. Kluge and C. P. Lillya, J. Org. Chem., 1971, 36, 1988–1995 Search PubMed; H. Toshima, S. Yoshida, T. Suzuki, S. Nishiyama and S. Yamamura, Tetrahedron Lett., 1989, 30, 6721–6724 CrossRef.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS; E. J. Corey, R. K. Bakshi, S. Shibata, C. P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925–7926 CrossRef CAS.
- S. Kanemasa, M. Nishiuchi, A. Kamimura and K. Hori, J. Am. Chem. Soc., 1994, 116, 2324–2339 CrossRef CAS; J. W. Bode, N. Fraefel, D. Muri and E. M. Carreira, Angew. Chem. Int. Ed., 2001, 40, 2082–2085 CrossRef CAS; J. W. Bode and E. M. Carreira, J. Am. Chem. Soc., 2001, 123, 3611–3612 CrossRef CAS; L. D. Fader and E. M. Carreira, Org. Lett., 2004, 6, 2485–2488 CrossRef CAS.
- H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092–2093 CrossRef CAS.
- R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5518–5526 CrossRef CAS.
- M. R. Mish, F. M. Guerra and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 8379–8380 CrossRef CAS; F. M. Guerra, M. R. Mish and E. M. Carreira, Org. Lett., 2000, 2, 4265–4267 CrossRef CAS; G. A. Whitlock and E. M. Carreira, J. Org. Chem., 1997, 62, 7916–7917 CrossRef CAS; H. Sasaki and E. M. Carreira, Synthesis, 2000, 135–138 CrossRef CAS.
- D. E. Frantz, R. Fässler and E. M. Carreira, J. Am. Chem. Soc., 2000, 122, 1806–1807 CrossRef CAS; D. E. Frantz, R. Fässler, C. S. Tomooka and E. M. Carreira, Acc. Chem. Res., 2000, 33, 373–381 CrossRef CAS; E. El-Sayed, N. K. Anand and E. M. Carreira, Org. Lett., 2001, 3, 3017–3020 CrossRef CAS; D. Boyall, D. E. Frantz and E. M. Carreira, Org. Lett., 2002, 4, 2605–2606 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- A. Melekhov and A. G. Fallis, Tetrahedron Lett., 1999, 40, 7867–7870 CrossRef CAS.
- M. Ishizaki, Y. Hara, S. Kojima and O. Hoshino, Heterocycles, 1999, 50, 779–790 CrossRef CAS.
- M. A. Blanchette, W. Choy, J. T. Davis, A. P. Essenfeld, S. Masamune, W. R. Roush and T. Sakai, Tetrahedron Lett., 1984, 25, 2183–2186 CrossRef CAS.
- J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS; J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852–860 CrossRef CAS; G. Mann, J. F. Hartwig, M. S. Driver and C. Fernandez-Rivas, J. Am. Chem. Soc., 1998, 120, 827–828 CrossRef CAS.
- D. M. T. Chan, K. L. Monaco, R. P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS; P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS.
- D. M. T. Chan, Tetrahedron Lett., 1996, 37, 9013–9016 CrossRef CAS.
- T. Hogberg, P. Strom, M. Ebner and S. Ramsby, J. Org. Chem., 1987, 52, 2033–2036 CrossRef.
- C. F. Hoyng, M. G. McKenna and D. L. Walters, Synthesis, 1982, 191–193 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434 CrossRef CAS.
- T. Sugimoto, J. Ishihara and A. Murai, Synlett, 1999, 541–544 CrossRef CAS; X. F. Ren, E. Turos, C. H. Lake and M. R. Churchill, J. Org. Chem., 1995, 60, 6468–6483 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.