Recent experimental and theoretical developments towards the observation of parity violation (PV) effects in molecules by spectroscopy
Jeanne
Crassous
*a,
Christian
Chardonnet
b,
Trond
Saue
c and
Peter
Schwerdtfeger
d
aLaboratoire de Chimie, École Normale Supérieure de Lyon, UMR CNRS 5182, 46, Allée d'Italie, F-69364, Lyon 07, France. E-mail: jeanne.crassous@ens-lyon.fr; Fax: +33 04 72 72 84 83; Tel: +33 04 72 72 83 95
bLaboratoire de Physique des Lasers, UMR 7538 CNRS–Université Paris 13, 99, avenue J.-B. Clément, 93430, Villetaneuse, France
cLaboratoire de Chimie Quantique et Modélisation Moléculaire, UMR 7551 CNRS-Université Louis Pasteur, 4, rue Blaise Pascal, F-67000, Strasbourg, France
dTheoretical Chemistry, Bldg. 44, Institute of Fundamental Sciences, Massey University (Albany Campus), Private Bag 102904, North Shore MSC, Auckland, New Zealand
First published on 4th May 2005
Abstract
Parity violation (PV) at the molecular level is known to be responsible for a tiny energy difference between the two enantiomers of a chiral molecule. This parity violation energy difference (PVED) has not yet been detected by experiment. In the last few years, the search for PV effects in molecules has made important steps ahead for several reasons. On one hand, very accurate infra-red spectroscopy measurements were performed by metrologists on bromochlorofluoromethane (CHFClBr) with a 10 Hz accuracy, which so far is the most precise. On the other hand, relativistic calculations were used for the evaluation of ΔEPV allowing for a screening of favorable molecules for future measurements. The synthesis of such chiral molecules with high parity violation effects is currently being investigated. In memory of Professor Jean-Bernard Robert.
 Jeanne Crassous Jeanne Crassous | Jeanne Crassous was born in Jarny, France, in 1968. She studied chemistry at the École Normale Supérieure de Lyon, and received her PhD in 1996 under the guidance of Prof. André Collet (ENS Lyon), working on the absolute configuration of bromochlorofluoromethane. After a one-year post-doctoral period studying the chirality of fullerenes in Prof. François Diederich's group (ETH Zurich), she received a CNRS research position (CR1). She is currently interested in the preparation of enantioenriched molecules for their chirality related properties. |
 Christian Chardonnet Christian Chardonnet | Christian Chardonnet is a former student of the École Normale Supérieure of Paris. He is now a research director at CNRS, team leader and former director of Laboratoire de Physique des Lasers-UMR CNRS Université Paris 13-Villetaneuse (France). His research interests are ultra-high resolution molecular spectroscopy and frequency metrology. |
 Trond Saue Trond Saue | Trond Saue was born in Harstad, Norway, in 1964. He received his PhD in theoretical chemistry at the University of Oslo, Norway, in 1996. After postdoctoral positions in Denmark and France as well as an assistant professorship at the university of Tromsø, Norway, he became CNRS assistant professor (CR1) at the Laboratoire de Chimie Quantique et de Modélisation Moléculaire at the Université Louis Pasteur in Strasbourg, France, in 2001. His research is centered on four-component molecular relativistic calculations, in later years with particular emphasis on density functional theory and molecular properties. He has published about 30 research papers and is a main author of the program system Dirac (http://dirac.chem.sdu.dk/). |
 Peter Schwerdtfeger Peter Schwerdtfeger | Peter Schwerdtfeger received his PhD from Stuttgart University in 1986. He became a Feodor-Lynen Alexander von Humboldt Fellow in 1987 at the University of Auckland, and a research fellow at the Australian National University in 1989. In 1991 he took up a lectureship at the University of Auckland, received his habilitation and venia legendi from the Philipps-University in Marburg in 1995, and in 1999 was awarded a personal chair at Auckland University. In 2001 he received a James Cook Fellowship. Since March 2004 he has held the chair in theoretical chemistry at Massey University in Auckland. His current research interests are in all aspects of electronic structure theory and relativistic quantum theory. He has been honoured with the Prince and Princess of Wales Science Award (1988), the SGS Prize (1994), the Hector Medal (2001), and is a Fellow of the Royal Society of New Zealand. |
Introduction
Chirality is commonly regarded as the property of geometrical objects that cannot be brought to coincide with their mirror-image. From the stereochemical point of view, chirality is the property of those stereomers that are called enantiomers. Note that in this context space reflection and space inversion are equivalent since the two symmetry operations differ by a π rotation about an axis perpendicular to the mirror plane, which does not change the handedness of the molecule. Although the existence of stable enantiomers at first sight seems trivial, this is not so from a quantum mechanical point of view. The standard Hamiltonian which describes the electromagnetic interactions between the constituent particles of a molecule is invariant under space inversion, and enantiomers cannot therefore correspond to eigenstates of this Hamiltonian since they do not have well-defined parity. This problem was addressed by Hund as early as 19271 who concluded that once a particular enantiomer has been prepared the barrier to interconversion typically gives tunneling times of million of years, in other words, the enantiomer is kinetically stable. However, this argument does not rule out the possibility of preparing a superposition of single molecule enantiomers, something which has never been observed by experiment so far.2 Interactions with the environment would subsequently lead to localization to a particular enantiomer and one would have to carry out experiments on the isolated molecule at very low temperatures. However, even under such conditions localization, or rather the oscillation of handedness, would occur with the introduction of parity non-conservation in the Hamiltonian. The total Hamiltonian H then consists of a parity even (H0) and odd (HPV) part,| H = H0 + HPV | (1) |
| P−1HP = H0 − HPV = H − 2 HPV ≠ H | (2) |
| ΔEPV = EPV,L − EPV,R = 2 EPV | (3) |
Among the four fundamental forces that rule the physical world, only the weak force4 does not conserve parity. Although parity violation has been observed in nuclear5 and atomic physics,6 it has never been unequivocally observed at the molecular level to date. Such a tiny energy difference (about 10−17kT at 300 K) should be measurable by any absorption spectroscopy provided a high resolution is reached.7 Several unsuccessful attempts have been made; for instance Mössbauer spectroscopy has been conducted on an iron complex,8 and circular dichroism was measured in recrystallized transition metal complexes.9 NMR has also been suggested as a possible technique to measure parity violation.10 Interestingly, infra-red (IR) spectroscopy has been the focus of several groups, following an original idea of Letokhov.11 In 1977, Glorieux et al. tried to measure differences in absorption energies of camphor enantiomers.12 The resolution reached (300 kHz) was too low to evidence PV effects (see below). Quack has proposed several types of spectroscopic experiments, among which are the measurement of time-dependent optical activity in molecules displaying tunneling splitting and PVED of the same magnitude and a direct measurement of ΔEPV using an intermediate spectroscopic level such as an excited achiral electronic state.13
In this paper we will provide a case study in the form of our joint efforts to detect PVED by infra-red spectroscopy. To evidence such tiny effects as PV in molecules, much care has to be taken to control and to improve the sensitivity and the resolution of the spectroscopy technique used. An experiment reaching a resolution of 10−13 for the relative frequency difference (ΔνPV/ν) has been carried out on bromochlorofluoromethane (CHFClBr) in Chardonnet’s laboratory and is described below. Preparation of enantioenriched molecules which are particularly favorable for PV observation is also most challenging. Chiral synthesis is currently being carried out by Crassous’s team and is also described in this paper. Finally, concomitant theoretical calculations (conducted at the relativistic level by Saue and Schwerdtfeger) aim at a better understanding of the origin of PV in molecules and guide the choice of chiral molecules for future experiments.
Theoretical considerations
Any measurement of PV effects between enantiomers of a chiral molecule requires strict verification by theory. Hence, a direct measurement of PV energy differences between enantiomers offers the best chance of both experimental and theoretical verification. Other suggested experiments like direct polymerization, condensation and crystallization14 will suffer from the lack of theoretical justification for such complex processes. Let us therefore briefly summarize some important theoretical aspects of the electroweak interaction between charged particles important for chiral molecular systems. A more detailed theoretical description has been given recently by Berger.15In quantum field theory electromagnetic interactions are viewed as an exchange of virtual photons. In molecular systems the weak interaction is dominated by the exchange of virtual Z0 bosons between electrons and nucleons. The corresponding Hamiltonian is expressed as16,17
 | (4) |
The nuclear spin dependent term H(2)PV shown in eqn. (4) is the dominant operator for Mössbauer and nuclear magnetic resonance transitions.18,10c The first term H(1)PV is the dominant operator for electronic or vibrational transitions and has been used in most theoretical investigations of PV effects. At the relativistic Hartree–Fock level the parity non-conserving energy shift EPV is obtained to first order as an expectation value.19
 | (5) |
It is currently a nontrivial issue to find the right compound for molecular PV measurements.21 In Quack's influential review article from 1989 on structure and dynamics of chiral molecules22 a number of chiral species was suggested which deserve further experimental and theoretical study. Most of the current theoretical work concentrates on vibrational transitions, although we mention here theoretical work on Mössbauer,8 NMR,10 and electronic spectroscopy.23 Molecular beam experiments using a high precision tuneable CO2 laser currently offer the best resolution for PV measurements. In fact, one of the first attempts to find PV differences in transition frequencies was made by Arimondo et al. in 1977 using the 1083.4788 cm−1 R(28) CO2 laser line for D- and L-camphor,12 which lies in the C–C*–CO bending mode range (C* denotes a chiral carbon). They concluded that PV effects in camphor must be below 300 kHz. A recent relativistic theoretical study shows that PV effects in camphor are probably below 10−5 Hz.24 It is therefore of no surprise that Rein stated in 1975 that the direct observation of energy differences between optical isomers is beyond the scope of present experimental facilities25 (and still it is). However, parity violation effects scale as Zn (n ≈ 5 for H(1)PV and n ≈ 3 for H(2)PV, Z is the nuclear charge),26 as shown in Fig. 1 in the case of H2X2 molecules for EPV (see eqn.(5)) and for the NMR frequency shift. This high Z-scaling behaviour implies that one has to introduce one or more heavy elements into the chiral molecular system, either as a central atom or as a ligand (or both because of the single-center theorem27), in order to reach the current experimental limit of around 1 Hz. The CO2 laser operates in the C–F vibrational frequency range and recent theoretical and experimental work therefore concentrated on chiral methane fluoride derivatives.
 | ||
| Fig. 1 Z n scaling behaviour for the Group 16 H2X2 systems for the PV energy contribution ΔEPV (in kJ mol−1) and for the PV line splitting ΔνPV (in Hz) where B0 (in T) is the static magnetic flux density of the NMR spectrometer. Data were taken from refs. 10c–20. | ||
Theoretical work on CHFClBr from three different research groups showed that the parity violation energy contribution EPV is around 0.03 Hz39 at the Hartree–Fock level of theory (1 au = 6.5797 × 1015 Hz = 2625.5 kJ mol−1), while a simple one-dimensional approach along the C–F stretching normal mode gave a PV difference ΔνRL = vPV,R − vPV,L of −1.7 mHz28 for the fundamental transitions (vPV,R and vPV,L defined in an analogous way to eqn. (3)). A few important facts should be mentioned however. First, anharmonicity effects in the C–F stretching mode cannot be neglected, including only harmonic effects gives the wrong sign for vPV,R or vPV,L.39a,29 Second, coupling with other modes can substantially alter ΔvRL as demonstrated in Quack's group.30 In fact, for CDFClBr a substantial enhancement in ΔvRL is predicted. This implies that future high precision theoretical work has to include possibly all nine normal modes in the vibrational analysis. Third, electron correlation is important for the determination of EPV contrary to what has been believed so far, and various density functionals give quite different results leading even to a change of sign for EPV.31 However, ΔvRL values seem to be typically an order or two in magnitude smaller than absolute energy differences and do not vary substantially between different methods.
Other potential candidates for vibrational PV measurements are CHFClI, CHFBrI and CFClBrI.19 Out of this series CHFBrI seems to be the most promising molecule with a calculated four-component relativistic Hartree–Fock value of ΔvRL = −50.8 mHz for the C–F stretching fundamental transition.28 Future investigations will need to improve this value using a multi-mode analysis and EPV calculated at the correlated level of theory for both CHFBrI and CDFBrI.
Faglioni and Lazzeretti recently considered BiHFBr and BiHFI.32 These systems are non-planar with a predicted high inversion barrier and are therefore chiral. The PV frequency shift ΔvRL is very high and possibly up to about 20 Hz, but these species are thermodynamically unstable and one needs to consider the first overtone of the H–Bi–X bending mode in order to reach the CO2 laser frequency range. On a similar basis, EPV values were reported for all Group 14 compounds EHFClBr (E = C, Si, Ge, Sn and Pb).39a However, these compounds become less thermodynamically stable with increasing nuclear charge on the central atom, and the important fundamental modes lie outside the CO2 laser frequency range.
A number of other compounds with relatively high parity violation energy differences were suggested in the past, namely Ge(CF3)ClBrI, Os(η5-C5H5)(CHPh)Cl(PiPr3), Re(η5-Cp*)(O)(CR3)Cl (Cp* = C5(CH3)5), (η5-C5H5)Re(CO)(NO)I, ClHgCHFBr, PR3AuCHFClBr and BiCH3I(κ2-CH2(CH)2NH2),33–34,35 some of them are known in the literature. The calculated Hartree–Fock EPV reported were in the 100 Hz region, but it is not clear how electron correlation might change these values. Nevertheless, for two model compounds, Os(η5-C5H5)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCl2)Cl(PH3) and Re(η5-Cp*)(O)(CH3)Cl the vibrational spectra have been calculated (Fig. 2), with the OsCCl2 and the ReO stretching modes both in the CO2 laser frequency range, and a calculated ΔvRL value of around 1 Hz.35 It is however questionable if these molecules are suitable for high precision vibrational spectroscopy.
CCl2)Cl(PH3) and Re(η5-Cp*)(O)(CH3)Cl the vibrational spectra have been calculated (Fig. 2), with the OsCCl2 and the ReO stretching modes both in the CO2 laser frequency range, and a calculated ΔvRL value of around 1 Hz.35 It is however questionable if these molecules are suitable for high precision vibrational spectroscopy.
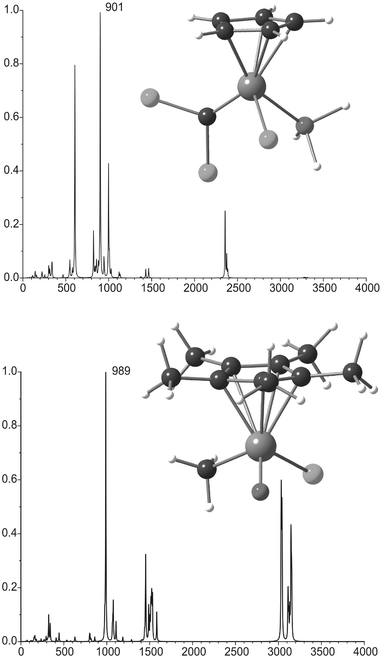 | ||
| Fig. 2 Predicted vibrational spectra for the model compounds Os(η5–C5H5)(CCl2)Cl(PH3) and Re(η5–Cp*)(O)(CH3)Cl. The most intense lines indicated belong to the OsC and ReO stretching modes respectively. Data taken from ref. 35. | ||
Accurate IR spectroscopy of bromochlorofluoromethane enantiomers: first set of experiments
In order to reach the best resolution to be able to observe parity violation in molecules, saturation spectroscopy (which suppresses the Doppler broadening, reducing the molecular linewidth from 50 MHz down to 10 kHz) together with a home-made ultrastable CO2 laser were used. These experiments were first performed on S-(+) and R-(−)-CHFClBr (1) having enantiomeric excesses (ee's) of 56.5 and 22%, respectively.36 Twin 3 m long Fabry-Perot cavities were each filled with one of the two enantiomers of CHFClBr and fed with the frequency-stabilized CO2 laser. One hyperfine component of the ν4 C–F stretching fundamental band of CHF37Cl81Br was selected and differences in the frequency absorption of this transition for the two enantiomers placed in the two cavities were searched for. Note that it is essential to have the enantiomers available because we perform the spectroscopy on the two species separately and the sensitivity of the comparison is limited by the precision with which we measure the line center and not by the resolution of the experiment which would be the case with a racemic mixture, the typical gain in sensitivity is about four orders of magnitude. Over 580 measurements a mean difference of 9.4 Hz was obtained, with a statistical uncertainty of 5.1 Hz and residual systematic uncertainties due to the instrumentation of 12.7 Hz,37νexp (R-(−)-1)–νexp (S-(+)-1) = 9.4 ± 5.1 ± 12.7 Hz. Consequently, no PV effect could be observed at a relative sensitivity of Δν/ν = 3.9 10−13, but an improvement of five orders of magnitude was achieved compared to the experiment done on camphor.12In another improved experimental set up, a stronger and narrower CHFClBr test transition was selected,38 and samples having higher ee's for (+) and (−)-1 (72% and 56%, respectively) were used. Over 771 measurements the final result obtained for the average frequency difference between the lines of the two samples of enantiomers was νexp (R-(−)-1)–νexp (S-(+)-1) = − 4.2 ± 0.6 ± 1.6 Hz (including statistical and systematic uncertainties due to the instrumentation), which corresponds to a relative sensitivity of Δν/ν = 4 × 10−14 for the frequency difference (the absolute frequency itself was also precisely measured: ν = 32 397 293 834.1 ± 3.0 kHz). The latter set of experiments also revealed that the significant frequency difference was proportional to pressure and cannot be a signature of a PV effect. This is probably due to uncontrollable residual gases present in the cells at a level below 5% for a typical pressure of 0.05–0.1 Pa. In parallel, quantum chemistry calculations on the expected vibrational frequency shift in the case of CHFClBr39 have concluded that the PV effect is three orders of magnitude smaller than the experimentally reached sensitivity.
Synthesis of chiral molecules for the observation of PV
As mentioned above, the first candidates for the observation of PV by the use of ultrastable CO2 were simple chiral molecules containing one asymmetric carbon and one C–F bond, displaying a C–F stretching mode around 1000 cm−1, namely bromochlorofluoromethane (CHFClBr) 1 and chlorofluoroiodomethane (CHFClI) 2. The synthesis of enantioenriched forms of such compounds that are devoid of chemical function requires original synthetic methods. In these two cases a decarboxylation reaction in a protic solvent of an appropriate diastereomerically enriched strychninium salt40 was used. In this way, it was possible to provide physicists with samples of (+)-1 and (−)-1 with respective ee's between 56–72% and 22–56% for PVED tests. Today it is possible to prepare (+) and (−)-1 with ee's up to 89% and 56% respectively by decarboxylation in ethyleneglycol at 120 °C of diastereomerically enriched salts {(+)-FClBrC–CO2H, (−)-strychnine} and {(−)-FClBrC–CO2H, (−)-strychnine}.41 Similarly (+)-2 and (−)-2 with ee's up to 63.3% and 23% respectively42 were prepared by decarboxylation of diastereomerically enriched salts {(+)-FClIC–CO2H, (−)-strychnine} and {(−)-FClIC–CO2H, (−)-strychnine}, in triethyleneglycol at 110 °C and under reduced pressure (see Scheme 1). Such reaction is known to take place with retention of configuration.43 Future work will also consider the deuterated species of 1 and 2. | ||
| Scheme 1 Synthesis of enantioenriched samples of S-(+)-2 and R-(−)-2 by decarboxylation of diastereomerically resolved strychninium salts. The ee's were measured by gas chromatography on a chiral stationary phase and by NMR of a complexation process. | ||
The enantiomeric purities of 1 and 2 could be obtained by two methods. A first method used low temperature analytical gas-chromatographic separation on an immobilized chiral stationary phase based on octakis(3-O-butanoyl-2,6-di-O-n-pentyl)-γ-cyclodextrin.42,44 In a second method a host–guest complexation process with enantioenriched chiral cryptophane hosts was used. For instance, (−)-cryptophane-C ((−)-3) enabled measurement of the enantiomeric purities of (+)-1 and (−)-1 samples.41,45 Indeed, diastereomeric host–guest complexes {(+)-1 @ (−)-cryptophane-C} and {(−)-1 @ (−)-cryptophane-C} could be visualized by 1H NMR in CDCl3 at 300 K. A thiomethylated cryptophane, namely (−)-cryptophane-E-(SCH3)6 ((−)-4),46 was used in the case of (−)-2 and gave an ee of 23 ± 3%. In this case, the diastereomeric host–guest complexes {(+)-1 @ (−)-cryptophane-E-(SCH3)6} and {(−)-1 @ (−)-cryptophane-E-(SCH3)6} were visualized by 19F NMR in C2D2Cl4 at 300 K (see Fig. 3). The question of the absolute configuration has been addressed by Raman optical activity (ROA) and molecular dynamics simulations for R-(−) and S-(+)-147, and more recently by optical rotation calculation42 and vibrational circular dichroism (VCD) for R-(−) and S-(+)-2.48
 | ||
| Fig. 3 Ee determination of (−)-2 by 19F NMR in C2D2Cl4 at 300 K using cryptophane (−)-4 as a chiral complexing agent. Two doublets corresponding to diastereomeric host–guest complexes {(+)-2 @ (−)-4} and {(−)-2 @ (−)-4}. | ||
As explained above, organometallic complexes containing heavier atoms display much higher PV effects. For this reason, we are now focused on the synthesis of chiral–at–metal complexes. These are, for example, oxorhenium complexes bearing chiral ligands which transfer their chirality to the metal atom. As mentioned above, the presence of the ReO bond is very important because its stretching mode at 900–1000 cm−1 can be brought to coincidence with the CO2 absorption lines. Compounds of this type have recently been described by Faller et al.49 Several grams of oxorhenium complexes TpReO(ephedrine) and TpReO(proline) (Tp = hydrotrispyrazolylborate) can be obtained in a few steps.
A complete determination of spectroscopic (rotational and rovibrational) constants of the racemic molecule is necessary because it enables one to identify a few rovibrational bands which could be favorable for a PV test. In this context, a supersonic molecular beam could easily be obtained with CHFClI, and microwave50 and rovibrational51 spectroscopy could be studied. In comparison, nothing is known about oxorhenium complexes and much effort will be made in the following months to investigate the rotational and vibrational spectroscopy of these chiral rhenium complexes for the purpose of a PV test based on the new experimental set-up described in the following section.
A new experimental set-up
Although more favorable molecules for the PV effect are very promising, the experimental set-up based on saturation spectroscopy in absorption cells already shows its limits. Molecular beam spectroscopy using a two-photon Ramsey fringe experiment, recently developed in Villetaneuse, seems to be very promising mainly because the collisional effects can be reduced by more than three orders of magnitude. The principle of this experiment is to generate a supersonic beam of chiral molecules which expands in a high-vacuum chamber (residual pressure below 10−4 Pa). This beam will interact with two standing waves generated in a four-mirror Fabry-Perot cavity. A Doppler-free two-photon transition between v = 0 and v = 2 vibrational levels occurs in each standing wave and an interference signal between the two excitation channels reveals Ramsey fringes with a resolution proportional to the inverse of the molecular time of flight between the two zones. With a supersonic beam of SF6 and a distance between zones of 1 m, a resolution of 100 Hz was obtained52 (see Fig. 4) which is three orders of magnitude higher than with the PV experiment in cells. The absolute frequency was measured very precisely at 28 412 764 347 322.1 ± 2.7 Hz. In a differential experiment that we plan to do, the uncertainty should be much lower since a lot of the systematic effects cancel out. The idea is to adapt this set-up for a new PV experiment: we will use a unique beam machine and feed it alternatively with the two enantiomers with a cycling time of 1–10 s to record separately the two spectra and directly compare the line centers. One essential advantage will be that the laser–molecule interaction conditions will be exactly the same. We can expect a sensitivity of 0.01 Hz, which should be enough to detect unambiguously the PV effect with the new generation of molecules. However, one must say that this expectation will be highly dependent on the quality of the signal that we obtain. This is related to the capability of feeding the beam with molecules which are solid at room temperature. Heating or laser techniques exist to sublimate molecules but must be checked on each particular case. | ||
| Fig. 4 Simplified block diagram of the two photon Ramsey fringes experiment with a supersonic beam of SF6. A first CO2 laser locked to the saturation signal of OsO4 is used as a local oscillator. A second laser is phase-locked to the first one and can be tuned with a RF synthesizer (not shown) through the Ramsey fringes. The detection of the population in the v = 2 level is performed by shifting the laser frequency on another optical path to reach the resonance with the transition v = 2→v = 1. The spectrum with a 100 Hz resolution and a signal to noise ratio of 45 over 1 s is displayed. | ||
Conclusion
In this paper we have described our attempts to observe parity violation in molecular systems using very accurate infra-red spectroscopy based on a CO2 laser. For this purpose, simple chiral molecules such as CHFClBr and CHFClI have been prepared in enantiomerically enriched forms and very accurate IR spectroscopy has been conducted on bromochlorofluoromethane enantiomers, improving the sensitivity by five orders of magnitude compared to a former test conducted on camphor. In parallel, highly accurate relativistic calculations have been done on the chiral halogenomethanes, but also on more favourable molecules such as transition metal complexes.In summary, we have shown the interplay between theory and experiment. Quantum chemical calculations serve to guide and validate experiment and may provide a deeper understanding of the effect of the molecular environment. The synthesis of candidate molecules in enantiomerically enriched forms is a non-trivial task. The detection of PV effects in molecules pushes spectroscopic techniques to their limits. Similarly, high precision calculations are needed to confirm these experiments.
If successful, these experiments allow tests of the standard model of the universe in the low-energy regime. They may answer important questions regarding the stability and reactivity of chiral molecules. Although there are a number of papers proposing that biomolecular chirality is, or may be, a consequence of PV,1,7b,53–60 there is currently, in our opinion, no proof that such a connection exists.7,61–66 It may, furthermore, be difficult to establish such a connection.67,68 For a nice recent review on the origin of homochirality in nature see Frank, Bonner and Zare.69
Acknowledgements
Radovan Bast is warmly thanked for providing us with a cover picture.References
- F. Hund, Z. Phys., 1927, 43, 805–826 CAS.
- J. A. Cina and R. A. Harris, J. Chem. Phys., 1994, 100, 2531–2536 CrossRef CAS.
- Y. Yamagata, J. Theor. Biol., 1966, 11, 495–498 CrossRef CAS.
- D. Lee and C. N. Yang, Phys. Rev., 1956, 104, 254–258 CrossRef CAS.
- C. S. Wu, E. Ambler, R. W. Hayward, D. D. Hoppes and R. P. Hudson, Phys. Rev., 1957, 105, 1413–1415 CrossRef CAS.
- (a) M. A. Bouchiat and C. C. Bouchiat, Phys. Lett. B, 1974, 48, 111–114 CrossRef CAS; (b) L. M. Barkov and M. S. Zolotorev, JETP, 1980, 52, 360–376; (c) M. A. Bouchiat and C. C. Bouchiat, Rep. Prog. Phys., 1997, 60, 1351–1396 CrossRef CAS; (d) C. S. Wood, S. C. Bennett, D. Cho, B. P. Masterson, J. L. Roberts, C. E. Tanner and C. E. Wieman, Science, 1997, 275(5307), 1759–1763 CrossRef CAS.
- Following the Salam hypothesis, calorimetry has also been used to attempt to detect PV in phase transitions in amino acid crystals. See: (a) A. Salam, in Chemical Evolution: Origin of Life, ed. C. Ponnamperuma and J. Chela-Flores, A Deepak Publishing, Hampton, VA, 1993, pp. 101–107 Search PubMed; (b) W. Wang, F. Yi, Y. Ni, Z. Zhao, X. Jin and Y. Tang, J. Biol. Phys., 2000, 26, 51–65 CrossRef CAS; (c) R. Sullivan, M. Pyda, J. Pak, B. Wunderlich, J. Thompson, R. Pagni, H. Pan, C. Barnes, P. Schwerdtfeger and R. Compton, J. Phys. Chem. A, 2003, 107, 6674–6680 CrossRef CAS.
- A. S. Lahamer, S. M. Mahurin, R. N. Compton, D. House, J. K. Laerdahl, M. Lein and P. Schwerdtfeger, Phys. Rev. Lett., 2000, 85, 4470–4473 CrossRef CAS.
- (a) A. Szabo-Nagy and L. Keszthelyi, Proc. Natl. Acad. Sci. USA, 1999, 96, 4252–4255 CrossRef CAS; (b) A. Szabo-Nagy, and L. Keszthelyi, in Advances in Biochirality, ed. P. Gyula, C. Zucchi and L. Caglioti, Elsevier, Amsterdam, 2002, pp. 367–376 Search PubMed.
- (a) A. L. Barra, J. B. Robert and L. Wiesenfeld, Phys. Lett. A, 1986, 115, 443–447 CrossRef CAS; (b) A. L. Barra and J. B. Robert, Mol. Phys., 1996, 88, 875–886 CrossRef CAS; (c) G. Laubender and R. Berger, ChemPhysChem., 2003, 4, 395–399 CrossRef CAS.
- (a) V. S. Letokhov, Phys. Lett., 1975, 53A, 275–277 Search PubMed; (b) O. N. Kompanets, A. R. Kukudzhanov, V. S. Letokhov and L. L. Gervits, Opt. Commun., 1976, 19, 414–416 CrossRef CAS.
- E. Arimondo, P. Glorieux and T. Oka, Opt. Commun., 1977, 23, 369–372 CrossRef CAS.
- M. Quack, Angew. Chem. Int. Ed., 2002, 41, 4618–4630 CrossRef CAS , and references therein.
- (a) L. Keszthelyi, J. Biol. Phys., 1994, 20, 241–245 CAS; (b) L. Keszthelyi, Mendeleev Commun., 2003, 13, 129–130 CrossRef; (c) L. Keszthelyi, Orig. Life Evol. Biosphere, 2001, 31, 249–256 CrossRef CAS.
- R. Berger, in Relativistic Electronic Structure Theory. Part 2: Applications, ed. P. Schwerdtfeger, Elsevier, Amsterdam, 2004, pp. 188-288 Search PubMed.
- S. A. Blundell, J. Sapirstein and W. R. Johnson, Phys. Rev. D, 1992, 45, 1602–1623 CrossRef CAS.
- J. Sapirstein, in Relativistic Electronic Structure Theory. Part 1: Fundamentals, ed. P. Schwerdtfeger, Elsevier, Amsterdam, 2002, pp. 468-522 Search PubMed.
- A. Soncini, F. Faglioni and P. Lazzeretti, Phys. Rev. A, 2003, 68, 33402 CrossRef.
- J. K. Laerdahl and P. Schwerdtfeger, Phys. Rev. A, 1999, 60, 4439–4453 CrossRef CAS.
- J. N. P. van Stralen, L. Visscher, C. V. Larsen and H. J. Aa. Jensen, Chem. Phys., 2005, 311, 81–95 CrossRef CAS.
- F. Faglioni and P. Lazzeretti, Phys. Rev. E, 2002, 65, 11904 CrossRef.
- M. Quack, Angew. Chem. Int. Ed. Engl., 1989, 28, 571–586 CrossRef.
- R. Berger, Phys. Chem. Chem. Phys., 2003, 5, 12–17 RSC.
- P. Schwerdtfeger, A. Kühn, R. Bast, J. K. Laerdahl, F. Faglioni and P. Lazzeretti, Chem. Phys. Lett., 2004, 383, 496–501 CrossRef CAS.
- D. W. Rein, J. Mol. Evol., 1974, 4, 15–22 CAS.
- M. A. Bouchiat and C. Bouchiat, J. Phys. (Paris), 1974, 35, 899 CAS; M. A. Bouchiat and C. Bouchiat, J. Phys. (Paris), 1974, 36, 493.
- R. A. Hegstrom, D. W. Rein and P. G. H. Sandars, J. Chem. Phys., 1980, 73, 2329–2341 CrossRef CAS.
- P. Schwerdtfeger, J. K. Laerdahl and Ch. Chardonnet, Phys. Rev. A, 2002, 65, 42508 CrossRef.
- M. Quack and J. Stohner, Chirality, 2001, 13, 745–753 CrossRef CAS.
- M. Quack and J. Stohner, J. Chem. Phys., 2003, 119, 11228–11240 CrossRef CAS.
- P. Schwerdtfeger, T. Saue, J. N. P. van Stralen and L. Visscher, Phys. Rev. A, 2005, 71, 12103 CrossRef.
- F. Faglioni and P. Lazzeretti, Phys. Rev. A, 2003, 67, 32101 CrossRef.
- P. Schwerdtfeger, J. Gierlich and T. Bollwein, Angew. Chem. Int. Ed., 2003, 42, 1293–1296 CrossRef CAS.
- R. Bast and P. Schwerdtfeger, Phys. Rev. Lett., 2003, 91, 23001 CrossRef.
- P. Schwerdtfeger and R. Bast, J. Am. Chem. Soc., 2004, 126, 1652–1653 CrossRef CAS.
- J. Crassous and A. Collet, Enantiomer, 2000, 5, 429–438 Search PubMed.
- (a) C. Daussy, T. Marrel, A. Amy-Klein, C. T. Nguyen, C. J. Bordé and C. Chardonnet, Phys. Rev. Lett., 1999, 83, 1554–1557 CrossRef CAS; (b) C. Chardonnet, C. Daussy, T. Marrel, A. Amy-Klein, C. T. Nguyen and C. J. Bordé, in Parity Violation in Atomic Physics and Electron Scattering, ed. B. Frois and M. A. Bouchiat, World Scientific, New-York, 1999, 325-355 Search PubMed; (c) C. Chardonnet, T. Marrel, M. Ziskind, C. Daussy, A. Amy-Klein and C. J. Bordé, J. Phys. IV, 2000, 10-Pr8, 45–54 Search PubMed.
- M. Ziskind, T. Marrel, C. Daussy and C. Chardonnet, Eur. Phys. J. D, 2002, 20, 219–225 Search PubMed.
- (a) J. K. Laerdahl, P. Schwerdtfeger and H. M. Quiney, Phys. Rev. Lett., 2000, 84, 3811–3814 CrossRef CAS; (b) M. Quack and J. Stohner, Zeit. Phys. Chem., 2000, 214, 675–703 CAS; (c) R. G. Viglione, R. Zanasi, P. Lazzeretti and A. Ligabue, Phys. Rev. A, 2000, 62, 52516-1-5.
- Diastereomerically enriched strychninium salt {(+)-FClBrC–CO2H, (−)-strychnine} and {(−)-FClBrC–CO2H, (−)-strychnine} were obtained with respective diastereomeric excesses (de) of 81% and 48–51%. Indeed, these diastereomeric salts displayed in methanol a ternary solubility diagram corresponding to a solid solution. For more details see J. Costante, N. Ehlinger, M. Perrin and A. Collet, Enantiomer, 1996, 1, 377–386 Search PubMed.
- J. Crassous, F. Monier, J.-P. Dutasta, M. Ziskind, C. Daussy, C. Grain and C. Chardonnet, ChemPhysChem., 2003, 4, 541–548 CrossRef CAS and references therein.
- J. Crassous, Z. Jiang, V. Schurig and P. Polavarapu, Tetrahedron: Asymmetry, 2004, 15, 1995–2001 CrossRef CAS.
- V. Schurig, M. Juza, B. J. Green, J. Horakh and A. Simon, Angew. Chem. Int. Ed. Engl., 1996, 35, 1680–1682 CrossRef CAS.
- H. Grosenick, V. Schurig, J. Costante and A. Collet, Tetrahedron: Asymmetry, 1995, 6, 87–88 CrossRef CAS.
- J. Canceill, L. Lacombe and A. Collet, J. Am. Chem. Soc., 1985, 107, 6993–6996 CrossRef CAS.
- C. Garcia, D. Humilière, N. Riva, A. Collet and J.-P. Dutasta, Org. Biomol. Chem., 2003, 1, 2207–2216 RSC.
- (a) J. Costante, L. Hecht, P. L. Polavarapu, A. Collet and L. D. Barron, Angew. Chem. Int. Ed., 1997, 36, 885–887 CrossRef CAS; (b) J. Costante-Crassous, T. J. Marrone, J. M. Briggs, J. A. McCammon and A. Collet, J. Am. Chem. Soc., 1997, 119, 3818–3823 CrossRef CAS.
- X. Cao, T. Freedman, L. Nafie and J. Crassous, to be published.
- J. W. Faller and A. R. Lavoie, Organometallics, 2000, 19, 3957–3962 CrossRef.
- A. Cuisset, J. R. Aviles Moreno, T. R. Huet, D. Petitprez, J. Demaison and J. Crassous, J. Phys. Chem. A, Search PubMed submitted.
- P. Soulard, J. Crassous, P. Asselin, to be published Search PubMed.
- A. Shelkovnikov, C. Grain, R. J. Butcher, A. Amy-Klein, A. Goncharov and C. Chardonnet, IEEE J. Quantum Electron., 2004, 40(8), 1023–1029 CrossRef CAS.
- A. Salam, J. Mol. Evol., 1991, 33, 105–113 CAS.
- T. Kitayama, T. Ushikubo, K. Morihashi and O. Kikuchi, J. Mol. Struct., 2002, 586, 1–7 CAS.
- R. Zanasi, P. Lazzeretti, A. Ligabue and A. Soncini, Phys. Rev. E, 1999, 59, 3382–3385 CrossRef CAS.
- A. J. Macdermott, Orig. Life Evol. Biosphere, 1995, 25, 191–199 CrossRef CAS.
- G. E. Tranter, Nature, 1985, 318, 172–173 CAS.
- (a) S. F. Mason, Nature, 1984, 311, 19–23 CAS; (b) S. F. Mason, Chem. Soc. Rev., 1988, 17, 347–359 RSC; (c) S. F. Mason, Biosystems, 1987, 20, 27–35 CrossRef CAS.
- A. J. MacDermott, Orig. Life Evol. Biosphere, 1995, 25, 191–199 CrossRef CAS.
- R. Berger, M. Quack and G. S. Tschumper, Helv. Chim. Acta, 2000, 83, 1919–1950 CrossRef CAS.
- W. A. Bonner, Orig. Life Evol. Biosphere, 1996, 26, 27–45 CAS.
- R. Berger and M. Quack, ChemPhysChem., 2000, 1, 57–60 CrossRef CAS.
- J. K. Laerdahl, R. Wesendrup and P. Schwerdtfeger, Chem. Phys. Chem., 2000, 1, 60–62 CrossRef CAS.
- R. Wesendrup, J. K. Laerdahl, R. N. Compton and P. Schwerdtfeger, J. Phys. Chem. A, 2003, 107, 6668–6673 CrossRef CAS.
- P. Schwerdtfeger and U. Müller-Herold, in Symmetry 2000, Part 1, ed. I. Hargittai and T. C. Laurent, Portland Press, London, 2002, pp. 317-328 Search PubMed.
- R. A. Hegstrom and D. K. Kondepudi, Sci. Am., 1990, 262, 108–115.
- W. A. Bonner, Chirality, 2000, 12, 114–126 CrossRef CAS.
- R. N. Compton and R. M. Pagni, Adv. At. Mol. Opt. Phys., 2002, 48, 219–261 Search PubMed.
- P. Frank, W. A. Bonner and R. N. Zare, in Chemistry for the 21st Century, ed. E. Keinan and I. Schechter, Wiley-VCH, Weinheim, 2001, pp. 173-208 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |