Amaryllidaceae and Sceletium alkaloids
Zhong Jin
Institute and State Key Laboratory of Elemento-organic Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: jinzhong2000@eyou.com
First published on 10th January 2005
Abstract
Covering: January 2003 to June 2004. Previous review: Nat. Prod. Rep., 2003, 20, 606
This review covers the morphology, isolation, total synthesis and biological activity of the naturally occurring alkaloids isolated from the Amaryllidaceae family, as well as the structurally related Sceletium alkaloids.
 Zhong Jin Zhong Jin | Born in Nanjin, P. R. China in 1973, Zhong Jin started to study chemistry at Nankai University in 1991. After obtaining his B.Sc., M.Sc. and Ph.D. degrees in organic chemistry from Nankai University, he joined the faculty of Nankai University in 2002. His research interests focus on the discovery of novel natural and unnatural biological molecules, the development of new selective and efficient synthetic methods, and the total syntheses of natural products, especially alkaloids. |
1 Introduction
Alkaloids are an important group of diversely distributed, chemically, biologically and commercially significant natural products. According to a recent analysis of the NAPRALERTSM database, about 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known alkaloids out of 150000 characterized natural products have been reported from a variety of sources including plants, fungi, marine microorganisms, mammals, etc., and the total number of plant-derived alkaloids is 22000 or so. Plants of the Amaryllidaceae family, a small group of monocotyledonous species, belong to the 20 most important alkaloid-containing plant families. Up to now, nearly 500 structurally diverse alkaloids have been isolated from the Amaryllidaceae family with a wide range of interesting biological activities and some of them have been approved for clinical therapy.
000 known alkaloids out of 150000 characterized natural products have been reported from a variety of sources including plants, fungi, marine microorganisms, mammals, etc., and the total number of plant-derived alkaloids is 22000 or so. Plants of the Amaryllidaceae family, a small group of monocotyledonous species, belong to the 20 most important alkaloid-containing plant families. Up to now, nearly 500 structurally diverse alkaloids have been isolated from the Amaryllidaceae family with a wide range of interesting biological activities and some of them have been approved for clinical therapy.2 Amaryllidaceae alkaloids
2.1 Morphology, isolation and structure
Alkaloid metabolism in the genus Galanthus has been investigated by GC/MS and TLC methods.1 Fourteen known alkaloids (trispheridine 1, galanthamine 2, buphanisine 3, norgalanthamine 4, norlycoramine 5, narwedine 6, crinine 7, maritidine 8, haemanthamine 9, tazettine 10, galwesine 11, lycorine 12, N-formylgalanthamine 13, and O-methylleucotamine 14) were identified out of twenty-one Amaryllidaceae alkaloids from sixteen Bulgarian Galanthus elwesii populations. The inter-population and intra-population variability in alkaloid metabolism showed remarkable differences in respect of their type of biosynthesis, the main alkaloid and the number of alkaloids. The alkaloids were, in the metabolic pathways, synthesized at least by three different types of intramolecular phenol oxidative coupling, that is, the coupling between the p–o′, p–p′, and o–p′ positions (phenol-O-methylcatechol) in O-methylnorbelladine (Scheme 1).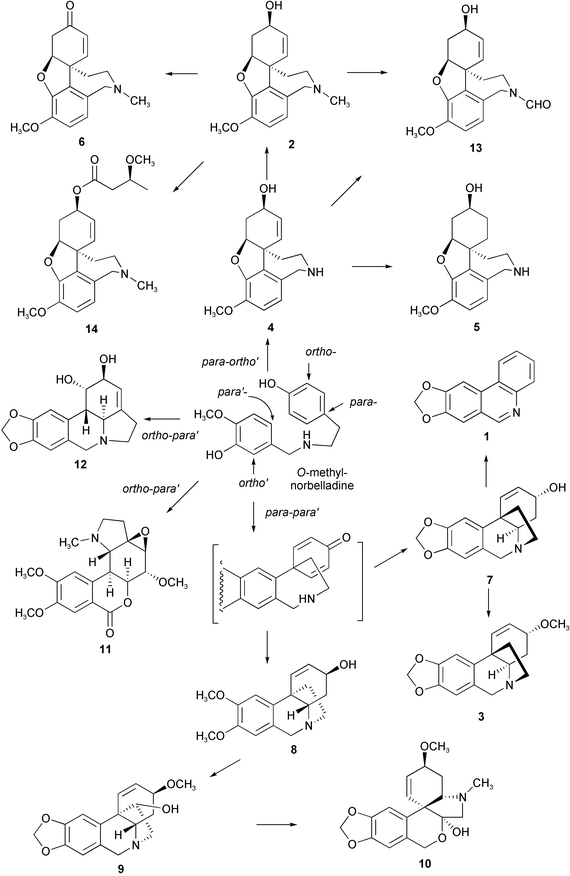 | ||
| Scheme 1 Biosynthetic pathways of the Amaryllidaceae alkaloids. | ||
Galanthamine 2, a quite widespread alkaloid in the Amaryllidaceae family, has become the most attractive of the alkaloids for its use in the treatment of Alzheimer's disease. It is a long-acting, selective, reversible and competitive acetylcholinesterase inhibitor, and produces beneficial effects even after the drug treatment has been terminated. Sixteen populations of Galanthus elwesii Hook fil and five populations of Galanthus nivalis L. growing in Bulgaria have been investigated for the presence of galanthamine by TLC and GC-MS.2 Galanthamine was found in two populations of G. elwesii. The distribution of galanthamine and four other alkaloids, N-formylnorgalanthamine 13, haemanthamine 9, homolycorine 15, and tazettine 10, in different organs (bulbs, leaves, stems, flowers and roots) of Narcissus confusus species, as well as the variations occurring during the ontogenic cycle of this plant species, have also been studied.3 The five alkaloids were found to be present in all the organs at every stage, with the exception of haemanthamine 9 in senescent flowers. A method for rapid qualitative and quantitative determination of galanthamine 2 in Amaryllidaceae plants using reverse-phase HPLC has been developed.4 The separation was achieved by a system consisting of a reversed phase C18 small pore (100 Å) column and TFA–water–acetonitrile (v/v/v 0.01 : 95 : 5) as the mobile phase.
The alkaloids cherylline 16, crinamidine 17, crinine 7, epibuphanisine 18, lycorine 12, powelline 19, undulatine 20, 1-epideacetylbowdensine 21, and 3-O-acetylhamayne 22 have been identified in bulblets of Crinum moorei propagated in vitro.5 Of the nine alkaloids identified, 22 was reported to occur in these bulblets for the first time. In addition, alkaloid production in C. moorei cultures was successfully triggered by manipulating the physical and nutritional environment under artificial conditions for growth.
A new indole alkaloid, galanthindole 23, which incorporated a non-fused indole ring, has been isolated from Galanthus plicatus ssp. byzantinus, a plant native to northwestern Turkey, along with two known alkaloids, hordenine 24 and 11-hydroxyvittatine 25.6 The isolation of hordenine from this plant is particularly interesting, since it probably provides the pendant phenylethyl moiety in the biosynthesis of plicamine 26, which constitutes a recently established novel subgroup of the Amaryllidaceae alkaloids.
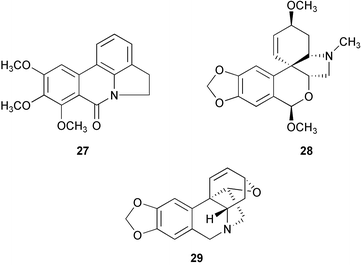
Thirteen alkaloids have been isolated and identified from dried bulbs and leaves of flowering Eucharis amazonica plants. Of these alkaloids, 7-methoxyoxoassoanine 27, 6-O-methylpretazettine 28, and apohaemanthamine 29, are reported for the first time from a natural source.7
Although, in nature, benzo[c]phenanthridine alkaloids found in Papaveraceae and Rutaceae plants do not belong to the Amaryllidaceae family, they are still discussed here because of their close relation to phenanthridine-type Amaryllidaceae alkaloids, as well as their promising biological profiles.
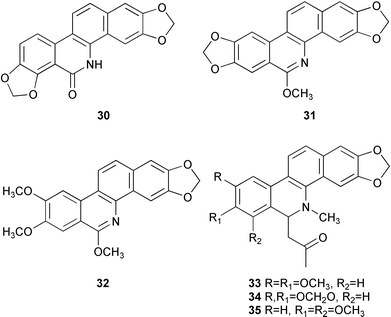
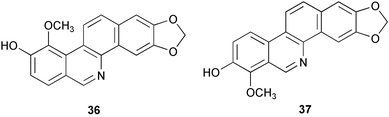
Fractionation of the CHCl3 extracts from the aerial part of Aregemone mexicana led to the isolation of two benzo[c]phenanthridine-type alkaloids, three benzylisoquinoline-type alkaloids, and six known non-alkaloidal compounds. Among them, N-demethyloxysanguinarine 30 was identified as a new alkaloid on the basis of their spectral data and chemical evidence.8 From the stem bark of Zanthoxylum rhoifolium, two new dihydrobenzophenanthridine-type alkaloids, rhoifolines A 31 and B 32, were isolated together with four other previously known benzophenanthridine alkaloids, 6-acetonyldihydronitidine 33 (= 8-acetonyldihydronitidine), 6-acetonyldihydroavicine 34 (= 8-acetonyldihydroavicine), 6-acetonyldihydrochelerythrine 35, and xanthoxyline 36.9 Further fractionation of the CHCl3 extracts of Formosan Argemone mexicana L. led to the isolation of four protopine-type alkaloids and eight known benzophenanthridine alkaloids together with three protoberberine-type alkaloids.10 The known benzo[c]phenanthridine alkaloid, (±)-6-acetonyldihydrochelerythrine 35, exhibited significant anti-HIV activity in H9 lymphocytes with EC50 and TI (Therapeutic Index) values of 1.77 µg mL−1 and 14.6, respectively. One known alkaloid, decarine 37, which showed antiplasmodial activity with IC50 values of 1.44 µM against Plasmodium falciparum D6 clone and 0.88 µM against P. falciparum W2 clone, has been isolated from the stem of Zanthoxylum syncarpum along with a known coumarin and a new norepinephrine derivative.11
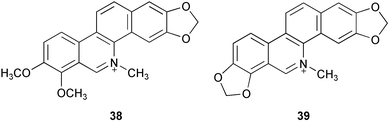
A comparative quantitative determination of the benzo[c]phenanthridine alkaloids chelerythrine 38 and sanguinarine 39 in the leaves of Macleaya microcarpa and Bocconia frutescens plants has been conducted to assess the possibility of using the latter species as a source of benzo[c]phenanthridine alkaloids.12 Furthermore, effects of the extractant type and the methodology of extraction on the total yield of these alkaloids have also been discussed. The maximum total yield of sanguinarine and chelerythrine was obtained using a CHCl3–C2H5OH (v/v 1 : 1) mixture for both M. microcarpa
(1.56%) and B. frutescens
(1.89%). A preparative HPLC method for the separation of sanguinarine and chelerythrine, which optimized the mobile phase focusing on a larger loading mass and volume, has been developed.13 The optimum HPLC mobile phase was 0.05 mol L−1 disodium hydrogen phosphate buffer and acetonitrile (v/v 70 : 30)
(pH = 3–3.5, adjusted by H3PO4).
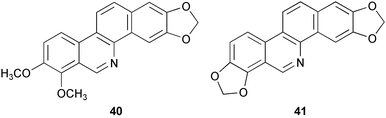
The 1H, 13C, and 15N NMR chemical shifts of two known benzo[c]phenanthridine alkaloids, norchelerythrine 40 and norsanguinarine 41, have been examined by gradient-selected 2D NMR spectroscopy and also extensive theoretical calculations.14 Chemical shifts assignments of the title alkaloids were based on NOE and multiple-bond chemical-shift correlation experiments (GSQMBC).
2.2 Total syntheses of alkaloids and their analogues
![Reagents and conditions: a) NaBH4, MeOH, r.t.; b)
(CH2OH)2, BF3·Et2O, THF, r.t.; c) Boc2O, t-BuOH–H2O, r.t.; d)
[Pd2(dba)3], dppe, TlOAc, CH3CN, reflux; e) 1 M HCl, THF, r.t.; f) SeO2, t-BuOH, AcOH, reflux; g) TFA, CH2Cl2, r.t.; h) NaBH4, CeCl3, MeOH, r.t.; i) MsCl, NEt3, CH2Cl2, r.t. then CsOAc, DMF, r.t., then K2CO3, MeOH, r.t.](/image/article/2005/NP/b316106b/b316106b-s2.gif) | ||
| Scheme 2 Reagents and conditions: a) NaBH4, MeOH, r.t.; b) (CH2OH)2, BF3·Et2O, THF, r.t.; c) Boc2O, t-BuOH–H2O, r.t.; d) [Pd2(dba)3], dppe, TlOAc, CH3CN, reflux; e) 1 M HCl, THF, r.t.; f) SeO2, t-BuOH, AcOH, reflux; g) TFA, CH2Cl2, r.t.; h) NaBH4, CeCl3, MeOH, r.t.; i) MsCl, NEt3, CH2Cl2, r.t. then CsOAc, DMF, r.t., then K2CO3, MeOH, r.t. | ||
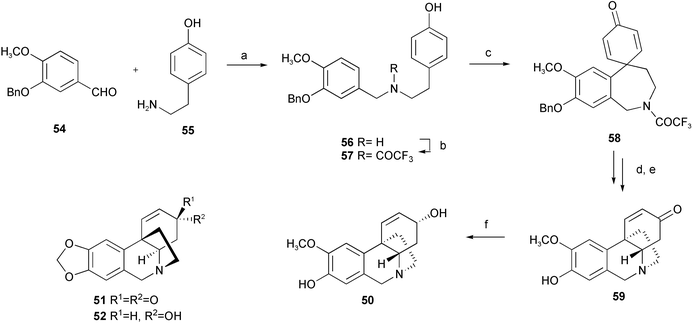
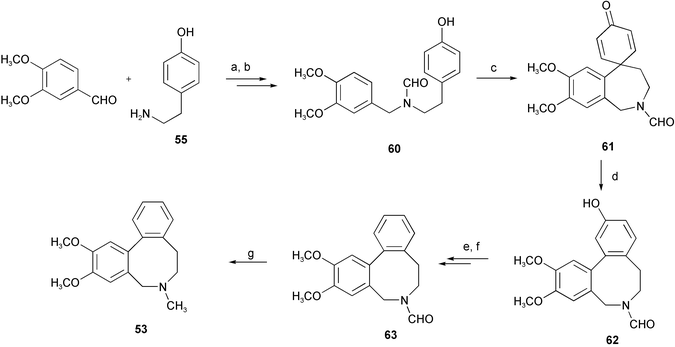
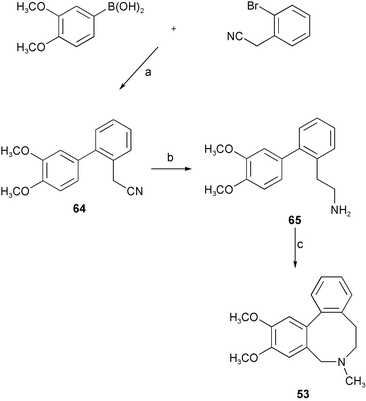
Mimicking the biosynthetic pathway of the Amaryllidaceae alkaloids, the crinine-type alkaloids (−)-siculine 50, (−)-oxocrinine 51, (+)-epicrinine 52, and a unique Amaryllidaceae alkaloid, buflavine 53 isolated from Boophane flava bulbs, have been synthesized by intramolecular biaryl phenol oxidative coupling (Scheme 3).16 Reductive amination of 3-O-benzylisovanillin 54 with tyramine 55 afforded phenethylbenzylamine 56. Protection of the amino group of 56 with the trifluoroacetyl group, followed by oxidation with phenyliodine(III) bis(trifluoroacetate) (PIFA) resulted in phenol coupling to yield dienone 58. Subsequent deprotection of the benzyl group and hydrolysis of the trifluoroacetamide gave the intramolecular Michael adduct 59. Reduction with L-Selectride® afforded alkaloid (±)-siculine 50. The alkaloids (±)-oxocrinine 51 and (±)-epicrinine 52 have been synthesized by similar strategy. With respect to the total synthesis of buflavine 53, attempted direct oxidative coupling to give eight-membered ring product 62 was not successful. Therefore, the seven-membered product 61 of the p–p′ oxidative coupling of 60 with PIFA was converted to 62 by dienone–phenol rearrangement with methanesulfonic acid as a catalyst. Subsequent deoxygenation of phenolic hydroxyl group and reduction of the formamide group gave target buflavine 53 in an overall yield of 64% (Scheme 4). Accordingly, it seems to be an acceptable hypothesis that buflavine might be also generated by the p–p′ coupling followed by a dienone–phenol rearrangement. Another concise and highly efficient total synthesis of buflavine 53 has been achieved from commercially available starting materials in three steps in an overall yield of 44% (Scheme 5).17 Initial Suzuki–Miyaura cross-coupling formed a biaryl ring skeleton. Pictet–Spengler cyclization then N-methylation of the biarylethylamine 65 in a one-pot reaction gave the eight-membered heterocyclic ring alkaloid 53.
 | ||
| Scheme 3 Reagents and conditions: a) NaBH4, r.t.; b) TFAA, pyridine, r.t.; c) PIFA, CF3CH2OH, −25 °C; d) BCl3, CH2Cl2, −78 °C; e) 10% aq. KOH ; f) L-Selectride, THF, −78 °C. | ||
 | ||
| Scheme 4 Reagents and conditions: a) NaBH4, r.t.; b) HCO2Et, reflux; c) PIFA, r.t.; d) MeSO3H, r.t.; e) Tf2O, 0 °C to r.t.; f) Pd(OAc)2, PPh3, Et3N, HCO2H, 60 °C; g) LiAlH4, r.t. | ||
 | ||
| Scheme 5 Reagents and conditions: a) Pd(PPh3)4, toluene–EtOH, H2O, K2CO3; b) NaBH4, CoCl2·6H2O; c) (CH2O)n/HCO2H, reflux. | ||
A new asymmetric total synthesis of (+)-vittatine 66 has been achieved starting from a readily available D-glucose derivative (Scheme 6).18 3-Deoxy-D-glucose derivative 67, prepared from commercially available methyl 4,6-O-benzylidene-α-D-glucopyranoside in two steps, was converted into the chiral cyclohexenone 68 using Ferrier's catalytic carbocyclization reaction as the key transformation. 1,2-Addition of 3,4-(methylenedioxy)phenylmagnesium bromide to 68 gave bicyclic adduct 69 as a diastereomeric mixture. Oxidation with PCC afforded cyclohexenone 70, then was reduced with Luche's reagent to give allyl alcohol 71. Claisen rearrangement successfully constructed the quaternary carbon centre of 72. Reduction with DIBAL-H followed by introduction of an amino function by Mitsunobu amination afforded 74. Treatment with Na–naphthalene removed the N-tosyl and O-benzyl groups to give 75. Construction of the perhydroindole skeleton was achieved by intramolecular aminomercuration–demercuration of 75 to provide 76. Sequential Chugaev reaction and deprotection of the N-Boc group gave 77. Finally Pictet–Spengler cyclization and deprotection of the O-TBS group furnished (+)-vittatine 66.
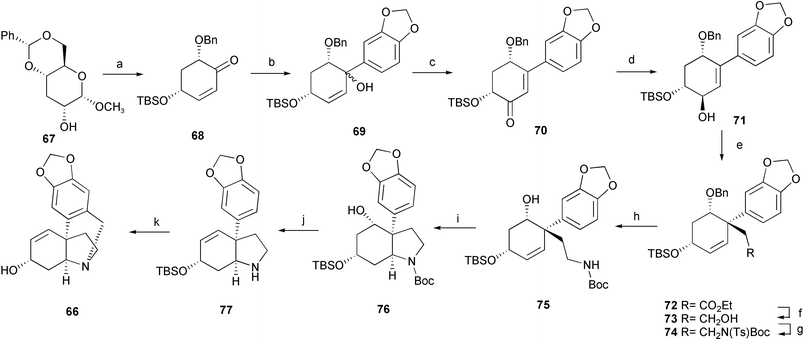 | ||
| Scheme 6 Reagents and conditions: a) (i) DIBAL-H, toluene, r.t., (ii) I2, Ph3P, imidazole, toluene, (iii) TBSCl, imidazole, DMF, (iv) t-BuOK, THF, (v) Hg(OCOCF3)2, then MsCl, Et3N, CH2Cl2; b) 3,4-(methylenedioxy)phenyl bromide, Mg, THF, −100 °C; c) PCC, 4 Å MS, CH2Cl2, r.t.; d) NaBH4, CeCl3·7H2O, MeOH–CH2Cl2 (1 : 1), −78 °C; e) CH3C(OEt)3, cat. EtCO2H, 4 Å MS, 130 °C; f) DIBAL-H, toluene, −78 °C; g) NH(Ts)Boc, PPh3, DEAD, THF, r.t.; h) Na–naphthalene, THF, −40 °C, 90 min; i) Hg(OCOCF3)2, THF, r.t., then NaBH4, 0.5 M aq. NaOH–MeOH, r.t., j) NaH, CS2, MeI, THF, then 1,2-dichlorobenzene, K2CO3, 4 Å MS, 160 °C, then BF3·Et2O, 4 Å MS, CH2Cl2, r.t.; k) formalin, 6 M aq. HCl–MeOH, 50 °C. | ||
Crinine-type alkaloids such as crinine 7, crinamine 78, haemanthamine 9, haemanthidine 79, tazettine 10, precriwelline 80, pretazettine 81, and 6a-epipretazettine 82 have a perhydroindole skeleton connected to an aromatic ring at a quaternary carbon ring junction. The antineoplastic activity exhibited, in particular by pretazettine 81, has stimulated interest in the synthesis of these compounds. Asymmetric total syntheses of (+)-crinamine 78, (−)-haemanthidine 79, and (+)-pretazettine 81 have been accomplished utilizing a palladium-catalyzed asymmetric allylic substitution and a carbonyl-ene reaction as key steps (Scheme 7).19 The starting cyclohexenylamine 85 was obtained by palladium-catalyzed asymmetric allylic amination of a 2-arylcyclohexenol derivative 83. After recrystallization from MeOH, enantiomerically pure 85 was obtained from the mother liquor. The intramolecular thermal carbonyl-ene reaction of (−)-86 proceeded in a highly stereoselective manner to give hexahydroindole derivative 87. Subsequently, oxidation with SeO2 afforded allyl alcohol 88, and methylation with trimethyl orthoformate in the presence of montmorillonite K-10 gave α-methoxylated compound 89a as a major product. The minor β-methoxylated compound 89b was detosylated, and methylenation with Eschenmoser's reagent afforded 90. After deacetylation, (+)-crinamine 78 was obtained. Detosylation of α-methoxylated compound 89a followed by treatment with methyl formate afforded formamide 91, which was treated with POCl3 followed by deacetylation to give alkaloid (−)-haemanthidine 79. The tazettine-type alkaloid (+)-pretazettine 81 was synthesized from (−)-79 according to a known method.
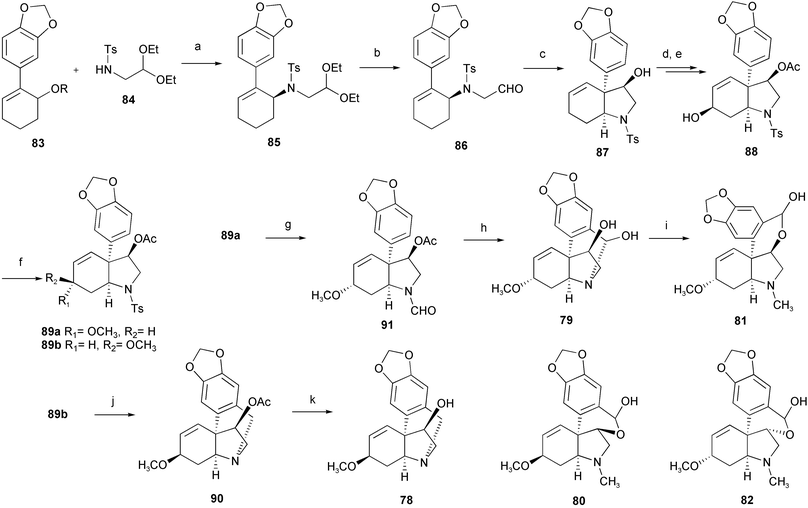 | ||
Scheme 7 Reagents and conditions: a) Pd2(dba)3, (S)-BINAPO, CHCl3; b) 10%, FeCl3–SiO2, acetone, r.t.; c) toluene, 230 °C; d) Ac2O, DMAP, CH2Cl2, 0 °C; e) SeO2, dioxane, reflux; f) Ms2O, Et3N, 0 °C, then MeOH; g) Na–naphthalene, THF, −100 °C, then HCO2Me, DMF, 90 °C; h) POCl3, 80 °C, then THF–H2O, r.t., then K2CO3, MeOH, r.t.; i) MeI, then HCl, then NaHCO3; j) Na–naphthalene, THF, −95 °C, then CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMe2I, MeCN, reflux; k) K2CO3, MeOH, r.t. NMe2I, MeCN, reflux; k) K2CO3, MeOH, r.t. | ||
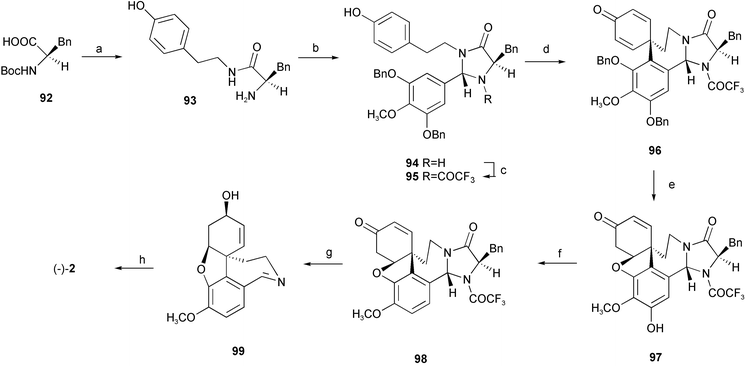 | ||
| Scheme 8 Reagents and conditions: a) tyramine, EDC·HCl, 1-hydroxy-1H-benzotriazole, THF, r.t., then MsOH, MeOH; b) (i) 3,5-dibenzyloxy-4-methoxybenzaldehyde, dioxane, r.t., (ii) HCl, r.t.; c) TFAA, pyridine, 0 °C; d) PIFA, CF3CH2OH, −40 °C; e) BCl3, CH2Cl2, −78 °C; f) (i) Tf2O, pyridine, 0 °C, (ii) Pd(OAc)2, PPh3, Et3N, HCOOH, DMF; g) (i) L-Selectride, THF, −78 °C, (ii) KOH, Bu4NBr, EtOH, 80 °C; h) LiAlH4, THF. | ||
Another synthesis of (−)-galanthamine involved oxidative cyclization of N-phenethylbenzamide 100 to yield (±)-9-bromo-8-oxonarwedine 101. A ketone protection–debromination–amide reduction–resolution sequence provided (−)-narwedine 102, and final stereoselective ketone reduction gave the target (−)-galanthamine (Scheme 9).21
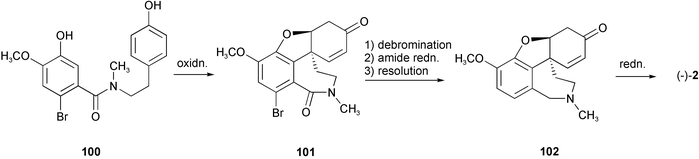 | ||
| Scheme 9 | ||
N-Chiral quaternary N-alkyl galanthaminium halides have been stereoselective prepared from (−)-galanthamine or its derivatives as second generation analogues.22 The stereoselective dealkylation of these quaternary N-alkyl galanthaminium halides has also been achieved using L-Selectride® or SuperHydride®.
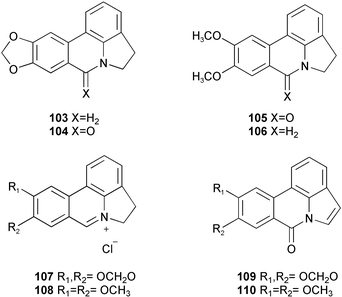
 | ||
| Scheme 10 | ||
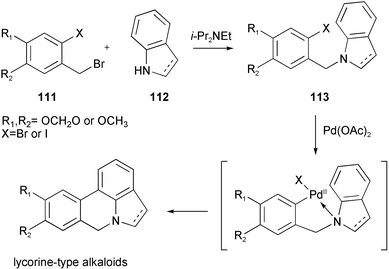
A novel total synthesis of the alkaloids anhydrolycorinone 104 and hippadine 109 has been accomplished using an iron-mediated oxidative alkylamine cyclization and an intramolecular palladium-mediated biaryl coupling as the key steps (Scheme 11).26 The iodopiperonylamine 118 was readily prepared on large scale in four steps from commercially available piperonyl alcohol 114. Cyclohexa-1,3-diene was almost quantitatively transformed to the complex salt 119 by an azadiene-catalyzed reaction with pentacarbonyliron and subsequent hydride abstraction using trityl tetrafluoroborate. The introduction of the side chain was achieved by nucleophilic addition of the lithium ester enolate 120 to afford complex 121. After reduction to aldehyde 122, sequential reductive amination with the hydrochloride of 118 using sodium cyanoborohydride gave the intermediate 123. Oxidative cyclization of complex 123 with ferricenium hexafluorophosphate afforded the tetrahydroindole complex 124. Demetalation with anhydrous trimethylamine N-oxide led to tetrahydroindole 125. Upon treatment with a stoichiometric amount of tetrakis(triphenylphosphine)palladium in the presence of air, a Heck-type coupling, with concomitant aromatization of the cyclohexadiene and oxidation of the benzylic position, occurred to give alkaloid 104. Oxidation of 104 with DDQ provided hippadine 109.
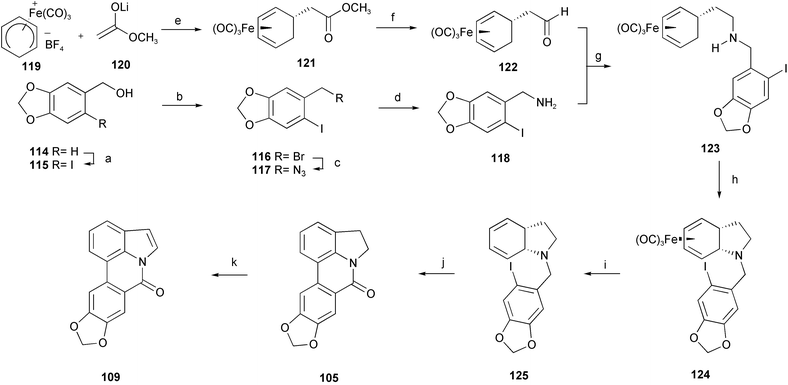 | ||
| Scheme 11 Reagents and conditions: a) I2, AgOCOCF3, CHCl3, −5 °C; b) PBr3, pyridine, Et2O, −20 °C; c) NaN3, DMF; d) PPh3, THF–H2O; e) THF, −78 °C; f) DIBAL-H, toluene, −78 °C; g) NaBH3CN, 4 Å MS; h) Cp2FePF6, Na2CO3, CH2Cl2, 25 °C; i) Me3NO, acetone, reflux; j) Pd(PPh3)4, DMF, 100 °C, air; k) DDQ, CH2Cl2, reflux. | ||
An efficient procedure for indole ring synthesis from 2-ethynylaniline derivatives catalyzed by Cu(II) salts has been used in a formal total synthesis of the alkaloid hippadine 109 (Scheme 12).27N-Boc-2-iodoaniline 127, prepared from commercially available 2-iodoaniline 126, was subjected to the Sonogashira coupling reaction with TMS-acetylene to afford 128. After regioselective lithiation with t-BuLi and subsequent lithium–iodine exchange, the palladium-catalyzed Suzuki–Miyaura coupling reaction between the resulting iodide 129 and the boronic ester 130 produced biaryl 131; removal of the TMS group then provided alkyne 132. Under catalysis by Cu(OAc)2, alkyne 132 was efficiently cyclized to form N-Boc indole 133. After deprotection and N-formylation with acetic formic anhydride, the resulting formamide 135 was subjected to Bischler–Napieralski cyclization with Tf2O, followed by an air oxidation reaction, to directly provide hippadine 109via the cyclic acetal 136.
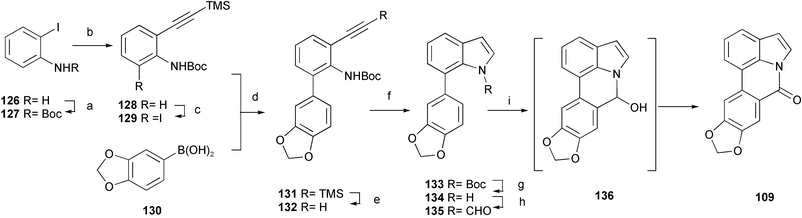 | ||
| Scheme 12 Reagents and conditions: a) Boc2O, THF, reflux; b) TMS-acetylene, CuI, PdCl2(PPh3)2, Et3N, reflux; c) t-BuLi, −20 °C, then ICH2CH2I, −100 °C; d) Pd(PPh3)4, Na2CO3, EtOH–benzene (3 : 1), reflux; e) K2CO3, MeOH; f) Cu(OAc)2, 1,2-dichloroethane, reflux; g) 3 M HCl, AcOEt; h) NaH, MeCO2CHO, DMF; i) Tf2O, DMAP, CH2Cl2. | ||
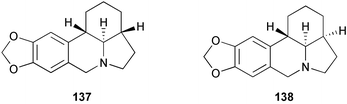
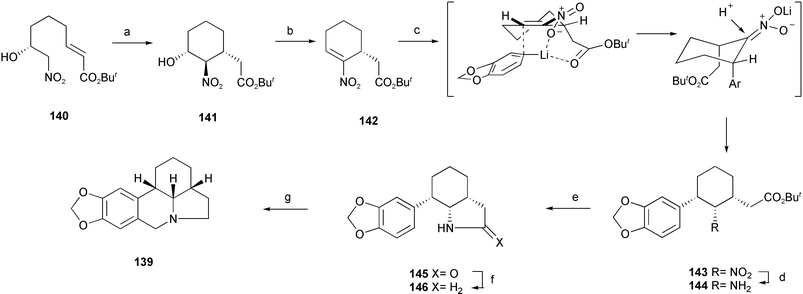
As the central core of lycorine-type Amaryllidaceae alkaloids, alkaloids α-lycorane 137, β-lycorane 138, and γ-lycorane 139 were characterized by the presence of the tetracyclic pyrrolo[d, e]phenanthridine ring system. Employing the stereoselective conjugate addition to nitroolefin and cyclization methodology, (±)-γ-lycorane 139 has been efficiently synthesized.28 Intramolecular nitro-Michael cyclization of the starting material 140, prepared by reported nitro-aldol procedures, gave adduct 141 highly stereoselectively. Dehydration of 141 under Mitsunobu conditions smoothly afforded nitroolefin 142. After stereoselective 1,4-conjugate addition of 3,4-methylenedioxyphenyllithium to 142 and subsequent reduction of the nitro group, an amine 144 was obtained as sole product in high yield. Upon treatment with sodium methoxide, 144 cyclized quantitatively to lactam 145, which was reduced. Pictet–Spengler cyclization then afforded (±)-γ-lycorane 139
(Scheme 13).
 | ||
| Scheme 13 Reagents and conditions: a) CsF, n-C13H27NMe3Br; b) DEAD, PPh3, HCO2H, THF, r.t.; c) 3,4-methylenedioxyphenyllithium, THF, −78 °C; d) Zn powder, 10% HCl, EtOH; e) MeONa, MeOH; f) BH3·THF, THF; g) 37% formalin, 6 M HCl–MeOH, r.t. | ||
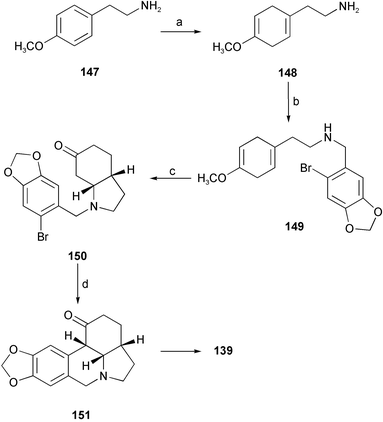
Another formal total synthesis of (±)-γ-lycorane 139, involving an intramolecular Michael addition and a Pd-mediated arylation of a ketone, has been reported.29 Starting from commercially available 2-(4-methoxyphenyl)ethylamine 147, a Birch reduction furnished the diene 148. Reductive amination with 6-bromopiperonal then afforded the secondary amine 149. Intramolecular Michael addition provided the octahydroindol-6-one 150. Finally, intramolecular arylation of 150 with Pd(0) and sodium tert-butoxide afforded the desired 151, which was converted to (±)-γ-lycorane 139 according to the literature (Scheme 14).
 | ||
| Scheme 14 Reagents and conditions: a) Na, NH3(l), THF, EtOH, −78 °C; b) (i) 2-bromo-4,5-methylenedioxybenzaldehyde, EtOH, 50 °C, (ii) NaBH4, EtOH, 0 °C; c) (i) 4 M HCl, MeOH, (ii) K2CO3, MeOH–H2O; d) Pd(dba)3, t-BuONa, toluene. | ||
An unusual five-step strategy for synthesis of lycorane tetracyclic skeleton has been developed using an intramolecular de Mayo reaction of an isocarbostyril substrate as the key step.30 The synthesis of the key precursor, isocarbostyril 155, began with a Michael addition followed by hydrolysis of the resulting ester 153. The acid chloride derived from 153 was used to acylate sodium tert-butyl acetoacetate resulting in the desired ester 154. Cleavage of the ester with an accompanying decarboxylation completed the synthesis of 155 in high yield. Intramolecular de Mayo photocyclization of 155 gave a single product 156 after purification by flash chromatography. Final aldol ring closure provided the lycorane analogue 157
(Scheme 15).
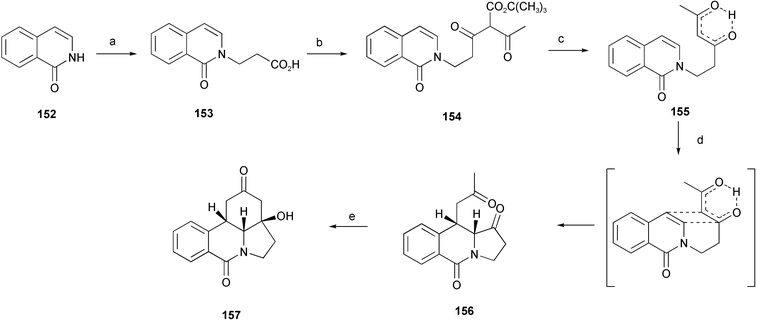 | ||
| Scheme 15 Reagents and conditions: a) methyl acrylate, cat. MeONa, then 1 M HCl reflux; b) SOCl2, then CH3COCH2CO2t-Bu, NaH; c) TsOH, benzene, reflux; d) hν; e) piperidine, toluene, reflux. | ||
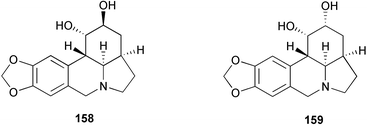
The lycorine-type alkaloids of the Amaryllidaceae family, dihydrolycorine 158, zephyranthine 159, and epi-zephyranthine 160, contain a 1,2-diol unit whose stereochemistry is a challenge for modern synthetic methodology. A novel total synthesis of epi-zephyranthine has been achieved, incorporating an intramolecular Diels–Alder reaction of a 2-imido-substituted furan and a Rh(I)-catalyzed ring opening of the resulting oxabicyclic adduct (Scheme 16).31 Addition of lithium carbamate 161 to the mixed anhydride 162 provided the expected imidofuran 163, which rapidly reacted to deliver the Diels–Alder cycloadduct 164. With catalysis by [Rh(COD)Cl]2 and DPPF in the presence of phenylboronic acid, the cis-diol boronate 165 was formed exclusively. Boronate 165 was cleaved to the corresponding diol which was protected as dioxolane 166 and converted to the corresponding benzamide 168 by removal of the N-Boc group followed by benzoylation with the acid chloride 167. Exposure of 168 to n-Bu3SnH in the presence of AIBN afforded the anticipated tetracyclic product 169. Reduction of 169 with BH3·THF followed by hydrolysis of the 1,3-dioxolane furnished epi-zephyranthine 160.
![Reagents and conditions: a) THF; b) 25 °C; c) PhB(OH)2, [Rh(COD)Cl]2, DPPF, THF; d) HCl, pinacol, then Me2C(OMe)2; e) Mg(ClO4)2, MeCN; f) AIBN, Bu3SnH, benzene, reflux; g) BH3·THF, then 3% HCl, MeOH.](/image/article/2005/NP/b316106b/b316106b-s16.gif) | ||
| Scheme 16 Reagents and conditions: a) THF; b) 25 °C; c) PhB(OH)2, [Rh(COD)Cl]2, DPPF, THF; d) HCl, pinacol, then Me2C(OMe)2; e) Mg(ClO4)2, MeCN; f) AIBN, Bu3SnH, benzene, reflux; g) BH3·THF, then 3% HCl, MeOH. | ||
 | ||
| Scheme 17 Reagents and conditions: a) Br2, AcONa, AcOH, r.t.; b) 1,3-propanediol, p-TsOH, toluene, reflux; c) t-BuLi, THF, then 4-O-benzylbenzaldehyde; d) PDC, CH2Cl2; e) Ph2P(O)H, toluene, reflux; f) CH2O, NaBH3CN, MeCN, r.t.; g) n-BuLi, THF, r.t. then NH4Cl; h) NaBH3CN, MeOH–HCl; i) p-TsOH, toluene, H2O, reflux, then HPLC, Chiralcel OD, ethanol–heptane; j) Pd/C, H2, MeOH, r.t.; k) p-TsOH, MeOH–toluene, reflux. | ||
 | ||
| Scheme 18 Reagents and conditions: a) n-BuLi, THF, −100 °C, then 2-isopropoxy-3-methoxybenzaldehyde; b) PDC, CH2Cl2; c) n-BuLi, THF; d) NaBH3CN, MeOH–HCl, then HPLC, Chiralcel OD, ethanol–heptane; e) H2, Pd(OH)2/C, MeOH; f) (CH2O)n, aq. HCl, EtOH, reflux; g) BCl3, CH2Cl2, 0 °C. | ||
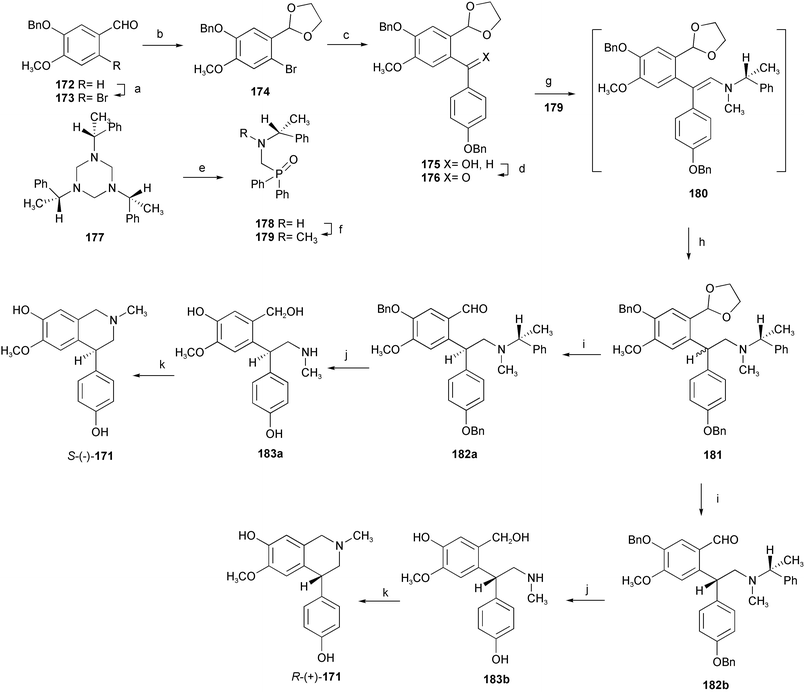
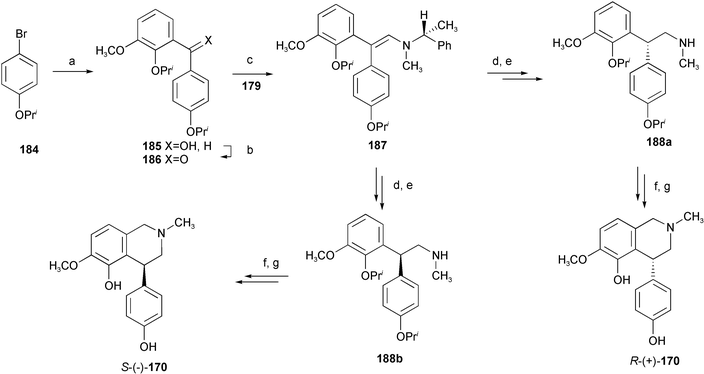
A novel and concise synthetic strategy for both enantiopure antipodes of alkaloid cherylline 171 has been devised relying upon the reduction of a polyprotected diarylenamine 180 bearing a chiral auxiliary.32 The desired diarylketone derivative 176 was prepared from benzyl-protected isovanillin 172 in four steps with a satisfactory yield. Subsequent Horner reaction between the diarylketone 176 and the phosphorylated methylamine 179 bearing a chiral auxiliary provided the rather unstable diarylenamine 180, which was reduced to saturated tertiary amine 181. Sequential regeneration of the formyl functionality and preparative HPLC separation furnished the pure diastereoisomers 182a and 182b. Deprotection by catalytic hydrogenation and final annulation reaction delivered the target alkaloid S-(−)-171 and its antipode (Scheme 17). The structurally related alkaloid (−)-latifine 170 and its (+)-antipode have also been prepared by an analogous strategy (Scheme 18).33
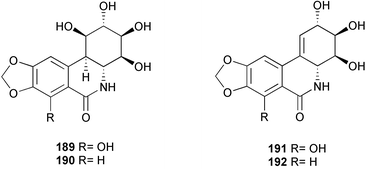
An elaborate total synthesis of pancratistatin 189 has been accomplished from readily available starting materials (Scheme 19).34 A Horner–Wadsworth–Emmons reaction between phosphonate 194, prepared from the known bromide 193, and acrolein dimer 195 afforded the desired (E)-olefin 196 with high stereoselectivity. A thermal Claisen rearrangement then provided the cis-disubstituted cyclohexene 197, the aldehyde group of which was oxidized to the corresponding carboxylic acid. Iodolactonization of acid 198 under two-phase conditions, followed by treatment of the resulting iodolactone 199 with DBU, led to the formation of the bicyclic lactone 200. Methanolysis of the lactone 200 and epimerization of the methoxycarbonyl group furnished trans-isomer 201. Saponification of the methyl ester, followed by a modified Curtius rearrangement of the resulting acid 202, gave a rather stable isocyanate intermediate which was further treated with MeONa–MeOH to generate the corresponding carbamate 203. After protection of the free hydroxyl group with benzoyl chloride to furnish 204, dihydroxylation of the Δ2,3-olefin with OsO4 afforded diol 205 smoothly. Regioselective elimination of the C-3 hydroxyl group of the diol 205 to generate the requisite Δ3,4-olefin 207 was achieved by employing the cyclic sulfate elimination reaction. Again, cis-dihydroxylation with OsO4 afforded highly functional intermediate 208. Peracetylation followed by a modified Bischler–Napieralski cyclization provided mainly the desired product 209. Finally, removal of the O-methyl group and other protecting groups afforded (±)-pancratistatin 189.
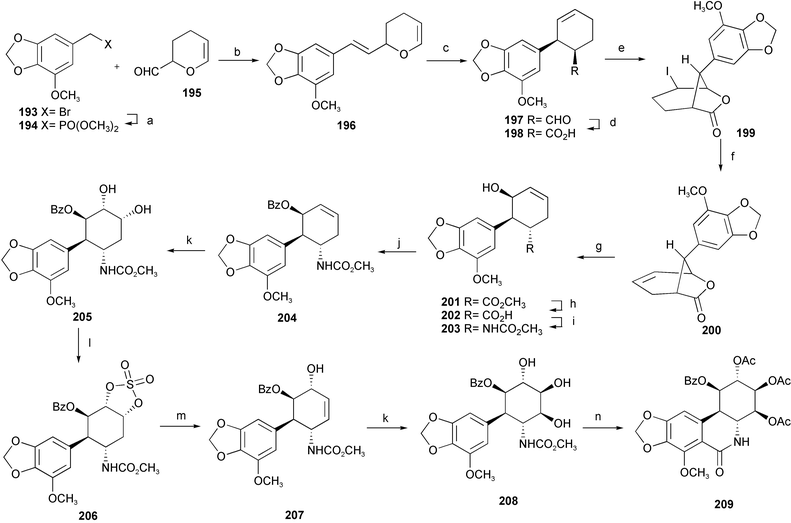 | ||
| Scheme 19 Reagents and conditions: a) P(OMe)3, sealed tube, toluene, 180 °C; b) n-BuLi, THF; c) toluene, sealed tube, 250 °C; d) 2-methylbutene, NaClO2, NaH2PO4, t-BuOH, H2O, 0 °C; e) KI3, NaHCO3, H2O, CH2Cl2; f) DBU, benzene, reflux; g) MeONa, MeOH, reflux; h) LiOH, THF, r.t.; i) DPPA, Et3N, toluene, reflux, then MeONa, MeOH, reflux; j) BzCl, Et3N, DMAP, CH2Cl2; k) OsO4, NMO, THF–H2O, r.t.; l) SOCl2, Et3N, CH2Cl2, then Oxone, RuCl3·3H2O, EtOAc–MeCN–H2O, r.t.; m) DBU, toluene, reflux, then H2SO4, THF–H2O, r.t.; n) Ac2O, DMAP, pyridine, CH2Cl2, r.t., then Tf2O, DMAP. | ||
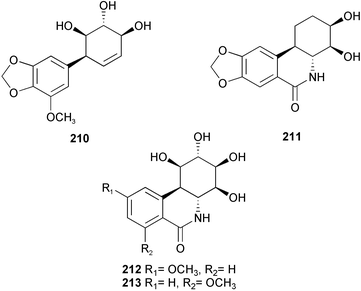
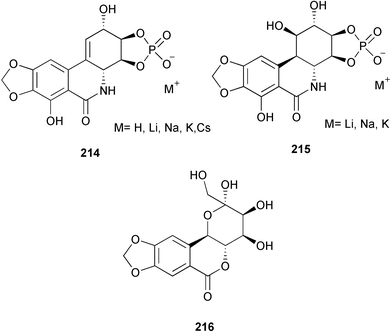
In an approach towards an efficient total synthesis of pancratistatin and its congeners, 1-aryl-1-deoxyconduritol F 210 has been prepared from the readily available monosaccharide D-xylose by a ring-closing metathesis and a diastereoselective aryl cuprate addition sequence.35 Some structural analogues of pancratistatin (211, 212, and 213) have been synthesized and evaluated in cancer cell line inhibition studies.36 From these studies, it seems that enhanced activity in pancratistatin-type compounds cannot be expected in compounds which do not have the completely functionalized cyclitol ring or that lack the full phenanthridone nucleus. Due to the low solubility properties of narciclasine and closely related pancratistatin, a series of phosphorylated derivatives 214 and 215 have been prepared using cation-exchange chromatography for preclinical development.37,38 In addition, for a systematic investigation to establish the minimum structure of the pharmacophore, a carbohydrate analogue 216 of pancratistatin has been stereospecifically synthesized in a synthesis involving three enzymic steps, the first being a dihydroxylation of 2,3-(methylenedioxy)naphthalene by a dioxygenase involved in microbial arene degradation.39
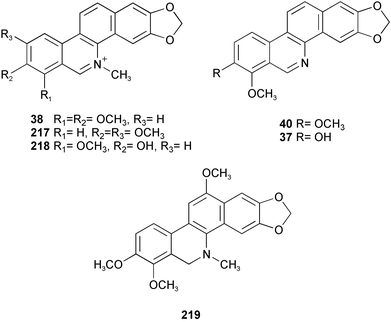
Naturally occurring benzo[c]phenanthridine alkaloids, such as chelerythrine 38, nitidine 217, fagaridine 218, nor-chelerythrine 40, decarine 37, and 12-methoxydihydrochelerythrine 219, have, in view of their significant biological activities, been approached synthetically by various methods. A short synthesis of fagaridine 218 and decarine 37 has been accomplished via an intramolecular Pd-mediated aryl–aryl coupling reaction (Scheme 20).40
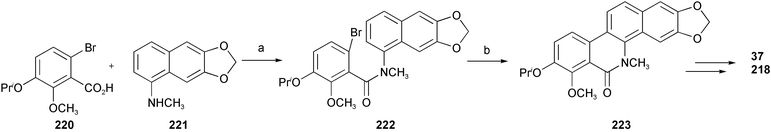 | ||
| Scheme 20 Reagents and conditions: a) oxalyl chloride, Et3N; b) Pd(OAc)2, P(o-Tol)3, DPPP. | ||
A concise total synthesis of 12-methoxydihydrochelerythrine 219 has been described featuring an efficient route to a 12-alkoxybenzo[c]phenanthridine skeleton via naphthoquinone monooxime as a key intermediate.41 Initial Friedel–Crafts acylation of 224 with homopiperonoyl chloride 225 afforded a deoxybenzoin derivative 226. Reformatsky reaction of 226 with ethyl bromoacetate gave the hydroxyester 227. Deoxygenation followed by alkaline hydrolysis quantitatively produced the acid 229. After cyclization, the resulting ketone 230 was aromatized into 1-naphthol 231 by DDQ and the phenolic group was protected by benzylation. Sequential oxidation with OsO4 and HIO4 followed by methylation with Me2SO4 afforded a methyl ester 233. After removal of the benzyl group, treatment of naphthol 234 with isoamyl nitrite followed by methylation with dimethyl sulfate afforded the desired quinone monooxime 235. Benzo[c]phenanthridine ring system 236 was smoothly constructed under reductive cyclization conditions. Mono-benzoylation of 236 and then 6-deoxygenation provided the base 237. Finally, double methylation at the N- and O-functions under reductive conditions followed by reoxidation afforded the target alkaloid 219 (Scheme 21).
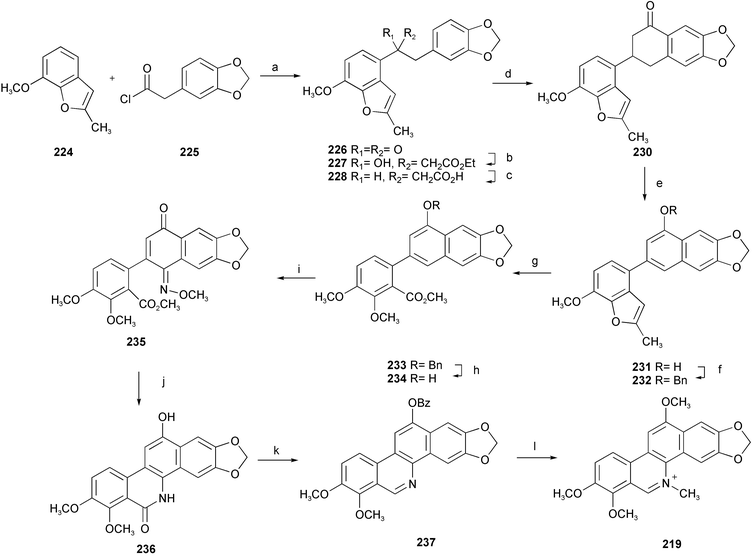 | ||
| Scheme 21 Reagents and conditions: a) SnCl4, CH2Cl2, −15 °C; b) BrCH2CO2Et, Zn, I2, THF, reflux; c) Et3SiH, TFA, CH2Cl2, 0 °C, then KOH, EtOH, reflux; d) POCl3, K2CO3, MeCN; e) TsOH, isopropenyl acetate, then DDQ, r.t., then NaOH, EtOH, 85 °C; f) BnBr, K2CO3, DMF, 50 °C; g) OsO4, pyridine, 30 °C, then Na2SO3, then HIO4, H2O–dioxane, r.t., then aq. NaOH, then Me2SO4, BnBu3NCl, aq. NaOH, benzene; h) Pd/C, H2, AcOH; i) i-C5H11ONO, K2CO3, DMF, then Me2SO4; j) Pd/C, H2, AcOH; k) Bz2O, then Tf2O, i-Pr2NEt, 0 °C, then Pd(OAc)2, dppp, Et3SiH, DMF; l) (i) MeONa, MeOH, (ii) Me2SO4, NaBH4, HMPA, (iii) DDQ, NaOH, benzene, then HCl. | ||
Employing a lithiated toluamide–benzonitrile cycloaddition as the key synthetic step, a concise total synthesis of benzo[c]phenanthidine alkaloids has been achieved.42,43 The cycloaddition between benzonitrile 241 and lithiated o-toluamides 238–240 afforded 3-arylisoquinolines 242–244, which were finally transformed into the benzo[c]phenanthidine alkaloids oxynitidine 245, oxysanguinarine 246 and oxyavcine 247 by sequential protection, deprotection and oxidation cyclization workup (Scheme 22).
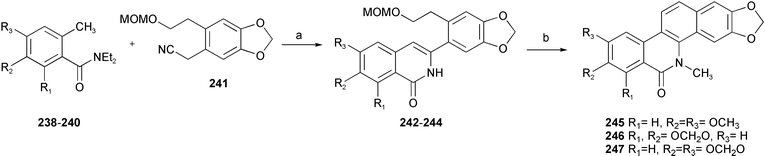 | ||
| Scheme 22 Reagents and conditions: a) (i) n-BuLi, THF, (ii) 10% HCl, THF; b) (i) K2CO3, MeI, DMF, (ii) 10% HCl, THF, (iii) PCC, NaOAc, CH2Cl2. | ||
2.3 Biological activity
From Crinum latifolium L. and Crinum asiaticum L. of the Amaryllidaceae family, mixtures containing all the alkaloids were obtained, from which seven known alkaloids (pratorinine 248, crinine 7, crinamidine 17, lycorine 12, 1-O-acetyllycorine 249, 1,2-di-O-acetyllycorine 250, and lycorine-1,2-di-O-β-D-glucoside 251) were isolated.44 The mixtures of all alkaloids showed strongly antimicrobial effects against Sarcina lutea, while pratorinine 248 was found to be active against Staphylococcus aureus. In addition, the mixture of all the alkaloids from C. latifolium demonstrated a significant cytotoxic response in the cell lines Hep-G2, RD, and Fl. Among the individual alkaloids evaluated, lycorine showed a very strong cytotoxic activity on Hep-G2, RD and Fl, and 1-O-acetyllycorine 249 exhibited a moderate activity on Hep-G2. The remaining alkaloids were found not to be cytotoxic in any of the cell lines tested.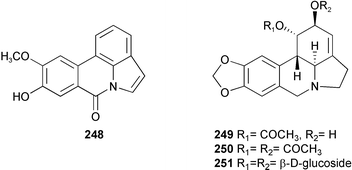
Investigation of the antimalarial potential of three Amaryllidaceae plants collected from the Pancratium maritimum L., Leucojum aestivum L., and Narcissus tazetta L. ssp. tazetta in Turkey has been undertaken. Plant extracts of the Amaryllidaceae species as well as their alkaloids, including four subtypes of alkaloids, namely lycorine-, crinine-, tazettine- and galanthamine-type, were evaluated in vitro for their ability to inhibit Plasmodium falciparum growth using a high-throughput screening method with a 96-well microtiter plate. All four groups of alkaloids exhibited antimalarial activity at different potencies. 6-Hydroxyhaemanthamine 252, haemanthamine 9 and lycorine 12 were found to the most potent alkaloids against chloroquine-sensitive P. falciparum
(T9.96), while galanthamine 2 and tazettine 10 had the least potent activity against chloroquine-resistant P. falciparum
(K1).45
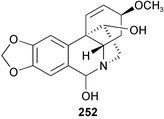
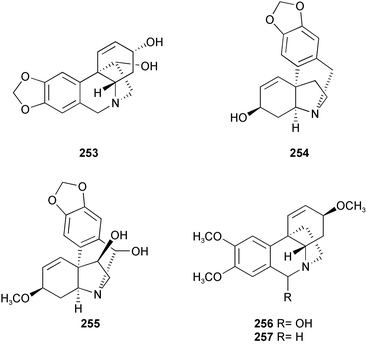
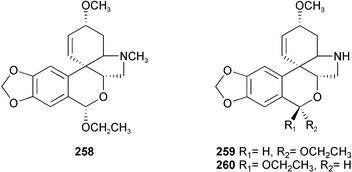
Twenty-three Amaryllidaceae alkaloids belonging to five ring types, isolated from Crinum moorei, C. macowanii, C. bulbispermum, and Cyrtanthus falcatus,46 have been evaluated for their acetylcholinesterase enzyme (AChE) inhibitory activity.47 Crinine 7, epibuphanisine 18, powelline 19, crinamidine 17, undulatine 20, hamayne 253, 3-O-acetylhamayne 22, epivittatine 254, crinamine 78, 6-hydroxycrinamine 255, 1-epideacetylbowdensine 21, papyramine 256
(mixture of two epimers), maritidine 8, and O-methylmaritidine 257 represent the crinine-type alkaloids. Tazettine 10, 8-α-ethoxyprecriwelline 258, N-desmethyl-8α-ethoxypretazettine 259, and N-desmethyl-8β-ethoxypretazettine 260 are tazettine-type alkaloids, while lycorine 12, 1-O-acetyllycorine 249, and 1,2-di-O-acetyllycorine 250 represent the lycorine-type alkaloids. Cherylline 16 and galanthamine 2, which was used as a positive control, represent the fourth and fifth ring types respectively. The lycorine-type alkaloids have proven to be the most active alkaloids, with 1-O-acetyllycorine 249 exhibiting inhibitory effects two times more potent than that of galanthamine 2. Although the mechanism of action of the alkaloids investigated in the study was unknown, a crystal structure has shown galanthamine 2 binding to the active-site gorge of AChE.48
Crinum giganteum, a medicinal plant of the Amaryllidaceae family, is widespread in the northern states of Nigeria. The aqueous extract of C. giganteum has shown central nervous system inhibitory activity.49
Visible delay of tepal senescence in cut Iris × hollandica (cv. Blue Magic) flowers has been observed by adding one cut daffodil flower (Narcisus pseudonarcissus, cv. Carlton) or mucilage exuded by daffodil stems to the vase water.50 Using LC/MS, GC/MS, 1H and 13C-NMR, alkaloid narciclasine 191 was identified as the active compound in the mucilage. The delay of senescence, either by mucilage or purified narciclasine 191, was correlated with a delayed increase in protease activity, and with a considerable reduction of maximum protease activity. Its effects on senescence and protease activity were similar to those of cycloheximide (CHX), another inhibitor of protein synthesis, but its effect was about 100 times higher than that of CHX.
From bulbs of fifteen wild Narcissus species growing in Spain, mannose-specific lectins (MSLs) have been extracted and assayed for their HIV-1 infection inhibitory activity in MT-4 cells. By comparison with the Narcissus pseudonarcissus agglutinin (NPA), the commercially available MSL obtained from daffodils, almost all the tested MSLs showed EC50 values similar to that of NPA, with some being comparable to those obtained with dextran sulfate and azidothymidine (AZT) without significant cytotoxicity.51
Galanthamine (Reminyl®) 2, as an approved treatment for Alzheimer's disease (AD), is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, and α6β4 nicotinic receptors (nAChRs). By comparison with expression on five CHO-SRE-Luci cell lines, it has been confirmed that galanthamine selectively modulated the activity of nAChRs and could not alter the activity of muscarinic acetylcholine receptors (M1–M5). The therapeutic action of galanthamine is mainly produced by its sensitizing action on nAChRs rather than by general cholinergic enhancement due to cholinesterase inhibition.52
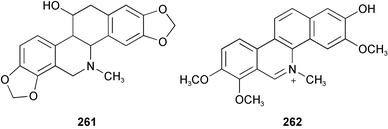
Benzo[c]phenanthridine alkaloids sanguinarine 39 and chelidonine 261, isolated from the plants Chelidonium majus L. and Macleaya cordata and microcarpa, showed irreversible inhibitory activity for the oxidative deamination reaction of serotonin and tyramine, catalyzed by mitochondrial monoamine oxidase (MAO).53 Quaternary benzo[c]phenanthridine alkaloids (QBA), such as fagaronine 262, chelerythrine 38, sanguinarine 39, and the QBA extract from Macleaya cordata plants exerted differential inhibitory effect on the hydrolytic activity of particular dipeptidyl peptidase (DPP)-like enzymes isolated from human blood plasma and from human and rat glioma cell lines.54 Bioassay-guided isolation of an alkaloid extract from the bark ash of Zanthoxylum clava-herculis led to the characterization of the benzo[c]phenanthridine alkaloid chelerythrine 38 as the major active principle. It displayed potent activity against strains of methicillin-resistant Staphylococcus aureus
(MRSA), which are highly resistant to clinically useful antibiotics by means of their multi-drug efflux mechanisms.55 From the rhizomes of Sanguinaia canadensis and the roots and rhizomes of Hydrastis canadensis, two plants used traditionally for the treatment of gastrointestinal ailments, chelerythrine 38 and sanguinarine 39 were identified and screened for in vitro antibacterial activity against fifteen strains of Helicobacter pylori. They inhibited the growth of the bacterium with an MIC50 value of 50.0 and 100.0 µg mL−1, respectively.56
3 Sceletium alkaloids
The Sceletium alkaloid mesembrine 263, which is a naturally occurring serotonin uptake inhibitor, is an octahydroindole skeleton natural product isolated from certain plants of the Sceletium genus of the Mesembryanthemaceae family. Employing a double Michael addition as the key step, a simple and efficient synthetic strategy for (±)-mesembrine has been developed.57 Initial double Michael addition of 3,4-dimethoxyphenylacetonitrile 264 to methyl acrylate 265 furnished diester 266. Sequential intramolecular Dieckmann condensation followed by decarboxylation yielded a cyclohexanone 268, the ketone group of which was protected to give a 1,3-dioxolane 269. The cyano group was then reduced to give the aldehyde 270 and then homologated by a Wittig reaction and hydroboration followed by oxidative work up to provide the alcohol 272, which was converted to a secondary amine 273 and then protected as its carbamate 274. Finally, after enone formation and deprotection, the octahydroindole skeleton was smoothly constructed by an intramolecular Michael addition (Scheme 23).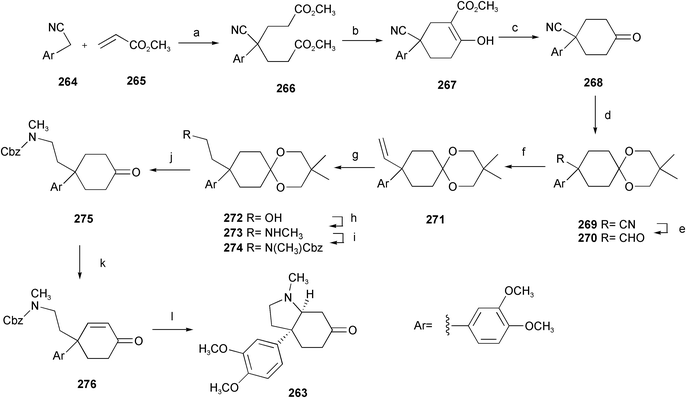 | ||
| Scheme 23 Reagents and conditions: a) 10% NaOH, TBAHSO4; b) NaH, DME, reflux; c) NaCl, DMSO, 140 °C; d) (CH3)2C(CH2OH)2, PPTS, benzene, reflux; e) DIBAL-H, CH2Cl2; f) Ph3PCH3I, NaNH2, Et2O–THF; g) BH3·SMe2, THF, then 30% H2O2, 30% NaOH; h) Et3N, MsCl, then 40% aq. MeNH2, THF–H2O, 80 °C; i) CbzCl, K2CO3, r.t.; j) acetone, H2O, cat. H2SO4; k) Et3N, TMSCl, MeCN, then NBS, THF, then Li2CO3, LiBr, DMF, 110 °C; l) BF3Et2O, Me2S, CH2Cl2, 0 °C to r.t. | ||
4 References
- S. Berkov, B. Sidjimova, L. Evstatieva and S. Popov, Phytochemistry, 2004, 65, 579 CrossRef CAS.
- B. Sidjimova, S. Berkov, S. Popov and L. Evstatieva, Pharmazie, 2003, 58, 935.
- S. Lopez, J. Bastida, F. Viladomat and C. Codina, Planta Med., 2003, 69, 1166 CrossRef CAS.
- N. R. Mustafa, J. K. Rhee and R. Verpoorte, J. Liq. Chromatogr. Relat. Technol., 2003, 26, 3217 CrossRef.
- C. W. Fennell, E. E. Elgorashi and J. van Staden, J. Nat. Prod., 2003, 66, 1524 CAS.
- N. Unver, G. I. Kaya, C. Werner, R. Verpoorte and B. Gozler, Planta Med., 2003, 69, 869 CrossRef CAS.
- F. Cabezas, A. Ramirez, F. Viladomat, C. Codina and J. Bastida, Chem. Pharm. Bull., 2003, 51, 315 CrossRef CAS.
- Y. Chang, F. Chang, A. T. Khalil, P. Hsieh and Y. Wu, Z. Naturforsch., C: Biosci., 2003, 58, 521 CAS.
- W. A. Conzaga, A. D. Weber, S. R. Glacomelli, I. I. Dalcol, S. C. S. Hoelzel and A. F. Morel, Planta Med., 2003, 69, 371 CrossRef.
- Y. Chang, P. Hsieh, F. Chang, R. Wu, C. Liaw, K. Lee and Y. Wu, Planta Med., 2003, 69, 148 CrossRef CAS.
- S. A. Ross, G. N. N. Sultana, C. L. Burandt, M. A. Elsohly, J. P. Marais and D. Ferreira, J. Nat. Prod., 2004, 67, 88 CrossRef CAS.
- (a) E. A. Abizov, O. N. Tolkachev, I. E. Kopylova and A. N. Luferov, Khim.-Farm. Zh., 2003, 37, 350 CAS; (b) E. A. Abizov, O. N. Tolkachev, I. E. Kopylova and A. N. Luferov, Chem. Abstr., 2004, 140, 292351.
- F. Zhang, B. Chen, X. Luo, X. Zhu and S. Yao, J. Liq. Chromatogr. Relat. Technol., 2004, 27, 1491 CrossRef CAS.
- J. Tousek, J. Dostal and R. Marek, J. Mol. Struct., 2004, 689, 115 CrossRef CAS.
- C. Bru, C. Thal and C. Guiliou, Org. Lett., 2003, 5, 1845 CrossRef CAS.
- S. Kodama, H. Takita, T. Kajimoto, K. Nishide and M. Node, Tetrahedron, 2004, 60, 4901 CrossRef CAS.
- P. Sahakitpichan and S. Ruchirawat, Tetrahedron Lett., 2003, 44, 5239 CrossRef CAS.
- M. Bohno, H. Imase and N. Chida, Chem. Commun., 2004, 1086 RSC.
- T. Nishimata, Y. Sato and M. Mori, J. Org. Chem., 2004, 69, 1837 CrossRef CAS.
- S. Kodama, Y. Hamashima, K. Nishide and M. Node, Angew. Chem., Int. Ed., 2004, 43, 2659 CrossRef CAS.
- D. A. Krikoryan, S. P. Parushev, and P. D. Mechkarova, Bulg. Pat. Appl. BG 104937; Chem. Abstr., 2004, 140, 423851 Search PubMed.
- M. Hirnschall, M. Treu, K. Mereiter, C. Hametner, J. Frohlich and U. Jordis, Tetrahedron: Asymmetry, 2003, 14, 675 CrossRef CAS.
- T. Harayama, H. Toko, A. Hori, T. Miyagoe, T. Sato, H. Nishioka, H. Abe and Y. Takeuchi, Heterocycles, 2003, 61, 513 CAS.
- T. Harayama, A. Hori, H. Abe and Y. Takeuchi, Heterocycles, 2003, 60, 2429 CAS.
- T. Harayama, A. Hori, H. Abe and Y. Takeuchi, Tetrahedron, 2004, 60, 1611 CrossRef CAS.
- H. Knolker and S. Filali, Synlett, 2003, 1752 CrossRef.
- K. Hiroya, S. Itoh and T. Sakamoto, J. Org. Chem., 2004, 69, 1126 CrossRef CAS.
- T. Yasuhara, E. Osafune, K. Nishimura, M. Yamashita, K. Yamada, O. Muraoka and K. Tomioka, Tetrahedron Lett., 2004, 45, 3043 CrossRef CAS.
- Z. Shao, J. Chem, R. Huang, C. Wang, L. Li and H. Zhang, Synlett, 2003, 2228 CAS.
- D. E. Minter and C. D. Winslow, J. Org. Chem., 2004, 69, 1603 CrossRef CAS.
- Q. Wang and A. Padwa, Org. Lett., 2004, 6, 2189 CrossRef CAS.
- S. Lebrun, A. Couture, E. Deniau and P. Grandclaudon, Org. Biomol. Chem., 2003, 1, 1701 RSC.
- A. Couture, E. Deniau, P. Grandclaudon and S. Lebrun, Tetrahedron: Asymmetry, 2003, 14, 1309 CrossRef CAS.
- H. Ko, E. Kim, J. E. Park, D. Kim and S. Kim, J. Org. Chem., 2004, 69, 112 CrossRef CAS.
- O. N. Nadein and A. Komienko, Org. Lett., 2004, 6, 831 CrossRef CAS.
- U. Rinner, H. L. Hillebrenner, D. R. Adams, T. Hudlicky and G. R. Pettit, Bioorg. Med. Chem. Lett., 2004, 14, 2911 CrossRef CAS.
- G. R. Pettit, N. Melody, M. Simpson, M. Thompson, D. L. Herald and J. C. Knight, J. Nat. Prod., 2003, 66, 92 CrossRef CAS.
- G. R. Pettit, N. Melody and D. L. Herald, J. Nat. Prod., 2004, 67, 322 CrossRef CAS.
- A. N. Phung, M. T. Zanetti, G. Whited and W. Fessner, Angew. Chem., Int. Ed., 2003, 42, 4821 CrossRef CAS.
- T. Harayama, T. Sato, Y. Nakano, H. Abe and Y. Takeuchi, Heterocycles, 2003, 59, 293.
- T. Watanabe, Y. Ohashi, R. Yoshino, N. Komano, M. Eguchi, S. Maruyama and T. Ishikawa, Org. Biomol. Chem., 2003, 1, 3024 RSC.
- T. N. Le, S. G. Gang and W. Cho, Tetrahedron Lett., 2004, 45, 2763 CrossRef CAS.
- T. N. Le, S. G. Gang and W. Cho, J. Org. Chem., 2004, 69, 2768 CAS.
- (a) T. S. Phan, D. B. Tran, G. M. Phan, T. M. Nguye, T. H. Hoang and H. M. Le, Tap Chi Duoc Hoc, 2003, 18 Search PubMed; (b) T. S. Phan, D. B. Tran, G. M. Phan, T. M. Nguye, T. H. Hoang and H. M. Le, Chem. Abstr., 2004, 140, 228463.
- B. Sener, I. Orhan and J. Satayavivad, Phytother. Res., 2003, 17, 1220 CrossRef CAS.
- E. E. Elgorashi and J. van Staden, S. Afr. J. Bot., 2003, 69, 593 Search PubMed.
- E. E. Elgorashi, G. I. Stafford and J. van Staden, Planta Med., 2004, 70, 260 CrossRef CAS.
- A. Zangara, Pharmcol., Biochem. Behav., 2003, 75, 675 Search PubMed.
- S. Amos, L. Binda, P. Akah, C. Wambebe and K. Gamaniel, Fitoterapia, 2003, 74, 23 CrossRef CAS.
- W. G. van Doorn, A. Sinz and M. M. Tomassen, Phytochemistry, 2004, 65, 571 CrossRef CAS.
- S. Lopez, M. Armand-Ugon, J. Bastida, F. Viladomat, J. A. Este, D. Stewart and C. Codina, Planta Med., 2003, 69, 109 CrossRef CAS.
- M. Samochocki, A. Hoffle, A. Fehrenbacher, R. Jostock, J. Ludwig, C. Christner, M. Radina, M. Zerlin, C. Ullmer, E. R. R. Pereira, H. Lubbert, E. X. Albuquerque and A. Maelicke, J. Pharmacol. Exp. Ther., 2003, 305, 1024 CrossRef CAS.
- (a) O. V. Yagodina, E. B. Nikoskaya and M. D. Faddeeva, Tsitologiya, 2003, 45, 1032 Search PubMed; (b) O. V. Yagodina, E. B. Nikoskaya and M. D. Faddeeva, Chem. Abstr., 2004, 140, 417224.
- (a) A. Sedo, R. Malik, J. Vicar, V. Simaek and J. Ulrichova, Physiol. Res. (Prague), 2003, 52, 367 Search PubMed; (b) A. Sedo, R. Malik, J. Vicar, V. Simaek and J. Ulrichova, Chem. Abstr., 2003, 139, 360768.
- S. Gibbons, J. Leimkugel, M. Oluwatuyi and M. Heinrich, Phytother. Res., 2003, 17, 274 CrossRef CAS.
- G. B. Mahady, S. L. Pendland, A. Stoia and L. R. Chadwick, Phytother. Res., 2003, 17, 217 CAS.
- S. P. Chavan, D. A. Khobragade, A. B. Pathak and U. R. Kalkote, Tetrahedron Lett., 2004, 45, 5263 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |