Mechanistic studies of La3+- and Zn2+-catalyzed methanolysis of aryl phosphate and phosphorothioate triesters. Development of artificial phosphotriesterase systems
Tony
Liu
,
Alexei A.
Neverov
,
Josephine S. W.
Tsang
and
R. Stan
Brown
*
Department of Chemistry, Queen's University, Kingston, Ontario, Canada K7L 3N6. E-mail: rsbrown@chem.queensu.ca; Fax: 613-533-6669; Tel: 613-533-2400
First published on 18th March 2005
Abstract
The methanolyses of a series of O,O-diethyl O-aryl phosphates (2,5) and O,O-diethyl S-aryl phosphorothioates (6) promoted by methoxide and two metal ion systems, (La3+)2(−OCH3)2 and 4:Zn2+:−OCH3
(4
= 1,5,9-triazacyclododecane) has been studied in methanol at 25 °C. Brønsted plots of the log![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) k2 values vs.sspKa for the phenol leaving groups give βlg values of −0.70, −1.43 and −1.12 for the methanolysis of the phosphates and −0.63, −0.87 and −0.74 for the methanolysis of the phosphorothioates promoted by the methoxide, La3+ and Zn2+ systems respectively. The kinetic data for the metal-catalyzed reactions are analyzed in terms of a common mechanism where there is extensive cleavage of the P–XAr bond in the rate-limiting transition state. The relevance of these findings to the mechanism of action of the phosphotriesterase enzyme is discussed.
k2 values vs.sspKa for the phenol leaving groups give βlg values of −0.70, −1.43 and −1.12 for the methanolysis of the phosphates and −0.63, −0.87 and −0.74 for the methanolysis of the phosphorothioates promoted by the methoxide, La3+ and Zn2+ systems respectively. The kinetic data for the metal-catalyzed reactions are analyzed in terms of a common mechanism where there is extensive cleavage of the P–XAr bond in the rate-limiting transition state. The relevance of these findings to the mechanism of action of the phosphotriesterase enzyme is discussed.
Introduction
Phosphorus triesters and phosphonate esters of general structure 1 (LG = leaving group, Z = alkyl, aryl, alkoxide or aryloxide) are widely used as insecticides, acaricides1,2 and OP-based chemical warfare (CW) agents.3 Owing to their toxicity and lingering effects on the environment, considerable effort has been directed toward methods of facilitating the controlled decomposition of these materials, particularly through hydrolysis and oxidation.3,4 Transition metal ions and lanthanides and certain mono- and di-nuclear complexes thereof are known to promote the hydrolysis of neutral phosphate and/or phosphonate esters,5,6 and Pt and Pd metallocycles are remarkably efficacious for thiophosphate pesticide hydrolysis.7
By contrast, metal-catalyzed alcoholysis reactions of organophosphates have received scant attention, although our recent work indicates that this is an effective strategy for the destruction of neutral organophosphate esters.8 For example, a methanolic solution of 2 mmol dm−3 in each of La3+ and NaOCH3 catalyzed the methanolysis of paraoxon (2, also a simulant for the phosphonofluoridate chemical weapon G-agents) by 109-fold relative to the background reaction at near-neutral sspH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9,10 and ambient temperature. La3+, and other lanthanides, also promote the ethanolysis of paraoxon11 and the 1,5,9-triazacyclododecane complexes of Zn2+ or Cu2+, as their monomethoxy forms (4:M2+:−OCH3), give excellent catalysis of the methanolysis of 2 and of the P
9,10 and ambient temperature. La3+, and other lanthanides, also promote the ethanolysis of paraoxon11 and the 1,5,9-triazacyclododecane complexes of Zn2+ or Cu2+, as their monomethoxy forms (4:M2+:−OCH3), give excellent catalysis of the methanolysis of 2 and of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S derivative, fenitrothion (3) for which La3+ is ineffective.12 A preliminary analysis of the effects of structural variation in the phosphate13 on the rates of catalyzed methanolysis of 2, its phenyl counterpart 5a, and the two corresponding O,O-diethyl S-phenyl- and O,O-diethyl S-(p-nitrophenyl)-phosphorothioate derivatives, 6a and 6b, indicated that the La3+-catalyzed methanolysis of the phosphorus esters is very sensitive to the nature of the leaving group, contrasting the relative insensitivity for La3+-catalyzed methanolysis of the corresponding aryl acetate esters.14,15 At the sspH optimum of 9 the second-order rate constants for La3+-catalyzed methanolysis of p-nitrophenyl acetate and phenyl acetate are very similar at 38 and 29 dm3 mol−1 s−1,14,15 while those for methanolysis of 2 and 5a vary by 1000-fold at ca. 2 dm3 mol−1s−1 and ca. 1.3 × 10−3 dm3 mol−1s−1 respectively.13
S derivative, fenitrothion (3) for which La3+ is ineffective.12 A preliminary analysis of the effects of structural variation in the phosphate13 on the rates of catalyzed methanolysis of 2, its phenyl counterpart 5a, and the two corresponding O,O-diethyl S-phenyl- and O,O-diethyl S-(p-nitrophenyl)-phosphorothioate derivatives, 6a and 6b, indicated that the La3+-catalyzed methanolysis of the phosphorus esters is very sensitive to the nature of the leaving group, contrasting the relative insensitivity for La3+-catalyzed methanolysis of the corresponding aryl acetate esters.14,15 At the sspH optimum of 9 the second-order rate constants for La3+-catalyzed methanolysis of p-nitrophenyl acetate and phenyl acetate are very similar at 38 and 29 dm3 mol−1 s−1,14,15 while those for methanolysis of 2 and 5a vary by 1000-fold at ca. 2 dm3 mol−1s−1 and ca. 1.3 × 10−3 dm3 mol−1s−1 respectively.13

The large leaving group effect on the rate of the La3+-catalyzed methanolysis of these phosphate triesters prompted the present investigation of the La3+- and Zn3+-catalyzed methanolysis of a series of phosphate (2,5) and phosphorothioate (6) esters. As will be shown, the mechanism of this reaction proceeds through a rate-limiting transition state having a very large degree of cleavage of the P-leaving group bond and is thus very different from that determined for the metal-catalyzed methanolysis reactions of the corresponding phenoxy acetate esters. These findings have important implications for the mechanism of action of phosphotriesterase, a dinuclear Zn2+-containing enzyme isolated from a soil bacterium, that effectively catalyzes the hydrolysis of paraoxon.16
Experimental
(a) Materials
3-Nitrophenol (99%), pentafluorophenol (99+%), 4-chlorophenol (99+%), 4-chloro-2-nitrophenol (98%), 4-methoxyphenol (99%), 4-methoxythiophenol (97%), 4-fluorothiophenol, NaHSO4, diazabicycloundecane (98%), and diethyl chlorophosphate (97%) were purchased from Aldrich and used as supplied. 4-Chlorothiophenol (98%, Acros Organics) and 3,5-dichlorothiophenol (98%, Matrix Scientific) were used as supplied.Phosphates 2 and 5a as well as phosphorothioates 6a and 6b were available from a previous study.13 All the other esters were prepared according to the following general protocol. CAUTION: phosphorothioates 6, and the phosphate triesters 2 and 5 are all acetylcholinesterase inhibitors, and should be synthesized and used with due attention to safety protocols.
Phosphates: 10 mmol of the appropriate phenol was added slowly to a solution of 1.6 mL diazabicycloundacene (DBU) in 20 mL of dry THF under argon and the resulting mixture was cooled in an ice bath while slowly adding 10 mmol diethylphosphoryl chloride. The mixture was then allowed to stir at room temperature for 3 h. The products were extracted three times with 20 mL of diethyl ether and washed twice with 20 mL of saturated NaHSO4, twice with 20 mL of 10% phosphate buffer and five times with 20 mL of water. The ether layer was dried with sodium sulfate and, following filtration, the volatiles were removed by rotary evaporation. Purification was achieved by flash chromatography over silica gel eluting with ethyl acetate–hexane (1 : 5 ratio) to produce the products in yields of around 60%.
The general method for the synthesis and purification of the phosphorothioate esters was the same as for the phosphotriesters except that the thiophenols, diethyl chlorophosphate and dry THF were mixed together before introducing DBU. The yields for the syntheses of phosphorothioate esters were around 55%.
All of the above esters had 1H NMR, 31P NMR, and mass spectra consistent with the structure.
(b) General UV/vis kinetics
Materials used to prepare solutions for kinetic determinations: sodium methoxide (0.5 mol dm−3), 1,5,9-triazacyclododecane (97%), tetrabutylammonium hydroxide (1.0 mol dm−3), La(OTf)3 (99.999%), anhydrous methanol (99.8%) and N-ethylmorpholine (99%) were all commercially available (Aldrich), while Zn(OTf)2 (98%) was obtained from Strem Chemical. Stock solutions (5 mmol dm−3) of each of the phosphate and phosphorothioate substrates were prepared in 99.8% anhydrous acetonitrile (Aldrich). La(OTf)3, sodium methoxide, and tetrabutylammonium hydroxide stock solutions were made in anhydrous methanol to 50 mmol dm−3. Zn(OTf)2 stock solutions in anhydrous methanol were formulated between 50 and 70 mmol dm−3 while the 1,5,9-triazacyclododecane stock solution in methanol was made between 30 and 50 mmol dm−3.For each kinetic run using Zn2+, the 4:Zn2+:−OCH3 catalyst was made in situ by adding known amounts of the Zn(OTf)3, 1,5,9-triazacyclododecane and tetrabutylammonium hydroxide stock solutions to anhydrous methanol such that the final volume was 2.5 ml with the final ratios of ligand, Zn2+ and −OCH3 being 1 : 1 : 0.5 to self-buffer the solution at sspH 9.1. The kinetics were followed by monitoring the change in absorbance of (0.5 to 2) × 10−4 mol dm−3 of 5d, 5b, 5e, 5c and 6a, 6c, 6e, 6b, 6f and 6d at 344, 276, 290, 428, 260, 266, 290, 277, 265, and 270 nm respectively. The absorbance vs. time data were fit to a standard first-order exponential equation to obtain the pseudo-first-order rate constants, kobs. The rates of reaction were monitored in duplicate at 4–7 different catalyst concentrations from 0.4 to 2.0 mmol dm−3. Second-order rate constants, k2obs, for the catalyzed reaction were obtained from plotting kobsvs. [4:Zn2+:−OCH3] and are presented in Tables 1 and 2.
| Aryloxy-phosphate | pKaa |
sspKab |
k 2 (OMe)/dm3 mol−1 s−1 |
k
2
Lac/dm3 mol−1 s−1 |
k 2 4:Zn:OMe/dm3 mol−1 s−1 |
|---|---|---|---|---|---|
| a pKa values in water from ref. 18. b sspKa values in methanol from refs. 10, 15, and 17. c k 2 La determined in a 17 mmol dm−3N-ethylmorpholine buffer at sspH 9.1. d k 2 values from ref. 13. e From duplicate initial rate measurements monitored by 1H NMR in CD3OD as described in the Experimental section. | |||||
| Pentafluoro (5b) | 5.53 | 8.84 | 0.201 ± 0.002 | 1070 ± 40 | 23.0 ± 0.6 |
| 4-Chloro-2-nitro (5c) | 6.32 | 10.64 | (6.4 ± 0.1) × 10−2 | 185 ± 7 | 11.4 ± 0.4 |
| p-Nitro (2) | 7.14 | 11.30 | (1.02 ± 0.03) × 10−2 | 23.2 ± 0.9d |
1.3 |
| m-Nitro (5d) | 8.39 | 12.41 | (6.0 ± 0.1) × 10−3 | 2.42 ± 0.07 | 0.58 ± 0.01 |
| p-Chloro (5e) | 9.38 | 13.59 | (6.3 ± 0.1) × 10−4 | (1.98 ± 0.07) × 10−2 | (8.04 ± 0.04) × 10−3 |
| p-H (5a) | 10.0 | 14.33 | 1.4 × 10−4d |
1.97 × 10−3d |
|
| p-Methoxy (5f) | 10.20 | 14.7 | (6.50 ± 0.01)
× 10−5e |
(2.2 ± 0.1)
× 10−4e |
(2.25 ± 0.05)
× 10−4e |
| ArylS-phosphorothioate | pKaa |
sspKab |
k 2 OMe/dm3 mol−1 s−1 | k 2 La/dm3 mol−1 s−1 | k 2 4:Zn:OMe/dm3 mol−1 s−1 |
|---|---|---|---|---|---|
|
a Aqueous pKa values from ref. 16e.
b
sspKa values in methanol computed from two-point linear regression pKa(MeOH)
= 1.2(pKa(water))
+ 2.83.
c Experimental sspKa values from ref. 19.
d Kinetic k2La values from ref. 13.
|
|||||
| p-Nitro (6b) | 4.61 | 8.4c |
0.12d |
12.4d |
0.84 ± 0.01 |
| 3,5-Dichloro (6d) | 5.07 | 8.9 | 0.152 ± 0.001 | 14.0 ± 0.5 | 0.97 ± 0.01 |
| p-Chloro (6c) | 5.97 | 10.1 | (1.88 ± 0.03) × 10−2 | 1.23 ± 0.05 | (11.6 ± 0.01) × 10−2 |
| p-Fluoro (6e) | 6.54 | 10.7 | (1.11 ± 0.02) × 10−2 | 0.46 ± 0.01 | (5.34 ± 0.06) × 10−2 |
| p-H (6a) | 6.68 | 10.9c |
4.8 × 10−3d |
0.48d |
(4.2 ± 0.1) × 10−2 |
| p-Methoxy (6f) | 6.95 | 11.2 | (2.22 ± 0.03) × 10−3 | (9.3 ± 0.2) × 10−2 | (1.46 ± 0.04) × 10−2 |
The La3+-catalyzed reactions were monitored by UV/vis spectrophotometry under buffered (N-ethylmorpholine, 17 mmol dm−3) conditions at three sspH values between 8.34 and 9.14 in the presence of (0.2 to 2.0) × 10−3 mol dm−3 La(OTf)3. This was done to ensure that the optimum rate of the reaction for all substrates plateaued by sspH 9.0. The rates of methanolysis of (0.5 to 2) × 10−4 mol dm−3 solutions of 5d, 5b, 5e, 5c and 6c, 6e, 6f, 6d were monitored at 348.6, 275, 286, 386.7, 235, 288, 239.6, and 281 nm respectively to obtain the pseudo-first-order rate constants (kobs) at each [La3+], and the gradients for the kobsvs. [La3+] plots were calculated to give the overall k2obs values for the La3+-catalyzed methanolysis at sspH 9.1 which are given in Tables 1 and 2. Kinetic data for 2, 5a, 6a and 6b were obtained from ref. 13. Initial rate kinetic data for La3+-catalyzed methanolysis of 5f were obtained by 1H NMR spectrometry by observing the rate of disappearance of starting material and the rate of formation of product for the first 10% of the reaction in CD3OD. Into two separate NMR tubes were placed 10 mmol dm−3 of substrate 5f along with 1.5 mmol dm−3 of catalyst (1 : 1 ratio of La3+ : −OCH3) in 1 mL of CD3OD. The tubes were immersed in a thermostated bath at 25 °C and the 1H NMR spectra were monitored periodically over 155 h. The integrations of the signals corresponding to the aromatic protons in the starting material and product were used to determine the initial rates which were converted into the second-order rate constants given in Table 1.
Kinetic data for Zn2+-catalyzed methanolysis of 5f were determined similarly in duplicate using 10 mmol dm−3 of substrate, 5 mmol dm−3 each of Zn(OTf)2 and 1,5,9-triazacyclododecane and 2.5 mmol dm−3 of Bu4NOCH3 in 1 mL of CD3OD, periodically monitored by 1H NMR over 45 h. The initial rate for the methoxide reaction with 5f was determined from duplicate reactions of 10 mmol dm−3 of substrate and 25 mmol dm−3 of sodium methoxide followed for 45 h to 23% completion.
The pseudo-first-order rate constants for methanolysis of the other phosphorus esters promoted by methoxide were determined by UV/vis (monitored at the same wavelength as for La3+-catalyzed methanolysis) in duplicate at four different [−OCH3] ranging from 0.01 to 0.04 mol dm−3. The second-order rate constants for these (k2OMe) were computed as the gradients of the kobsvs. [methoxide] plots and are given in Tables 1 and 2.
Results
Given in Tables 1 and 2 are the second-order rate constants for the methanolysis of the phosphates and phosphorothioates promoted by −OCH3, La3+ and 4:Zn2+:−OCH3. In all cases, these were determined from the gradients of the plots of kobsvs. [methoxide] or [metalx+]total. In the case of 4:Zn2+, the solutions were self-buffered at sspH 9.1 through half neutralization of the catalyst ([4:Zn2+:−OCH3]/[4:Zn2+:HOCH3] = 1). Earlier work with 4:Zn2+ showed that the catalytic activity was entirely due to the methoxide form, 4:Zn2+:−OCH3,12b and the k24:Zn:OMe values reported in Tables 1 and 2 were calculated based on the concentration of the catalytically active species. La3+catalysis for all substrates was determined by observing the kobsvs. [La3+]total at the sspH value of 9.14 in a 17 mmol dm−3N-ethylmorpholine buffer system. Earlier work on the La3+-catalyzed methanolysis of paraoxon had shown that the catalytic rate constant plateaus between sspH ca. 8.3 and ca. 9.2,8 so it was important to see that the same trend exists for all the other phosphate and phosphorothioate esters, which is confirmed in this work. Given in Tables 1 and 2 are the observed second-order rate constants determined at sspKa 9.1, where earlier analysis8 indicated that >90% of the catalysis occurs through a single form of the catalyst, namely La3+2(−OCH3)2, vide infra.Also given in Tables 1 and 2 are the sspKa values for the phenols and thiophenols in methanol. While experimental methanol sspKa values for most of the phenols are available17,18 or can be calculated from their water values by the linear regressions given in refs. 10 and 17
(pKaMeOH
= 1.12pKaH2O
+ 3.56), to our knowledge the only two available thiol sspKa values in methanol are thiophenol (10.9) and p-nitrothiophenol (8.4).19 Using these two values, a computed linear regression of sspKa(MeOH)
= 1.2(pKa(water))
+ 2.83 is proposed to relate the aqueous and methanolic pKa values for the unknown aryl thiols.
In Figs. 1, 2 and 3, as aids to visualizing the data, are presented Brønsted plots of the second-order rate constants for methoxide-, La3+- and 4:Zn2+:−OCH3-catalyzed methanolysis of the phosphate and phosphorothioate esters vs. the sspKa of the conjugate acids of the ArO− or ArS− leaving groups. Linear regressions for the methoxide reactions of the phosphates (except 5b which falls well under the Brønsted plot in Fig. 1) and phosphorothioates (except 6b) are given in eqns. (1) and (2) respectively.
| logk2OMe(phosphate)
=
(6.20 ± 0.70)
−
(0.70 ± 0.05) sspKaHOAr | (1) |
| logk2OMe(phosphorothioate)
=
(6.00 ± 0.86)
−
(0.76 ± 0.08) sspKaHSAr | (2) |
| logk2La(phosphate)
=
(17.60 ± 1.07)
−
(1.43 ± 0.05) sspKaHOAr | (3) |
| logk24:Zn:OMe(phosphate)
=
(13.05 ± 1.59)
−
(1.12 ± 0.13) sspKaHOAr | (4) |
| logk2La(phosphorothioate)
=
(8.93 ± 1.01)
−
(0.87 ± 0.10) sspKaHSAr | (5) |
| logk24:Zn:OMe(phosphorothioate)
=
(6.64 ± 0.69)
−
(0.74 ± 0.06) sspKaHSAr | (6) |
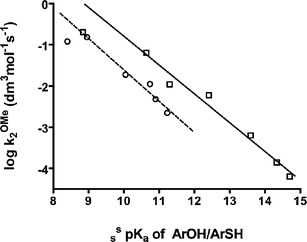 | ||
| Fig. 1 Brønsted plots of the log second-order rate constant for methoxide attack on phosphates 2 and 5 as well as on phosphorothioates 6 (except 6b) vs. sspKa values for the corresponding phenols or thiophenols; linear regressions for the phosphate (except 5b) and phosphorothioate (except 6b) data give slopes of −0.70 ± 0.05 (solid line, □) and −0.76 ± 0.08 (dashed line, ○) respectively. | ||
 | ||
| Fig. 2 Brønsted plot of the log second-order rate constant for La3+- and 4:Zn2+:−OCH3-catalyzed methanolysis of phosphates 2 and 5 vs. sspKa values for the corresponding phenols; linear regressions through the La3+ and 4:Zn2+:−OCH3 data (except 5b) give gradients of −1.43 ± 0.08 (solid line, □) and −1.12 ± 0.13 (dashed line, ○) respectively. Note that the points on lower right for the p-methoxy derivative (5f) are coincident. | ||
 | ||
| Fig. 3 Brønsted plot of the log second-order rate constant for La3+- and 4:Zn2+:−OCH3-catalyzed methanolysis of phosphorothioates 6 vs. sspKa values for the corresponding thiophenols; linear regressions through the La3+ and 4:Zn2+:−OCH3 data give gradients of −0.87 ± 0.10 (solid line, □) and −0.74 ± 0.06 (dashed line, ○) respectively. | ||
Shown in Figs. 4 and 5 are alternative presentations of the data where the methoxide rate constants are plotted vs. the La3+ or 4:Zn2+:−OCH3 rate constants for all the phosphate and phosphorothioate substrates. The linear regressions for these are given in eqns. (7) and (8), and (9) and (10) respectively.
| logk2La(phosphate)
=
(1.94 ± 0.10)logk2OMe(phosphate)
+
(4.65 ± 0.26) | (7) |
| logk24:Zn:OMe(phosphate)
=
(1.49 ± 0.11)logk2OMe(phosphate)
+
(2.78 ± 0.27) | (8) |
| logk2La(phosphorothioate)
=
(1.15 ± 0.11)logk2OMe(phosphorothioate)
+(2.10 ± 0.18) | (9) |
| logk24:Zn:OMe(phosphorothioate)
=
(0.99 ± 0.06)logk2OMe(phosphorothioate)
+
(0.79 ± 0.11) | (10) |
 | ||
| Fig. 4 Plots of logk2OMe vs. logk2La (□) or logk24:Zn:OMe (○) for the methanolysis of phosphate esters at 25 °C: slope for La3+ plot is 1.94 ± 0.10; slope for 4:Zn:OMe plot is 1.49 ± 0.11 all data included. Note that the data points for the p-methoxy derivative (5f) in lower left corner are coincident. | ||
 | ||
| Fig. 5 Plots of logk2OMe vs. logk2La (□) or logk24:Zn:OMe (○) for the methanolysis of phosphorothioate esters at 25 °C: slope for La3+ plot is 1.15 ± 0.10; slope for 4:Zn:OMe plot is 0.99 ± 0.06. | ||
Discussion
The various second-order rate constants for the reactions of the phosphates and phosphorothioates are presented in Tables 1 and 2 along with the leaving group phenol sspKa values in methanol and water. For the aryloxy esters the HOAr sspKavalues are all known or can be computed from known linear regressions relating the methanol and aqueous pKa values.10,15,17,18 As far as we are aware, the HSAr sspKavalues in methanol for the arylthiols are only known for the thiophenol and for p-nitrothiophenol, and if we assume that there is also a linear dependence of these methanol and aqueous pKa values one can compute a two-point linear regression of sspKa(MeOH) = 1.2(pKa(water))
+ 2.83 from which the sspKavalues for the other thiols may be estimated. Although the gradient of this linear regression is quite similar to that reported for the phenols (1.12),17 these computed thiol sspKavalues are viewed as estimates only for the construction of various Brønsted plots for the phosphorothioate esters.
In constructing the various Brønsted plots for the La3+-catalyzed processes, we used the second-order rate constants obtained as the gradients of plots of Δkobs/Δ[La3+] under buffered conditions at sspKa 9.1, so the k2La values are presented per La3+ ion even though we have determined that the actual catalytic species are dimers (La3+)2(−OCH3)2,3,4 the relative importance of which depend on the sspKaand the [dimer].8,13,20 For the La3+ catalytic study, the aryloxy phosphate esters encompass a range of 106 in leaving group acidity and about 107 in k2La but only about three orders of magnitude in k2OMe. The second-order rate constants for the 4:Zn2+:−OCH3-catalyzed reactions were determined under self-buffered conditions at sspKa 9.1 through half neutralization of the catalyst, and the k24:Zn:OMe values reported in Tables 1 and 2 are computed on the basis of the active form.
(a) Uncatalyzed reaction of methoxide
The Brønsted plots of logk2OMe data for methoxide reactions of the phosphate and phosphorothioate esters vs. the sspKa values shown in Fig. 1 provide reasonable linear correlations, the respective βlg values being −0.70 ± 0.05 and −0.76 ± 0.08. In both series the derivatives having the lowest sspKavalues of the leaving phenol/thiophenol (5b and 6b) are less reactive than predicted on the basis of the equilibrium sspKa data, and cursory inspection reveals that the plots have some curvature, with the latter points lying below the indicated lines. Non-linear Hammett and Brønsted behaviours have been observed for the hydroxide reactions of substituted aryl benzoates21 and acetates22 and Schowen23 reported that logk2 values for methoxide reactions of aryl acetates and carbonates in methanol are not linearly related to the sspKa values for ionization of the corresponding phenols. Our previous study of the methanolysis of aryl acetates also showed some discontinuity in the Brønsted plot of logkOMevs. phenol sspKa for the most acidic phenols such as pentafluoro- and 2,4-dinitro-phenol.15 Such curvature, if observed, does not result from a change in mechanism or rate-limiting step in the reaction but is related to a greater importance of the resonance and inductive interactions in the equilibrium acid dissociation constants (on which the Hammett and sspKa values are based) than in the kinetic processes where far less charge development generally occurs in the rate-limiting TS.21–23 In the present study we have chosen to omit the data for the pentafluorophosphate and phosphorothioate derivatives, 5b and 6b, from calculation of the gradients of the Brønsted plots. The βlg values of −0.70 and −0.76 respectively for the methoxide reaction of esters 2 and 5
(except 5b) and 6
(except 6b) can be compared with the corresponding hydroxide βlg values of −0.41 to −0.35 for 2-aryloxy-2-oxo-1,3-dioxaphosphorinanes24,25
−0.43 and −0.44 for hydroxide attack on diethyl aryl phosphate triesters,16e,26 and −0.42 for hydroxide attack on diethyl S-aryl phosphorothiolates.16e
Khan and Kirby,24 and later Rowel and Gorenstein,25 discussed the reaction of oxyanions with some 2-aryloxy-2-oxo-dioxaphosphorinanes concluding that, for strong nucleophiles such as hydroxide and methoxide, the mechanism was consistent with, although not uniquely so, a two-step process that proceeds via a five-coordinate intermediate as in eqn. (11).
 | (11) |
However, as the nucleophiles became weaker, the βlg values increased, supporting either a two-step process where the breakdown became more rate limiting24 or a change in mechanism to a concerted, SN2 displacement at P.25
More recently, Williams and co-workers provided convincing evidence for a single transition state for the transfer of the diphenylphosphoryl group between phenoxide anions in water.27 In that case, a Brønsted plot of the second-order rate constants for reaction of the phenoxides with p-nitrophenyl diphenyl phosphate against the pKa of the nucleophile's conjugate acids gave a straight line with βnuc = 0.66 with no evidence of a break which would have been required if there was an intermediate produced. In the case of the HO− nucleophile reacting with diethyl aryloxy phosphates, they judged, by analogy with the above reactions, that HO− displacement of aryloxy leaving groups was probably concerted, although with little cleavage of the ArO–P bond.26 This conclusion is supported by the 18O-phenoxy kinetic isotope effect of 1.006 for hydroxide-promoted cleavage of paraoxon 2 which was interpreted28 as being consistent with a bond order of 0.75 for the P–OAr bond in the “SN2-like transition state of an associative mechanism with concerted, asynchronous departure of the leaving group”.
Our data for methanolysis of the aryl phosphates 2 and 5 and diethyl S-aryl phosphorothioates 6 indicate that the βlg values are more negative by ca. 0.3 units than is the case for the hydroxide reactions.16e,24,25 Following the “effective charge treatment” described by Jencks29 and Williams,30 the Brønsted βlg value for nucleophilic attack of methoxide on the aryloxyphosphates suggests a process where the rate-limiting transition state has appreciable changes in the P–OAr bond. This could be viewed as resulting from either a two-step process with the attack step largely rate limiting due to the fact that the methoxide nucleophile is a far poorer leaving group than any of the aryloxy anions,31 or more likely with a concerted process as shown in Scheme 1. The extent of breaking of the P–OAr bond in the TS can be measured by the Leffler parameter, α, which measures the change in the Brønsted βlg for the TS relative to the βeq for equilibrium transfers of acyl or phosphoryl groups between oxyanion nucleophiles. In the case of the transfer of the (EtO)2PO group, the βeq value is −1.8726 with the O–Ar oxygen in the starting material having a net effective charge of +0.87. When methoxide is the nucleophile, the Leffler parameter of βlg/βeq
= 0.37 suggests that the P–OAr cleavage is 37% of the way from starting material to product.
 | ||
| Scheme 1 | ||
For phosphorothioates 6 reacting with methoxide, a similar sort of analysis for the reaction proceeding through a two- or one-step reaction can be invoked. In this case one has no exact measure of the effective charge on the S-atom in the ArS–P unit, but based on comparison of the known effective charges on the S and O atoms of ArS–C(O)CH3 and ArO–C(O)CH3 of 0.4 and 0.7 respectively,30a one might expect that S is less positive than O in the case of the ArX–P unit. Assuming the effective charge on S is 0.5–0.6, the Leffler α for a concerted P–SAr cleavage can be computed as ca. 0.45–0.50.
(b) La3+- and 4:Zn2+:−OCH3-catalyzed methanolysis
Given in Figs. 2 and 3 are the respective Brønsted plots for the metal-catalyzed methanolysis of the phosphate and phosphorothioates. For phosphates 2 and 5, the respective βlg values for La3+ and 4:Zn2+:−OCH3 are large and negative, being −1.43 ± 0.08 and −1.12 ± 0.13 respectively if we exclude the pentafluoro derivative 5b which deviates downward from both plots, probably due to the known difficulties in plotting kinetic data against ionization data of phenols.23 The La3+ and 4:Zn2+:−OCH3 Brønsted plots for the phosphorothioates given in Fig. 3 have βlg values of −0.87 ± 0.10 and −0.74 ± 0.06 respectively.The large negative βlg values for the metal-ion-catalyzed methanolysis of the phosphate esters suggests a process where there is considerable cleavage of the leaving group in the transition state, far more than is the case for the methoxide reaction. In our previous study of the La3+ and 4:Zn2+:−OCH3-promoted methanolysis of acetate esters having aryloxy and some alkoxy leaving groups,15 we determined that both metal systems exhibited Brønsted plots with a break at ca.sspKa 14.7, consistent with a catalyzed process comprising a pre-equilibrium binding of the metal ion followed by kinetic steps proceeding through a reversibly-formed intermediate as shown in Scheme 2. With good leaving groups the internal attack of metal-coordinated methoxide on the transiently bound ester was rate limiting (βlg ≈ 0), but with poor leaving groups the breakdown of the metal-coordinated anionic tetrahedral intermediate was rate limiting (βlg = −0.71). In that case, it appeared that metal ion coordination actually stabilized the tetrahedral intermediate, but the situation with the present phosphate/phosphorothioate esters is clearly different because there is a large dependence of the rate on the leaving group in the sspKa region where βlg is zero for the reaction of the carboxylate esters. Accordingly we suggest that, for the metal-catalyzed methanolysis of these phosphates/phosphorothioates, there is no evidence for a change in rate-limiting step for aryloxy or arylthio leaving groups and, by inference, no evidence for an intermediate.
 | ||
| Scheme 2 MX+–−OCH3 refers to (La3+)2(−OCH3)2 or 4:Zn2+:−OCH3. | ||
Although we have never observed evidence of saturation kinetics, it is difficult to envision any mechanism for the metal-promoted reactions where the ion does not bind to the phosphate to provide Lewis activation toward attack. Indeed, phosphate complexation of lanthanides and actinides is well known32 and structures are known for Zn2+ complexes of phosphine oxides33 and tritoluoyl phosphate.34
We analyze the present Brønsted plots as being consistent with the mechanism in eqns. (12) and (13)
 | (12) |
 | (13) |
| k2obs
=
Kbk1
=
CbC110(βb
+
β1)pKa | (14) |
| logk2obs
=
{logCb
+ logC1}
+
(βb
+
β1)pKa | (15) |
O) compounds correlates with the Taft σ* values of the substituents.32d,e These considerations indicate that for the La3+- or 4:Zn2+:−OCH3-promoted methanolysis of the phosphates, the observed βlg may be a lower limit because it will be reduced by the positive βb.
Presented in Scheme 3 is a proposed mechanism of reaction for the La3+-catalyzed reaction of the phosphate which earlier studies have shown is largely (>90%) attributable to the involvement of a bis-methoxy-bridged dimer, (La3+)2(−OCH3)2, 7 in Scheme 3.8 In Scheme 3, for the sake of visual clarity, we have omitted the methanols of solvation on each La3+ as well as any possible associated triflates about which we have no information. It seems unlikely that a methoxy group bound between two La3+ ions will be sufficiently nucleophilic to attack the phosphate,37 so we propose that the pre-equilibrium binding of phosphate to form 8 induces opening of one of the methoxy bridges to reveal a kinetically active form (9) with one of the La3+ ions acting as a Lewis base, the other serving to deliver the methoxide intramolecularly. Since the Leffler parameter, α, for the La3+-catalyzed reaction is βlg/βeq
=
−1.43/−1.87 = 0.76, the transition state for this reaction (10) has extensive cleavage of the P–OAr bond, and this large value is most consistent with a concerted reaction within the complex. Catalytic turnover requires a final dissociation of the diethyl methyl phosphate with the reformation of 7. In the case of the 4:Zn2+:−OCH3-catalyzed reaction a similar mechanism is envisioned but this time the transition structure (shown below as structure 12)

 | ||
| Scheme 3 When X = O, α = 0.76. | ||
While one might expect that the positively charged metal ion might stabilize an anionic penta-coordinate intermediate, the far more negative βlg values observed for the metal-ion-catalyzed reactions, compared to those for the methoxide reactions, suggest that the metal ions do not stabilize sufficiently any putative intermediate to direct the reaction through a stepwise pathway. The reason why the La3+ reaction exhibits a significantly more negative βlg than does the Zn2+ reaction is not immediately obvious, but could result from better attack/departure angles in the 6-membered La3+ TS relative to the 4-membered Zn2+ TS or from a greater electrostatic stabilization of the transition structure in the case of the more highly charged La3+ system. Given the fact that the catalysis becomes better with the aryloxy anion leaving groups with lower sspKa values for their conjugate acids, it does not seem likely in either the La3+ or Zn2+ cases that direct coordination of the leaving group to the metal ion is a significant factor in the catalysis.
In the case of La3+- and Zn2+-catalyzed methanolysis of the phosphorothioate esters the observed βlg values of −0.87 and −0.74 also signify an associative mechanism with some departure of the leaving group, but it is difficult to assign the extent of the bond cleavage since the βeq value is not known for the phosphoryl transfer between thiol and oxygen nucleophiles. It is highly instructive to consider the graph shown in Fig. 6 which is a plot of the logk2 constants for the La3+- and 4:Zn2+−OCH3-catalyzed reactions for the entire series of phosphate and phosphorothioate esters. The fact that the slopes for each series of substrate are very similar and without breaks over some seven orders of magnitude for the La+3 reaction and five orders of magnitude for the Zn2+ reaction indicates that there is no change of mechanism throughout either series. Furthermore, the slopes of 1.30 ± 0.05 and 1.17 ± 0.04 for the phosphate and the phosphorothioate reactions are some measure of the greater efficacy of the La3+ reaction which may relate to the higher net positive charge or better geometry for the La3+-catalyzed processes. Finally, the fact that both the phosphate and phosphorothioate esters lie essentially on the same line indicates that there cannot be any special catalytic assistance to the departure of the leaving group since one would expect a different effect for assistance of departure of the softer SAr group by Zn2+ relative to the ‘hard’38 La3+. This is drastically different to a situation we have previously reported where, for the methanolysis of PS and PO phosphates, La3+ and Zn2+ or Cu2+ show very different catalytic effects implying that in these materials coordination to the phosphoryl O and S are required components of catalysis.12 Thus on the basis of the current evidence, it is reasonable that the phosphorothioate methanolysis promoted by both metal-containing systems is concerted and proceeds analogously to the process depicted in Scheme 3.
 | ||
| Fig. 6 Plots of logk2La vs. logk24:Zn:OMe for the methanolysis of phosphate (■) and phosphorothioate (□) esters at 25 °C: the linear regression for the phosphate ester plot is logk2La = (1.30 ± 0.05)logk24:Zn:(OMe) + (1.03 ± 0.10), r2 = 0.9930; the linear regression for the phosphorothioate esters is logk2La = (1.17 ± 0.04)logk24:Zn:(OMe) + (1.18 ± 0.05), r2 = 0.9952. | ||
(c) Comparision of k2OMe and k24:Zn(OMe) or k2La
Comparison of the La3+ and 4:Zn2+:−OCH3 kinetic data in Tables 1 and 2 with the corresponding k2OMe kinetic data indicates that the metal ion systems are more effective than methoxide for promoting the methanolysis of this series of phosphates and phosphorothioates. For the phosphates 2 and 5, the k2La/k2OMe ratio varies from 5000 for the pentafluorophenoxy derivative 5b to ca. 3 for the phenoxy derivative 5a. Over the same series, the k24:Zn:OMe/k2OMe ratio varies only by 40-fold (115 to ca. 3). On the other hand, the phosphorothioates show very little variation of the k2La/k2OMe or k24:Zn:OMe/k2OMe ratios with leaving groups, the values being 40–100 throughout the arylthio series for the former and 5–8 for the latter.Shown in Figs. 4 and 5 are the plots of logk2OMevs. logk2La or logk24:Zn:OMe for the phosphates and phosphorothioates. As was the case in our earlier study of the metal-catalyzed methanolysis of esters,15 we assume here that the mechanism of methoxide-promoted methanolysis is similar enough for all the phosphate esters with the nucleophilic addition being either rate limiting or concerted with leaving group departure that the rate constant, k2OMe, can be used as an empirical measure of the composite effects of structural changes that incorporate both electronic and steric effects. This approach has advantages over the Brønsted or Hammett plots since the latter rely on substituent-induced changes to equilibrium pKa processes which may not be appropriate for how a substituent influences a particular reaction where the rate-limiting step(s) have less charge development on the leaving group. An additional benefit is that this treatment does not rely on experimental or estimated sspKa values for a given substituent which, if in error, may influence the conclusions based on the slopes of Brønsted plots.
The slopes of the lines in Fig. 4 for methanolysis of the phosphates are 1.94 and 1.49 for La3+ and the Zn2+ complex respectively, indicating that catalytic enhancement of the methanolysis reaction by the metal ion gets better as the leaving group gets better. The data also suggest that for leaving groups having sspKa values larger than 15, the second-order rate constant for the methoxide reaction will be larger than those for either metal-catalyzed reaction. The slopes of the lines in Fig. 5 for the phosphorothioates are, within experimental uncertainty, essentially unity for both the La3+- and Zn2+-catalyzed reactions. Importantly, the fact that all the plots in Figs. 4 and 5 are linear with no break over the range of substrates studied indicates that there is no change in mechanism or rate-limiting step for one of the reactions which is not manifested in the other.
(d) Relevance to the phosphotriesterase enzyme
The above study has a direct bearing on the mechanism of action of phosphotriesterase (PTE), a Zn2+-contaning enzyme found in the soil-dwelling bacterium Pseudomonas diminuta which has been shown to degrade pesticides such as paraoxon and parathion.15 X-Ray diffraction shows that the active site comprises two Zn2+ ions separated by 3.4 Å, one of which is coordinated to the protein by two His imidazoles and an aspartate, and the second by two histidines: both metal ions are bridged by the oxygens of N-carboxy lysine and an −OH or water having a kinetic pKa of 5.8–5.916c,39 which is thought to be the active nucleophile.16a,39,40 X-Ray diffraction studies also showed that inhibitors such as triethyl phosphate and di-iso-propyl methylphosphonate bind to the more solvent-exposed Zn2+ion with PO coordination.41 The metal ions can be removed from the wild-type enzyme and replaced by Cd2+, Ni2+, Co2+ and Mn2+, and all reconstituted enzymes show activity. The native enzyme has been shown to react with paraoxon in 18O-labelled water to produce the (EtO)2P(O)18OH product, and also to react with chiral O-ethyl phenylphosphonothioic acid to give a product that is inverted.42 Detailed kinetic studies of the hydrolysis of a series of diethyl aryl phosphates and phosphorthioates conducted on the (Zn2+)2 wild-type enzyme by Hong and Raushel indicated that the respective Brønsted βlg values of Vmaxvs. pKa of aryl phosphates and phosphorothioates in water were −2.2 and −1.0 respectively, the high value for the phosphates being interpreted as arising from a significantly dissociative mechanism with a quite product-like transition state.16e
There is much in common between the reactivity of the enzyme for both phosphates and phosphorothioates and that which is operative for our very simple dinuclear (La3+)2(−OCH3)2 and mono-nuclear 4:Zn2+:−OCH3 systems. The most obvious similarity comes from comparison of the large Brønsted βlg we see for both sets of substrates and those exhibited by the wild-type enzyme. However, very much higher βlg values are reported for the Cd2+- and Mn2+-enzymes (−3.0 and −4.3 respectively) which clearly are far greater than one expects for simple P–OAr bond cleavage in water.16e The latter βlg values are obtained by plotting the rate data against the aqueous pKa values which likely do not reflect faithfully the solvation and electrostatic stabilization of the leaving anion in the active site. In fact, if the pKa values in the lower dielectric constant solvents MeOH or DMSO were used, which tends to expand the pKa scales of phenols,19 this would lessen considerably the βlg values. Nevertheless, the kinetic data do indicate a very large degree of cleavage of the P–OAr and P–SAr bond in the rate-liming step of the enzymatic phosphoryl transfer, similar to what we have observed in our simple catalytic system which we have interpreted in terms of a concerted mechanism. All the other data from the enzyme studies, for example the inversion of configuration in hydrolysis of an optically active starting material and 18O-incorporation of solvent water to the hydrolytic product42 are consistent with a concerted mechanism for the enzyme.
Conclusions
The above study indicates that metal-catalyzed methanolyses of these series of phosphate and phosphorothioate triesters proceed by a mechanism that involves considerable cleavage of the leaving group in the rate-limiting step, in fact more cleavage than in the analogous methoxide attack on the same esters. The data indicate that while the dinuclear (La3+)2(−OCH3)2 system has a large and negative βlg for phosphate methanolysis (−1.43), that of the mononuclear complex 4:Zn2+:−OCH3 is slightly less at −1.15. These large negative values are best interpreted in terms of a concerted displacement mechanism with considerable departure of the leaving group bond in the rate-limiting step. The reason for the difference in βlg of the La3+ and Zn2+ complexes may be a result of differences in total (+) in the vicinity of the transition structure, or geometric differences where the La3+-containing transition structure is more optimally aligned for internal methoxide displacement of the leaving group. It is interesting to note that, despite the fact that metal-catalyzed hydrolysis of phosphate triesters has been extensively investigated for many years,5,6 in no case of which we are aware has such a significant catalytic effect over such a wide series of substrates been observed. A significant aspect of the efficacy of the La3+ and Zn2+ catalysts in methanol must arise from a medium or solvent effect since in general the metal hydroxo counterparts in water are significantly less reactive than our metal methoxide catalysts in methanol and even less reactive than hydroxide itself. Given that the phosphotriesterase enzyme contains a dinuclear Zn2+ active site, it would be interesting to compare the efficacy for alcoholysis promoted by an appropriate dinuclear Zn2+-containing catalyst with that of 4:Zn2+:−OCH3, a study which is currently underway in these laboratories.Acknowledgements
The authors gratefully acknowledge the financial assistance of the Natural Sciences and Engineering Research Council of Canada, Queen's University and the United States Department of the Army, Army Research Office, Grant No. W911NF-04-1-005743and the Defense Threat Reduction Agency, Joint Science and Technology Office (06012384BP). T. L. thanks the Summer Student Work Experience Program at Queen's University.References and notes
- A. Toy and E. N. Walsh, Phosphorus Chemistry in Everyday Living, American Chemical Society, Washington, DC, 2nd edn., 1987, ch. 18–20 Search PubMed; L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley, New York, 2000 Search PubMed; M. A. Gallo and N. J. Lawryk, Organic Phosphorus Pesticides. The Handbook of Pesticide Toxicology, Academic Press, San Diego, CA, 1991 Search PubMed; P. J. Chernier, Survey of Industrial Chemistry, VCH, New York, 2nd edn., 1992, pp. 389–417 Search PubMed.
- K. A. Hassall, The Biochemistry and Uses of Pesticides, VSH, Weiheim, 2nd edn., 1990, pp. 269–275 Search PubMed.
- Y.-C. Yang, J. A. Baker and J. R. Ward, Chem. Rev., 1992, 92, 1729 CrossRef CAS; Y.-C. Yang, Acc. Chem. Res., 1999, 32, 109 CrossRef CAS; Y.-C. Yang, Chem. Ind. (London), 1995, 334 CAS.
- H. Morales-Rojas and R. S. Moss, Chem Rev., 2002, 102, 2497 CrossRef CAS and refs. cited therein.
- S. H. Gellman, R. Petter and R. Breslow, J. Am. Chem. Soc., 1986, 108, 2388 CrossRef CAS; R. S. Brown and M. Zamkanei, Inorg. Chim. Acta, 1985, 108, 201 CrossRef CAS; R. S. Kenley, R. H. Flemming, R. M. Laine, D. S. Tse and J. S. Winterle, Inorg. Chem., 1984, 23, 1870 CrossRef CAS; B. S. Cooperman, in Metal Ions in Biological Systems, H. Sigel, ed., Marcel Dekker, New York, 1976, vol. 5, p. 79 and refs. cited therein Search PubMed; F. M. Menger, L. H. Gan, E. Johnson and H. D. Durst, J. Am. Chem. Soc., 1987, 109, 2800 Search PubMed; F. M. Menger and T. Tsuno, J. Am. Chem. Soc., 1989, 111, 4903 CrossRef CAS; P. Scrimin, P. Tecilla and U. Tonellato, J. Org. Chem., 1991, 56, 161 CrossRef CAS and refs. cited therein; F. Tafesse, Inorg. Chim. Acta, 1998, 269, 287 CrossRef CAS; P. Scrimmin, G. Ghinlanda, P. Tecilla and R. A. Moss, Langmuir, 1996, 12, 6235 Search PubMed; C. A. Bunton, P. Scrimmin and P. Tecilla, J. Chem. Soc., Perkin Trans. 2, 1996, 419 CrossRef CAS; Y. Fujii, T. Itoh and K. Onodera, Chem. Lett. Jpn., 1995, 305 CrossRef CAS; S. J. Oh, C. W. Yoon and J. W. Park, J. Chem. Soc., Perkin Trans. 2, 1996, 329 RSC; T. Berg, A. Simeonov and K. Janda, J. Comb. Chem., 1999, 1, 96 CAS; J. R. Morrow and W. C. Trogler, Inorg. Chem., 1989, 28, 2330 RSC; R. W. Hay and N. Govan, J. Chem. Soc., Chem. Commun., 1990, 714 CrossRef CAS; T. C. Bruice, A. Tsubouchi, R. O. Dempsy and L. P. Olson, J. Am. Chem. Soc., 1996, 118, 9867 CrossRef CAS; J. A. A. Ketelar, H. R. Gersmann and M. M. Beck, Nature, 1956, 177, 392 RSC; D. Kong, A. E. Martell and J. Reibenspies, Inorg. Chim. Acta, 2002, 333, 7 CrossRef CAS; R. W. Hay and N. Govan, Polyhedron, 1998, 17, 463; R. W. Hay and N. Govan, Polyhedron, 1998, 17, 2079 CrossRef CAS; R. W. Hay, N. Govan and K. E. Parchment, Inorg. Chem. Commun., 1998, 1, 228 CrossRef CAS; B. L. Tsao, R. J. Pieters and J. Rebek, Jr., J. Am. Chem. Soc., 1995, 177, 2210 CrossRef CAS; M. Yamami, H. Furutachi, T. Yokoyama and H. Ōkawa, Inorg. Chem., 1998, 37, 6832 CrossRef CAS; C. M. Hartshorn, A. Singh and E. L. Chang, J. Mater. Chem., 2002, 12, 602 CrossRef; V. Chandrasekhar, A. Athimoolan, S. G. Srivatsan, P. S. Sundaram, S. Verma, A. Steiner, S. Zacchini and R. Butcher, Inorg. Chem., 2002, 41, 5162 CrossRef CAS; M. Rombach, C. Maurer, K. Weis, E. Keller and H. Vahrenkamp, Chem. Eur. J., 1999, 5, 1013 RSC.
- L. Barr, C. J. Easton, K. Lee, S. F. Lincoln and J. S. Simpson, Tetrahedron Lett., 2002, 7797 CrossRef CAS; W. H. Chapman and R. Breslow, J. Am. Chem. Soc., 1995, 117, 5462 CrossRef; T. Koike and E. Kimura, J. Am. Chem. Soc., 1991, 113, 8935 CrossRef CAS; M. M. Ibrahim, K. Ichikawa and M. Shiro, Inorg. Chim. Acta, 2003, 353, 187 CrossRef CAS; M. D. Santana, G. Garcia, A. A. Lozano, G. López, J. Tudela, J. Pérez, L. García, L. Lezama and T. Rojo, Chem. Eur. J., 2004, 10, 1738 CrossRef CAS; F. Mancin, E. Rampazzo, P. Tecilla and U. Tonellato, Eur. J. Org. Chem., 2004, 281 CrossRef CAS; K. Yamaguchi, F. Agaki, S. Fujinami, M. Suzuki, M. Shionoya and S. Suzuki, Chem. Commun., 2001, 375 RSC.
- G. M. Kazankov, V. S. Sergeeva, L. A. Efremenko, S. D. Varfolomeev and A. D. Ryabov, Angew. Chem., Int. Ed., 2000, 39, 3117 CrossRef CAS; G. M. Kazankov, V. S. Sergeva, A. A. Borisenko, A. L. Zatsman and A. D. Ryabov, Russ. Chem. Bull., Int. Ed., 2001, 50, 1844 CrossRef CAS.
- J. S. W. Tsang, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2003, 125, 7602 CrossRef CAS.
- For the designation of pH in non-aqueous solvents we use the forms described by Bosch and co-workers10 based on the recommendations of the IUPAC Compendium of Analytical Nomenclature. Definitive Rules 1997, Blackwell, Oxford, UK, 3rd edn., 1998 Search PubMed. If one calibrates the measuring electrode with aqueous buffers and then measures the pH of an aqueous buffer solution, the term sspKa is used; if the electrode is calibrated in water and the ‘pH’ of the neat buffered methanol solution then measured, the term sspKa is used; and if a correction factor of −2.24 (in the case of methanol) is subtracted from the latter reading, then the term sspKais used.
- Given that the autoprotolysis constant of methanol is 10−16.77 (mol dm−3)2, the neutral sspKa in methanol is 8.4; see E. Bosch, F. Rived, M. Rosés and J. Sales, J. Chem. Soc., Perkin Trans. 2, 1999, 1953 Search PubMed.
- R. S. Brown, A. A. Neverov, J. S. W. Tsang, G. T. T. Gibson and P. J. Montoya-Pelaez, Can. J. Chem., 2005, 82, 1.
- (a) A. A. Neverov and R. S. Brown, Org. Biomol. Chem., 2004, 2, 2245 RSC; (b) W. Desloges, A. A. Neverov and R. S. Brown, Inorg. Chem., 2004, 43, 6752 CrossRef CAS.
- J. A. W. Tsang, A. A. Neverov and R. S. Brown, Org. Biomol. Chem., 2004, 2, 3457 RSC.
- A. A. Neverov, T. McDonald, G. Gibson and R. S. Brown, Can. J. Chem., 2001, 79, 1704 CrossRef CAS.
- A. A. Neverov, N. E. Sunderland and R. S. Brown, Org. Biomol. Chem., 2005, 3, 65 RSC.
-
(a) M. M. Benning, J. M. Kuo, F. M. Raushel and H. M. Holden, Biochemistry, 1995, 34, 7973 CrossRef CAS; M. M. Benning, J. M. Kuo, F. M. Raushel and H. M. Holden, Biochemistry, 1994, 33, 15001 CrossRef CAS;
(b) D. P. Dumas, S. R. Caldwell, J. R. Wild and F. M. Raushel, J. Biol. Chem., 1989, 264, 19659 CAS;
(c) G. A. Omburo, J. M. Kuo, L. S. Mullins and F. M. Raushel, J. Biol. Chem., 1992, 267, 13278 CAS;
(d) J. L. Vanhooke, M. M. Benning, F. M. Raushel and H. M. Holden, Biocehmistry, 1996, 35, 6020 Search PubMed;
(e) S.-B. Hong and F. M. Raushel, Biochemistry, 1996, 35, 10904 CrossRef CAS.
- F. Rived, M. Rosés and E. Bosch, Anal. Chim. Acta, 1998, 374, 309 CrossRef CAS.
- V. A. Palm, Tables of Rate and Equilibrium Constants of Heterocyclic Reactions, Vol. 1 (1), Proizbodstvenno-Izdatelckii Kombinat Biniti, Moscow, 1975 Search PubMed; V. A. Palm, Tables of Rate and Equilibrium Constants of Heterocyclic Reactions, Suppl. Vol. 1, issue 3, Tartuskii gosudarsvennii Universitet, Tartu, 1985 Search PubMed.
- B. W. Clare, D. Cook, E. C. F. Ko, Y. C. Mac and A. J. Parker, J. Am. Chem. Soc., 1966, 88, 1911 CrossRef CAS.
- G. T. T. Gibson, A. A. Neverov and R. S. Brown, Can. J. Chem., 2003, 81, 495 CrossRef CAS ; We have determined that the maximal activity for the La3+-catalyzed methanolysis of both carboxylate esters and phosphate triesters lies in the sspKaregion between 8.7 and 9.1, where 95% of the activity is attributed to La3+2(−OCH3)2. Thus, the conditional observed k2La rate constants reported in Tables 1 and 2 are ca. half of the k2 of the La3+2(−OCH3)2 active form.
- Z. S. Chaw, A. Fischer and D. A. R. Happer, J. Chem. Soc. B, 1971, 1818 RSC; J. Kirsch, A. Clewell and A. Simon, J. Org. Chem., 1968, 33, 127 CrossRef CAS; A. A. Humffray and J. J. Ryan, J. Chem. Soc. B, 1967, 468 RSC.
- J. J. Ryan and A. A. Humffray, J. Chem. Soc. B, 1966, 842 RSC; T. C. Bruice and M. Mayahi, J. Am. Chem. Soc., 1960, 82, 3067 CrossRef CAS; J. F. Kirsch and W. P. Jencks, J. Am. Chem. Soc., 1964, 86, 837 CrossRef CAS.
- C. G. Mitton, R. L. Schowen, M. Gresser and J. Shapley, J. Am. Chem. Soc., 1969, 91, 2036 CrossRef CAS.
- S. A. Khan and A. J. Kirby, J. Chem. Soc. B, 1970, 1172 RSC.
- R. Rowell and D. G. Gorenstein, J. Am. Chem. Soc., 1981, 103, 5894 CrossRef CAS.
- S. A. Ba-Saif and A. Williams, J. Org. Chem., 1988, 63, 2204 CrossRef.
- S. A. Ba-Saif, M. A. Waring and A. Williams, J. Am. Chem. Soc., 1990, 112, 8115 CrossRef CAS.
- S. R. Caldwell, F. M. Raushel, P. M. Weiss and W W Cleland, Biochemistry, 1991, 30, 7444 CrossRef CAS.
- W. P. Jencks and M. Gilchrist, J. Am. Chem. Soc., 1968, 90, 2622 CrossRef CAS; D. J. Hupe and W. P. Jencks, J. Am. Chem. Soc., 1977, 99, 451 CrossRef CAS; J. M. Sayer and W. P. Jencks, J. Am. Chem. Soc., 1977, 99, 464 CrossRef CAS; M. J. Gresser and W. P. Jencks, J. Am. Chem. Soc., 1977, 99, 6963 CrossRef CAS.
- (a) S. Thea and A. Williams, Chem. Soc. Rev., 1986, 15, 125 RSC; (b) A. Williams, Acc. Chem. Res., 1984, 17, 425 CrossRef CAS; (c) A. Williams, Concerted Organic and Bio-organic Mechanisms, CRC Press, Boca Raton, FL, 2000 Search PubMed.
- Given that the autoprotolysis constant of methanol is 10−16.77 (mol dm−3)2 (ref. 10) one can compute an sspKa of methanol of 18.13 on the mol dm−3 scale.
-
(a) J. Petrova, S Momchilova, E. T. K. Haupt, J. Kopf and G. Eggers, Phosphorus, Sulfur, Silicon Relat. Elem., 2002, 177, 1337 CrossRef CAS;
(b)
D. R. Peterman, R. V. Fox and H. W. Rollins, Abstract of papers, 223rd ACS National Meeting, Orlando, FL, April 7–11, 2002, NUCL-131;
(c) J. R. Ferraro, A. W. Herlinger and R. Chiarizia, Solvent Extr. Ion Exch., 1998, 16, 775 CrossRef CAS;
(d) R. Du Preez and J. S. S. Preston, South Afr. J. Chem., 1986, 39, 89 Search PubMed;
(e) R. Du Preez and J. S. S. Preston, South Afr. J. Chem., 1986, 39, 137 Search PubMed;
(f) D. F. Peppard, G. W. Mason, W. J. Driscoll and S. J. McCarty, J. Inorg. Nucl. Chem., 1959, 12, 141 CrossRef CAS;
(g) E. N. Lebedeva, M. G. Zaitseva, O. V. Galaktionova, L. V. Bystrov and S. S. Korovin, Koord. Khim., 1981, 7, 870 (
Chem. Abstr.
, 1981
, 95
, 87058
) CAS;
(h) O. V. Galaktionova, E. N. Lebedeva, V. V. Yastrebov and S. S. Korovin, Zh. Neorgan. Khimii, 1980, 25, 2660 (
Chem. Abstr.
, 1980
, 93
, 226562
) Search PubMed;
(i) A. K. Pyartman, A. A. Kopyrin, E. A. Puzikov and K. B. Bogatov, Zhu. Neorgan. Khimii, 1996, 41, 347 (
Chem. Abstr.
, 1996
, 125
, 124905
) Search PubMed;
(j) A. K. Pyartman, A. A. Kopyrin, E. A. Puzikov and K. B. Bogatov, Zhu. Neorgan. Khimii, 1996, 41, 686 (
Chem. Abstr.
, 1996
, 126
, 11927
) Search PubMed;
(k) A. K. Pyartman, V. A. Keskinov, S. V. Kovalev and A. A. Kopyrin, Radiochemistry (Moscow)
(Translation of Radiokhimiya), 1997, 39, 142 (
Chem. Abstr.
, 1997
, 127
, 210889
) Search PubMed.
- C. A. Kosky, J.-P. Gayda, J. F. Gibson, S F. Jones and D. J. Williams, Inorg. Chem., 1982, 21, 3173 CrossRef CAS.
- C. M. Mikulski, L. L. Pytlewski and N. M. Karagannis, Inorg. Chim. Acta, 1979, 32, 263 CrossRef CAS.
- R. Schurhammer, V. Erhart, L. Troxler and G. Wipf, J. Chem. Soc., Perkin Trans. 2, 1999, 2423 RSC and refs. cited therein.
- D. M. Rackham, Spectrosc. Lett., 1980, 13, 513 CrossRef CAS.
- N. H. Williams, W. Cheung and J. Chin, J. Am. Chem. Soc., 1998, 120, 8079 CrossRef CAS; D. Wahnon, A.-M. Lebuis and J. Chin, Angew. Chem., Int. Ed. Engl., 1995, 34, 2412 CrossRef CAS.
- T. L. Ho, Hard and Soft Acids and Bases Principle in Organic Chemistry, Academic Press, New York, 1977 Search PubMed.
- S. D. Aubert, Y. Li and F. M. Raushel, Biochemistry, 2004, 43, 5707 CrossRef CAS.
- M. M. Benning, H. Shim, F. M. Raushel and H. M. Holden, Biochemistry, 2001, 40, 2712 CrossRef CAS.
- M. M. Benning, S.-B. Hong, F. M. Raushel and H. M. Holden, J. Biol. Chem., 2000, 275, 30556 CrossRef CAS.
- V. E. Lewis, W. J. Donarski, J. R. Wild and F. M. Raushel, Biochemistry, 1988, 27, 1591 CrossRef.
- The content of the information does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
| This journal is © The Royal Society of Chemistry 2005 |