“Click chemistry” en route to pseudo-starch
Laurence
Marmuse
a,
Sergey A.
Nepogodiev
*a and
Robert A.
Field
*ab
aCentre for Carbohydrate Chemistry, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: s.nepogodiev@uea.ac.uk
bDepartment of Biological Chemistry, John Innes Centre, Norwich, UK NR4 7UH. E-mail: r.a.field@uea.ac.uk
First published on 11th May 2005
Abstract
Rapid assembly of starch fragment analogues was achieved using “click chemistry”. Specifically, two hexadecasaccharide mimics containing two parallel maltoheptaosyl chains linked via [1,2,3]-triazoles to a maltose core were synthesized using Cu(I)-catalyzed [3 + 2] dipolar cycloaddition of azido saccharides and 6,6′- and 4′,6′-dipropargylated p-methoxyphenyl maltoside.
Detailed knowledge of structural features is essential for understanding the biosynthesis of starch components and their assembly into the starch granule. Through this understanding, rational alteration of the pathway of starch biosynthesis using genetic manipulation promises to deliver a new generation of novel raw materials.1 Starch is composed of two polysaccharides: an essentially linear α-(1→4)-D-glucan amylose and the highly branched macromolecule amylopectin, consisting of relatively short (1→4)-D-glucan chains attached through α-(1→6)-linkages to an α-(1→4)-D-glucan backbone.2 It is believed that local organization of these branched chains of amylopectin is responsible for the ordered semi-crystalline structure of the starch granule in which crystalline regions alternate with amorphous zones. The short branches of amylopectin form double helices that are stacked in the crystalline regions, whereas amorphous zones are occupied by amylopectin fragments incorporating (1→6)-branch points. To gain an insight into the role of such branch points in the propagation of double helices, computer modelling3,4 and NMR spectroscopic studies4 have been undertaken on amylopectin fragments. However, obtaining experimental information about these fragments is hampered by the fact that they represent only a small fragment of the total polysaccharide material. Therefore synthetic, well-defined fragments of amylopectin incorporating α-(1→6)-branch points would be useful tools for physicochemical and biochemical studies.
Synthesis of oligosaccharides related to starch have been reported in the literature,5 but poor stereoselectivity in the 1,2-cis-glucosylation reaction is known6 to be a serious obstacle in the assembly of the large branched fragments. To enforce interactions of two parallel oligosaccharide chains, they need to be attached to a template, as in the case of a cellulose II mimic developed by Vasella and co-workers.7 Application of the template concept to the construction of amylopectin fragment analogues requires development of a simple and efficient strategy for conjugation of long-chain maltooligosaccharides to a template. One of the reactions that can satisfy these requirements is Huisgen's 1,3-dipolar cycloaddition8 of azides and terminal acetylenes, yielding triazoles. The potential of this reaction has been recently enhanced by the discovery9 that Cu(I) catalyzes formation of a single regioisomer of substituted 1,2,3-triazoles, making this reaction one of the most powerful “click chemistry”10 transformations. In carbohydrate chemistry this methodology has been successfully applied for the synthesis of multivalent saccharides11 and cyclodextrin analogues.12
Continuing our efforts on generating synthetic amylopectin fragments6 we describe herein an approach to the construction of amylopectin analogues composed of two linear maltoheptaose chains attached to a maltose template through heterocyclic bridges. Two types of molecules, with attachment points at the 4′,6′ and 6,6′ positions of a maltose template, were selected as targets. The strategy for the introduction of a matching pair of reactive groups, suitable for 1,3-dipolar cycloaddition, requires the simple and efficient introduction of azide and alkyne groups into suitable building blocks. The branching template chosen was a dipropargylated maltose derivative, whereas linear chains containing an azido group in the reducing terminal anomeric positions comprised the cycloaddition partner.
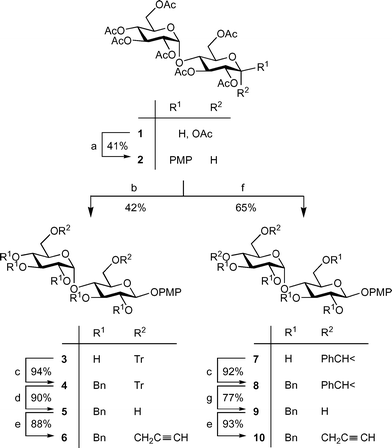
Isomeric di-O-propargyl derivatives 6 and 10 were synthesized starting from readily available13 maltose peracetate 1 (Scheme 1). Glycosidation of 1 with p-methoxyphenol in the presence of BF3·OEt2 gave an α,β mixture of aryl glycosides from which pure β-anomer 2 was isolated by crystallisation in 41% yield. Deacetylation of 2 followed by selective protection of primary OH groups via alkylation with TrCl in pyridine afforded 6,6′-di-O-trityl derivative 3 in 42% overall yield. For the synthesis of 4′,6′ di-O-propargyl maltoside 10, glycoside 2 was deacetylated and selectively benzylidenated to produce acetal 7 in 65% overall yield. After benzylation of the remaining hydroxy groups in 3 and 7, acid-labile temporary triphenylmethyl and benzylidene groups in 4 and 8 were removed to give diols 5 and 9 in 90% and 77% yield, respectively. Reactions of dialkoxides prepared in situ from diols 5 and 9 with propargyl bromide led to target di-O-propargyl maltosides 6 and 10 in 88% and 93% yield, respectively.
 | ||
Scheme 1 Reagents and conditions: (a) p-methoxyphenol, CH2Cl2, BF3·OEt2; (b) 1. MeOH, NaOMe 2. TrCl, pyridine; (c) BnBr, NaH; (d) TsOH, MeOH–CH2Cl2; (e) CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2Br, NaH, THF–DMPU; (f) 1. NaOMe, MeOH, 2. PhCH(OMe)2, TsOH, DMF; (g) 90% AcOH. CCH2Br, NaH, THF–DMPU; (f) 1. NaOMe, MeOH, 2. PhCH(OMe)2, TsOH, DMF; (g) 90% AcOH. | ||
A series of peracetylated β-glycosyl azides was synthesized by reaction14 of Me3SiN3 with glucosyl bromide 11, maltotriosyl bromide 12,15 and maltoheptaosyl bromide 1315 in the presence of Bu4NF. Thus glycosyl azides 14,1415, and 16 were prepared in 65–83% yield (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions: Me3SiN3, TBAF, THF. | ||
The results of 1,3-dipolar cycloaddition of dipropargylated maltosides and azidoglucosides are shown in Scheme 3. All reactions were carried out using (Ph3P)3·CuBr as a catalyst in the presence of DIPEA as a base as described previously,11a except that, instead of microwave irradiation, a longer reaction time (12 h) at room temperature was applied. The yields of cycloaddition reactions varied between 65 and 27%, decreasing when increasing length of the azidooligosaccharide chain.
 | ||
| Scheme 3 Synthesis of templated bis-glucopyranoside (17), bis-maltotrioside (18), and bis-maltoheptaosides (19 and 20). Reagents and conditions: (a) (Ph3P)3CuBr, DIPEA, toluene, 12 h, room temperature. | ||
The structure of cycloaddition products was confirmed by NMR spectroscopy and mass spectrometry. Both 1H and 13C NMR spectra of 17–20 revealed very close but distinguishable resonances (δHca. 5.7–5.9 and δCca. 85–86) corresponding to the anomeric center of the glucopyranose residues attached to N-1 of the triazole unit. From the rest of the considerably overlapping resonances of acetylated glucoparanose residues, only clusters corresponding to signals of α-anomeric (δCca. 95–96) and C-6 (δCca. 61.5–62) carbon atoms were reliably assignable. Characteristic signals of anomeric carbon atoms (δC-1ca. 102 and δC-1′ca. 97), as well as resonances corresponding to p-methoxyphenyl group (δOMeca. 55.5 and aromatics δca. 115 and δCca. 118) were observed in the 13C NMR spectra of compounds 17–20. The [1,2,3]-triazole unit was evident from 1H NMR spectra by the presence of two separate resonances (δHca. 7.7). Therefore, NMR data clearly indicated formation of a single isomer in each case, which for the copper(I)-catalyzed cycloaddition reaction is known to be the 1,4-substituted [1,2,3]-triazole.9b Regioselectivity of cycloaddition in the synthesis of 6,6′-di-substituted derivatives 17–19 also followed from the observation of only one pair of doublets of aromatic protons (δHca. 6.9 and δHca. 7.0) belonging to the anomeric p-methoxyphenyl group in the 1H NMR spectra. We noted previously6 that the chemical shifts of these signals are highly sensitive to the stereochemistry of a substituent at the 6 position of a p-methoxyphenyl β-maltoside unit. All triazole-bridged products were analyzed by MALDI-TOF MS, giving the expected sodium adducts of molecular ions: 1744.7 (17), 2897.1 (18), 5202.7 (19), and 5207.6 (20).
In summary, we have described the first application of “click chemistry” based on cycloaddition of substituted azide and alkynes to the synthesis of well-defined branched oligosaccharide mimics. Starting from dipropargylated maltoside and azido maltooligosaccharides this modular approach allowed the construction of a number of [1,2,3]-triazole-based analogues of amylopectin fragments in one simple coupling step. These analogues include two isomeric hexadecasaccharide analogues which have potential for templating formation of double helixes between two parallel maltoheptaosyl chains attached to a core maltose unit. Studies to investigate such assembly processes are ongoing.
Acknowledgements
We thank the EPSRC and the Weston Foundation for financial support. The EPSRC Mass Spectrometry Service Centre, Swansea are acknowledged for invaluable support.References
- (a) A. M. Smith, Curr. Opin. Plant Biol., 1999, 2, 223–229 CrossRef CAS; (b) C. J. Slattery, I. H. Kavakli and T. W. Okita, Trends Plant Sci., 2000, 5, 291–298 CrossRef CAS; (c) A. G. Heyer, J. R. Lloyd and J. Kossmann, Curr. Opin. Biotechnol., 1999, 10, 169–174 CrossRef CAS.
- (a) D. J. Gallant, B. Bouchet and P. M. Baldwin, Carbohydr. Polym., 1997, 32, 177–191 CrossRef CAS; (b) R. F. Tester, J. Karkalas, in Biopolymers: Polysaccharides II, vol. 6, Wiley-VCH, Weinheim, 2002, pp. 383–438 Search PubMed; (c) A. Buleon, P. Colonna, V. Planchot and S. Ball, Int. J. Biol. Macromol., 1998, 23, 85–112 CrossRef CAS; (d) A. Imberty, A. Buleon, V. Tran and S. Pérez, Starch/Staerke, 1991, 43, 375–384 Search PubMed.
- (a) A. Imberty and S. Pérez, Int. J. Biol. Macromol., 1989, 11, 177–185 CrossRef CAS; (b) A. Buleon and V. Tran, Int. J. Biol. Macromol., 1990, 12, 345–352 CrossRef CAS; (c) A. Neszmelyi and J. Hollo, Starch/Staerke, 1989, 41, 1–3 Search PubMed.
- F. Corzana, M. S. Motawia, C. Hervé du Penhoat, F. van den Berg, A. Blennow, S. Pérez and S. B. Engelsen, J. Am. Chem. Soc., 2004, 126, 13144–13155 CrossRef CAS.
- (a) M. S. Motawia, C. E. Olsen, K. Enevoldsen, J. Marcussen and B. L. Møller, Carbohydr. Res., 1995, 277, 109–123 CrossRef CAS; (b) I. Damager, C. E. Olsen, B. L. Møller and M. S. Motawia, Carbohydr. Res., 1999, 320, 19–30 CrossRef CAS; (c) M. S. Motawia, K. Larsen, C. E. Olsen and B. L. Møller, Synthesis, 2000, 11, 1547–1556 CrossRef; (d) I. Damager, C. E. Olsen, B. L. Møller and M. S. Motawia, Synthesis, 2002, 418–426 CrossRef CAS; (e) I. Damager, C. E. Olsen, A. Blennow, K. Denyer, B. L. Møller and M. S. Motawia, Carbohydr. Res., 2003, 338, 189–197 CrossRef CAS.
- L. Marmuse, S. A. Nepogodiev and R. A. Field, Tetrahedron: Asymmetry, 2005, 16, 477–485 CrossRef CAS.
- B. Bernet, J. W. Xu and A. Vasella, Helv. Chim. Acta, 2000, 83, 2072–2114 CrossRef CAS.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, vol. 1, ed. A. Padwa, Wiley, 1984, pp. 1–176 Search PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tørnoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS; (c) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS.
- C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- (a) F. Perez-Balderas, M. Ortega-Munoz, J. Morales-Sanfrutos, F. Hernández-Mateo, F. G. Calvo-Flores, J. A. Calvo-Asin, J. Isac-García and F. Santoyo-González, Org. Lett., 2003, 5, 1951–1954 CrossRef CAS; (b) S. Chittaboina, F. Xie and Q. Wang, Tetrahedron Lett., 2005, 46, 2331–2336 CrossRef CAS.
- (a) K. D. Bodine, Y. Gin and M. S. Gin, J. Am. Chem. Soc., 2004, 126, 1638–1639 CrossRef CAS; (b) B. Hoffmann, B. Bernet and A. Vasella, Helv. Chim. Acta, 2002, 85, 265–287 CrossRef CAS.
- K. P. R. Kartha, M. Aloui and R. A. Field, Tetrahedron Lett., 1996, 37, 8807–8810 CrossRef.
- E. D. Soli, A. S. Manoso, M. C. Patterson, P. DeShong, D. A. Favor, R. Hirschmann and A. B. Smith, III, J. Org. Chem., 1999, 64, 3171–3177 CrossRef CAS.
- E. Farkas, L. Jánossy, J. Harangi, L. Kandra and A. Lipták, Carbohydr. Res., 1997, 303, 407–415 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |