Molecular rearrangements of diynes coordinated to triosmium carbonyl clusters: reactions of [Os3(μ-H)2(CO)10] and [Os3(CO)10(MeCN)2] with 1,4-dipyridylbuta-1,3-diyne
Lionel P.
Clarke
a,
Jacqueline M.
Cole
a,
John E.
Davies
a,
Alexandra
French
a,
Olivia F.
Koentjoro
b,
Paul R.
Raithby
*bc and
Gregory P.
Shields†
a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, UK BA2 7AY. E-mail: p.r.raithby@bath.ac.uk
cCCLRC Daresbury Laboratory, Daresbury, Warrington, UK WA4 4AD
Abstract
Reaction of [Os3(μ-H)2(CO)10] with 1,4-dipyridylbuta-1,3-diyne yields two clusters, [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}]
1 and [Os3(μ-H)(CO)10-{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}]
2, in which the diyne has rearranged to form a substituted indolizine ring system. Complex 1 converts slowly to 2 at room temperature, and may be decarbonylated to yield [Os3(μ-H)(CO)9{μ-η1:η2:η1-(C8H5N)–C–(C5H4N)}]
3. An analogous reaction involving [Os3(CO)10(MeCN)2] generates three products, [Os3(μ-H)(CO)10{μ-η1:η1-(NC5H3)–C2C2–(C5H5N)}]
4 and [{Os3(μ-H)(CO)10}2{μ-η1:η1-(NC5H3)–C2–}2]
5, both coordinated via orthometallated pyridyl rings, and a minor product [{Os3(CO)10}2{μ3-η1:η1:η1-C2-(NC5H4)}2]
6, coordinated via
μ-carbene and σ-N interactions, the linking ligand retaining its central C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond. Complex 4 reacts with [Os3(μ-H)2(CO)10] to form the linked cluster [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}]
7, also forming an indolizine ring system. The structures of 1–3, 6, 6·2[CH2Cl2] and 7 have been established by X-ray crystallography.
C bond. Complex 4 reacts with [Os3(μ-H)2(CO)10] to form the linked cluster [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}]
7, also forming an indolizine ring system. The structures of 1–3, 6, 6·2[CH2Cl2] and 7 have been established by X-ray crystallography.
Introduction
It is well known that transition metals mediate cycloaddition reaction of alkynes in organic synthesis reactions. The results range from the formation of small rings such as cyclopropenes to furans and benzene to larger rings such as cyclooctatetraenes.1 Most of the transition metal complexes utilized in reactions of this type are mononuclear, with the exception of the Pauson–Khand reaction which employs [Co2(CO)8] as a catalyst.2–4 However, there is an appealing analogy between the nature of the interactions between unsaturated organic ligands on a cluster and those of organic molecules on a catalytic metal surface,5,6 fuelling the field of cluster–polyyne study. Unfortunately, for most cluster-catalysed reactions, there is little direct evidence for the participation of cluster intermediates.The chemistry of alkynes when coordinated to transition metal carbonyl clusters is well documented,7–16 along with analogous reactions involving polyynes. Both alkynes and polyynes display a wide range of coordination modes when coordinated to polynuclear carbonyl clusters, and their reactivity is characterised by transition-metal mediated carbon–carbon bond formation and cyclisations of unsaturated hydrocarbons.11–16
Recently, the reactions of 1,3-conjugated diynes with ruthenium and osmium clusters have attracted considerable interest because of the unusual transformations that these molecules undergo when they are attached to the cluster core. For the osmium systems, observed chemistry includes intramolecular rearrangement or cyclisation of the ligand under mild conditions17–23 and carbon–carbon bond rupture in thermolysis reactions,24 and related results are observed for ruthenium cluster systems.25 The result of the ligand rearrangement generally depends essentially on the nature of the terminal substituents of the diyne.
The activated cluster [Os3(CO)10(MeCN)2] typically reacts with diynes (RCC–CCR′) to form 48-electron clusters [Os3(μ3-η2-RC2CCR′)(μ-CO)(CO)9] in which the diyne is coordinated by one alkyne unit, with the possibility of two isomers in instances where the substituent on the diyne is inequivalent.18,22,26,27 The reaction can also lead to the formation of linear 50-electron clusters [Os3(μ3-η4-RC2C2R)]
(R = Fc [ferrocenyl], 2-C4H3S)27–29 in which both alkyne units are coordinated. Upon reacting [Os3(CO)10(MeCN)2] with (MeCC–CCMe) further metallocyclic products incorporating two diynes and CO incorporating one or two Os3 units are also obtained.20 Reactions involving [Os3(CO)10(MeCN)2] have also been reported for 1,8-bis(ferrocenyl)-octatetrayne30 to give Os3(CO)10(μ3-η2-Fc–C2–CC–CC–CC–Fc), Os3(CO)10(μ3-η2-Fc–CC–C2–CC–CC–Fc), and Os6(CO)20(μ6-η4-Fc–CC–C2–CC–C2–Fc).
The diyne ligands in the cluster of the type [Os3(μ3-η2-RC2CCR′)(μ-CO)(CO)9] may be cleaved on heating to form the acetylide cluster [Os3(μ-η1-C2R)(μ3-η2-C2R)(CO)9]22,26,28 or alternatively the cluster can be reacted with water to form ethynyl complexes [Os3(μ3-η3-RC3CHR)(μ-H/μ-OH)(CO)9]
(R = Fc, Me, Ph).20,31 The cluster [Os3(μ3-η2-RC2CCR′)(μ-CO)(CO)9]
(R = H) can also undergo CO loss and transform to the hydrido acetylide complex [Os3(μ-H)(μ3-η2-C2CCR′)(CO)9]. The free alkyne group in [Os3(μ3-η2-RC2CCR′)(μ-CO)(CO)9]
(R = H,18 R′
= SiMe3,18 R = R′
= Me20) can be coordinated to a Co2(CO)8 moiety to give clusters of the type [Os3(μ3-η2-||-RCC)(μ2-η2-CCR′)(μ-CO)(CO)9(Co2(CO)6)].
Hydrido clusters exhibit different reactivity with diynes as hydrogen is usually transferred from the cluster to the diyne. For example, [Os4(μ-H)4(CO)12] can react with RCC–CCR′
(R = R′
= Fc) to give an ethynyl cluster [Os4(μ-H)3(μ-η2-FcCCHCCFc)(CO)11], with the diyne being hydrogenated at the second carbon atom.32 Reactions of diynes with [Os3(μ-H)2(CO)10] are thought to proceed via an abstraction of a hydrogen atom attached to the β-carbon atom of the diyne which leads to the formation of a bond between the β-carbon atom and the third carbon of the –C2–C2– diyne group.22,23 The result of the ligand rearrangement generally depends essentially on the nature of the terminal substituents of the diyne. This process, accompanied by the elimination of H2O, leads to the formation of a cyclic product incorporating a furan ring in the case of HOH2CCC–CCCH2OH.17 The [HOs3(CO)10(η1:η1-OC4H2CCH3] has been shown to undergo an aldol condensation reaction with aromatic aldehydes.33 Similar cyclisation products have also been observed in the reaction of [Os3(μ-H)2(CO)10] with substituted diynes RCC–CCR′
(R = Ph, R′
= CH2NHPh; R = Ph, R′
= CH2NHCH2Ph; R = R′
= CH2NHPh and R = R′
= Ph).24,34 Reactions involving [Os3(μ-H)2(CO)10] and Me3SiCC–CCSiMe3 do not result in cyclisation products due to the 1,2-shift of the SiMe3 stabilising the ethynyl ligand.19 Similarly, a 1,2-shift of one of the ferrocenyl groups along the butadiyne chain has been suggested to account for the products of the reaction of FcC2C2Fc with [Os3(μ-H)(CO)10(μ-η2-NC5H4)].28 More recently, the reaction of 1,8-bis-(ferrocenyl)-octatetrayne with [Os3(μ-H)2(CO)10] has been shown to yield products involving trans-hydrogenation and cyclisations with the incorporation of CO.21
In this paper, we extend these studies to the reactions of 1,4-dipyridylbuta-1,3-diyne with the activated clusters [Os3(μ-H)2(CO)10] and [Os3(CO)10(MeCN)2].
Results and discussion
Synthesis of 1 and 2
The reaction of [Os3(μ-H)2(CO)10] with 1,4-dipyridylbuta-1,3-diyne at room temperature affords [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}] 1 as a major dark red product in 90% yield and [Os3(μ-H)(CO)10-{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}] 2 as a minor dark blue product in 8% yield (Scheme 1). Both products were characterised by spectroscopic and crystallographic methods.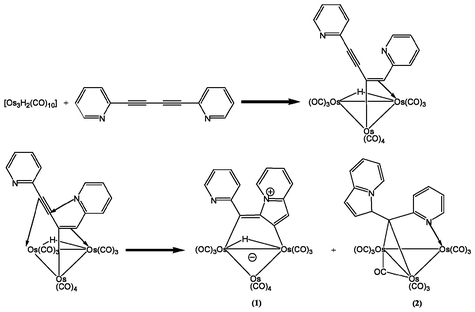 | ||
| Scheme 1 | ||
The FAB-MS spectrum of 1 exhibited the molecular ion at m/z 1058, while the IR spectrum was characterised solely by absorptions for terminal carbonyl groups (Table 1). The relatively low absorption values for these carbonyls are consistent with the zwitterionic formulation for the complex (Scheme 1). The 1H and 13C NMR data collected were consistent with a hydrido cluster containing both an indolizinyl and pyridyl ring. The solid state molecular structure of 1 (Fig. 1, Table 2) was obtained from stable single crystals grown by evaporation of a CH2Cl2–hexane solution. The crystal structure of 1 has two independent but structurally similar molecules in the asymmetric unit of formula. Both molecules exhibit an indolizinyl ring system formed by H-transfer from the hydrido osmium cluster to the first carbon atom of the diyne followed by nucleophilic attack of one of the pyridyl nitrogen atoms on the third atom of the diyne (Scheme 1). A mechanism involving elimination of H2O and aniline has been previously proposed for the reactions of [H2Os3(CO)10] with 2,4-hexadiyne-1,6-diol yielding [Os3(μ-H)(CO)10{μ-η1:η1-(OCHCHCC)–C–CH3}] and in similar reactions with [PhNHCH2C2C2CH2NHPh] as the diyne yielding [Os3(μ-H)(CO)10{μ-η1:η1-(Ph-(NCNCHCC)–C–CH3)}].24 In the case of 1,4-dipyridylbuta-1,3-diyne the mechanism is simpler as no hydrogen migration or elimination occurs, although a similar product is obtained in each case.
![The molecular structure of [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}]
1.](/image/article/2005/NJ/b412578a/b412578a-f1.gif) | ||
| Fig. 1 The molecular structure of [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}] 1. | ||
| Compound | Analysis (C, H, N) | IR (νCO/cm−1) (CH2Cl2) | 1H NMR (δ) (CDCl3) | 13C NMR (δ) (CDCl3) | MS Obs. (Calc. 190Os) |
|---|---|---|---|---|---|
| a Spectrum recorded at 400 MHz. b Spectrum recorded at 500 MHz. | |||||
| (1) |
C-27.10 (27.22)
H-1.08 (0.95) N-2.58 (2.64) |
2094(s), 2054(vs), 2045(vs), 2012(ms) 1996(ms), 1984(sh) | b8.82 (d, 1H, 3J = 4.82 Hz), 7.77 (m, 1H), 7.46 (m, 1H), 7.18 (dd, 1H, 3J = 5.02, 6.94 Hz), 7.11 (d, 1H, 3J = 8.37 Hz), 7.02 (s, 1H), 6.83 (d, 1H, 3J = 7.87 Hz), 6.73 (d, 1H, 3J = 6.43 Hz), 6.35 (m, 1H), −15.01 (s, 1H, Os–H–Os) | 175.90–190.36 (10s, 10CO), 151.52, 160.55, 163.81, 169.61, 173.12 (5s, 5C), 150.41 (1s, 1CH, five-membered ring), 114.55–137.65 (8s, 8CH, six-membered rings) | 1058 (1058) |
| (2) |
C-27.07 (27.22)
H-1.24 (0.95) N-2.76 (2.64) |
2091(ms), 2049(vs), 2011(ms), 1995(sh) 1846(m, br) | b8.91 (d, 1H, 3J = 5.80 Hz), 7.37 (d, 1H, 3J = 8.86 Hz), 7.31 (m, 1H), 6.67 (d, 1H, 3J = 4.04 Hz), 6.65 (m, 2H), 6.61 (d, 1H, 3J = 8.85 Hz), 6.43 (m, 1H), 6.31 (d, 1H, 3J = 4.00 Hz), 5.74 (d, 1H, 3J = 8.31 Hz) | 186.36 (s, 3CO), 177.52 (s, 3CO), 171.09 (s, 3CO), 156.83, 135.09 (2s, 2CH, five-membered ring), 98.21–123.72 (8s, 8CH, six-membered rings) | 1030 (1058) |
| (3) |
C-27.50 (26.79)
H-1.09 (0.97) N-2.78 (2.71) |
2088(s), 2063(vs), 2036(vs), 2007(s), 1993(s), 1975(sh) | b8.64 (d, 1H, 3J = 4.21 Hz), 7.72 (m, 1H), 7.26 (m, 4H, pyridyl ring), 6.67 (d, 1H, 3J = 7.12 Hz), 6.61 (s, 1H), 6.17 (m, 1H), −18.32 (s, 1H, Os–H–Os) | 181.02, 167.78 (s, 2C), 175.78 (s, 9CO), 148.14 (s, CH, five-membered ring), 110.38–136.40 (8s, 8CH, six-membered rings) | 1029 (1030) |
| (4) |
C-26.71 (27.27)
H-0.73 (0.75) N-2.34 (2.65) |
2103(m), 2063(vs), 2051(s), 2019(s), 2007(s), 1991(ms), 1971(sh) | a8.65 (d, 1H, 3J = 4.36 Hz), 7.72 (m, 1H), 7.61 (d, 1H, 3J = 7.22 Hz), 7.35 (m, 1H), 7.26 (dd, 3H = 7.30, 4J = 1.18 Hz), 7.12 (m, 1H), 7.01 (dd, 3J = 7.81, 4J = 1.25 Hz), −14.65 (s, 1H, Os–H–Os) | 166.12–183.57 (10s, 10CO), 150.65, 144.90, 141.46 (3s, 3C), 124.30–139.91 (6s, 6CH), 85.49, 83.67, 78.98, 72.15 (4s, 4C, diyne) | 1056 (1056) |
| (5) |
C-23.95 (21.38)
H-1.02 (0.41) N-1.82 (1.46) |
2103(w), 2072(sh), 2061(vs), 2052(s), 2021(s), 2007(m), 1992(s) | a7.35 (dd, 2H, 3J = 7.97, 4J = 1.35 Hz), 7.12 (m, 2H), 7.07 (dd, 2H, 3J = 7.81, 4J = 1.32 Hz), −14.56 (s, 2H, Os–H–Os) | — | 1878 (1906) |
| (6) |
C-21.49 (21.38)
H-0.69 (0.41) N-1.52 (1.46) |
2103(w), 2085(m), 2061(sh), 2053(vs), 2042(s), 2014(sh), 2007(sh), 1980(sh), 1843(br) | a8.86 (d, 2H, 3J = 5.60 Hz), 7.68 (m, 2H), 7.41 (dd, 2H, 3J = 8.24, 4J = 1.39 Hz), 6.78 (m, 2H) | — | 1878 (1906) |
| (7) |
C-21.50 (21.28)
H-0.60 (0.52) N-1.72 (1.46) |
2101(sh), 2095(m), 2061(vs), 2051(s), 2018(s), 1999(s), 1975(sh) | a8.71–7.06 (m, 7H), 7.10 (s, 1H), −13.71 (s, 1H, Os–H–Os), −14.33 (s, 1H, Os–H–Os) | 117.49–143.73 (8s, 8CH, pyridyl) | 1909 (1908) |
| 1 | 2 | 3 | 6·2[CH2Cl2] | 7 | [Os3(μ-H){μ-η1:η1-CH3CC–C(H)C(H)–NPh}]24 | [Os3(μ-H){μ3-η1:η3:η1-CH3–CC–C(H)C(H)–NPh}]24 | [Os3(μ-H)(CO)9{μ3-η1:η3:η1-Ph(C)C9H6}]34 | |
|---|---|---|---|---|---|---|---|---|
| Os(1)–Os(3) | 2.9486(9), 2.9493(9) | 2.7954(9), 2.8122(9) | 2.9782(14) | 2.8176(9) | 2.693(2) | 2.9424(4) | 2.9653(2) | 2.9601(11), 2.9446(11) |
| Os(1)–Os(2) | 2.8977(11), 2.9086(11) | 2.8201(9), 2.8328(9) | 2.8417(11) | 2.8137(12) | 2.899(2) | 2.8991(4) | 2.8138(2) | 2.8365(10), 2.8460(10) |
| Os(2)–Os(3) | 2.9189(11), 2.9071(12) | 2.7468(10), 2.7460(10) | 2.8229(10) | 2.7541(9) | 2.916(2) | 2.9022(4) | 2.8050(2) | 2.8349(11), 2.8271(11) |
| Os(1)–C(49) | 2.108(14), 2.103(14) | — | 2.082(12) | — | 2.09(4) | 2.095(7) | 2.078(4) | 2.27(2), 2.10(2) |
| Os(3)–C(47) | 2.130(14), 2.126(13) | 2.25(2), 2.172(14) | 2.110(10) | 2.197(9) | 2.11(4) | 2.132(7) | 2.106(5) | 2.11(2), 2.16(2) |
| Os(2)–C(47) | — | 2.215(13), 2.203(13) | 2.229(10) | 2.220(10) | — | — | 2.281(4) | 2.29(2), 2.28(2) |
| Os(2)–C(48) | — | — | 2.311(11) | — | — | — | 2.384(4) | 2.28(2), 2.29(2) |
| Os(2)–C(49) | — | — | 2.670(10) | — | — | — | 2.359(4) | 2.27(2), 2.24(2) |
| Os(1)–N(41) | — | 2.159(11), 2.136(12) | — | 2.161(8) | — | — | — | — |
| N(41)–C(42) | 1.34(2), 1.33(2) | 1.34(2), 1.33(2) | 1.32(2) | 1.328(14) | 1.34(4) | — | — | — |
| N(41)–C(46) | 1.31(2), 1.36(2) | 1.36(2), 1.37(2) | 1.342(15) | 1.392(12) | 1.45(4) | — | — | — |
| C(46)–C(47) | 1.51(2), 1.49(2) | 1.45(2), 1.48(2) | 1.497(14) | 1.467(14) | 1.49(5) | — | — | 1.53(2), 1.53(2) |
| C(47)–C(48) | 1.33(2), 1.33(2) | 1.50(2), 1.53(2) | 1.442(14) | 1.448(13) | 1.40(5) | 1.374(10) | 1.425(6) | 1.42(2), 1.33(2) |
| C(48)–C(49) | 1.43(2), 1.46(2) | 1.37(2), 1.36(2) | 1.453(14) | 1.19(2) | 1.44(5) | 1.439(10) | 1.466(6) | 1.38(2), 1.31(2) |
| C(49)–C(50) | 1.37(2), 1.37(2) | 1.39(2), 1.43(2) | 1.37(2) | — | 1.37(5) | 1.405(10) | 1.439(6) | 1.53(2), 1.63(2) |
| C(50)–C(51) | 1.39(2), 1.41(2) | 1.35(3), 1.37(3) | 1.40(2) | — | 1.43(5) | 1.380(11) | 1.341(6) | 1.47(3), 1.50(3) |
| C(51)–C(52) | 1.41(2), 1.41(2) | 1.41(3), 1.41(2) | 1.42(2) | — | 1.41(5) | — | — | — |
| C(52)–C(53) | 1.39(2), 1.37(2) | 1.38(3), 1.37(3) | 1.35(2) | — | 1.42(5) | — | — | — |
| C(53)–C(54) | 1.40(2), 1.40(2) | 1.39(3), 1.42(2) | 1.41(2) | — | 1.28(5) | — | — | — |
| C(54)–C(55) | 1.33(2), 1.36(2) | 1.35(2), 1.35(2) | 1.34(2) | — | 1.40(5) | — | — | — |
| C(55)–N(56) | 1.37(2), 1.36(2) | 1.38(2), 1.41(2) | 1.366(14) | — | 1.34(5) | — | — | — |
| N(56)–C(48) | 1.48(2), 1.47(2) | 1.40(2), 1.37(2) | 1.435(13) | — | 1.49(4) | 1.455(9) | 1.416(5) | 1.49(2), 1.52(2) |
| N(56)–C(51) | 1.36(2), 1.37(2) | 1.39(2), 1.40(2) | 1.407(13) | — | 1.31(4) | 1.337(7) | 1.385(6) | 1.43(3), 1.35(3) |
| Os(2)–(μ-CO) | — | 2.16(2), 2.02(2) | — | 2.105(11) | — | — | — | — |
| Os(3)–(μ-CO) | — | 2.08(2), 2.35(2) | — | 2.173(11) | — | — | — | — |
| Os(4)–Os(6) | — | — | — | — | 2.901(2) | — | — | — |
| Os(4)–Os(5) | — | — | — | — | 2.887(2) | — | — | — |
| Os(5)–Os(6) | — | — | — | — | 2.887(2) | — | — | — |
| Os(4)–C(42) | — | — | — | — | 2.19(4) | — | — | — |
| Os(6)–N(41) | — | — | — | — | 2.18(3) | — | — | — |
The indolizinyl ring system in 1 is essentially planar and approximately perpendicular to the Os3 triangle, and the free pyridyl ring is rotated in such a way as to minimize steric hindrance between the perihydrogens of the aromatic rings in the cluster. The ligand formally acts as a three electron donor to the 48-electron cluster core. The hydride ligand was not located, but potential energy calculations place the hydride as bridge along the longest Os(1)–Os(3) edge, which is also bridged by the organic indolizine ligand. Both the C(47)–C(48) and C(49)–C(50) exhibit a distance [of 1.33(2) Å and 1.37(2) Å] that is considerably shorter than the adjacent C(48)–C(49) contact of 1.43(2) Å, suggesting the localisation of double bonds next to a formal single bond. In other organic substituted indolizines, such as methyl 3-bis(trimethylsilyl)aminoindolizine-2-carboxylate, 2,8-dimethyl-3-nitroindolizine, and 2-methyl-3-nitroindolizine, the carbon atoms connected to the carbon adjacent to the nitrogen in the indolizine ring have distances in the range of 1.383–1.406 Å.35,36
The 1H NMR spectrum of 2 exhibited resonances in the aromatic region, with no hydride proton detected. The presence of only three resonances in the CO region of the 13C NMR spectrum indicates localized CO fluxionality of the molecule at room temperature, and poor solubility precluded a variable temperature 13C NMR study. The aromatic region of the 13C NMR spectrum demonstrated resonances consistent with the presence of an indolizine ring. However, no resonance was detected for the quaternary carbon bound to the cluster. Only one resonance was observed to arise from the pyridyl ring, which can be explained in terms of the bonding position of the pyridyl ligand to the cluster core. The FAB-MS spectrum did not exhibit the molecular ion; however, a fragment ion at m/z 1030 arising from the loss of one carbonyl group from the cluster was observed. The IR spectrum was characterised by absorptions for both terminal and bridging carbonyl groups.
The solid state molecular structure of 2 was determined by single-crystal X-ray diffraction from single crystals grown from a CH2Cl2–hexane solution at −20 °C (Fig. 2, Table 2) and indicates that further hydrogen transfer from the metal cluster, with respect to 1, has occurred to generate a non-coordinated indolizine ring. The crystal structure of 2 again shows two independent but structurally similar molecules in the asymmetric unit. The molecular structure consists of a closed metal triangle bridged by an η1-alkylidene ligand between Os(1)–Os(3). The carbonyl bridged Os(2)–Os(3) bond is the shortest metal–metal edge in the cluster. A second bonding interaction comes from the adjacent pyridyl ligand bound to the alkylidene bridge such that the ligand acts as a 4-electron donor. The plane running through all six atoms of the pyridyl moiety lies perpendicular to the Os(2)–Os(3) edge explaining the presence of only one resonance for the pyridyl ligand in the 13C NMR spectrum. This μ3-η1:η1:η1 bonding mode of a RC–C(R)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR ligand has previously been observed in other clusters.37–42
NR ligand has previously been observed in other clusters.37–42
![The molecular structure of [Os3(μ-H)(CO)10{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}]
2.](/image/article/2005/NJ/b412578a/b412578a-f2.gif) | ||
| Fig. 2 The molecular structure of [Os3(μ-H)(CO)10{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}] 2. | ||
When a dichloromethane solution of 1 is subjected to a nitrogen purge for 48 h, 2 is produced in 40% yield. This suggests that 2 is the thermodynamic product, while 1 is the kinetic product; the bridging carbene cluster 2 being stabilized by the nitrogen donor bond.
The room temperature absorbance spectrum for 1 in dichloromethane displayed absorptions with λmax of 529 and 376 nm. The absorbance peak at low energy is representative of an inter-valence charge transfer, while the absorbance peak at higher energy indicates a ligand–ligand charge transfer. In contrast, the absorbance spectrum of 2 only shows λmax at 446 nm, indicative of the ligand–ligand charge transfer. The disappearance of the inter-valence charge transfer band is consistent with the fact that the indolizine moiety is no longer coordinated to the osmium cluster in the latter cluster.
It is interesting to compare this cluster reaction with 1,4-dipyridylbuta-1,3-diyne to that of [Os3(μ-H)2(CO)10] with 1,4-diphenylbutadiyne,34 which produces a substituted indane ring system via similar cyclisation of the diyne. In the latter reaction, the isomeric products are 50-electron open triangles which exhibit an allylic η1:η3:η1 coordination mode involving the indane ring system rather than the η1:η1:η1 manner occurring for 1.
Thermolysis of 1
Thermal decarbonylation of 1 in heptane under reflux results in the loss of a single carbonyl ligand from the Os(2) vertex to produce [Os3(μ-H)(CO)9{μ3-η1:η2:η1-(C5H4N)–C–(C8H5N)}] 3 as the single red product in quantitative yield (Scheme 2). | ||
| Scheme 2 | ||
The carbonyl region of the 13C NMR spectrum of 3 revealed only one resonance indicating carbonyl fluxionality, and poor solubility precluded a variable temperature 13C NMR study. Not all the expected resonances were observed in the aromatic region due in part to the long relaxation time associated with the quaternary carbon centres in the cluster. The 1H NMR spectrum demonstrated that the resonance due to the bridging hydride has shifted to δ −18.32 ppm from δ −15.01 ppm in 1. The FAB-MS spectrum exhibited the [M − H]+ ion at m/z 1029, while the IR spectrum was characterised again by terminal ν(CO) absorptions, the relatively low values of which are consistent with the formulation of this cluster as a zwitterions (Scheme 2) as observed for 1.
The solid state molecular structure of 3 was determined by single-crystal X-ray diffraction from single crystals grown from a CH2Cl2–hexane solution at −20 °C (Fig. 3). The molecular structure of 3 illustrates that the organic ligand has a superficially similar bonding mode to that found in the pentagonal pyramidal nido clusters [Os3(μ-H)(CO)9{μ3-η1:η3:η1-CH3CC–C(H)C(H)–NPh}], [Os3(μ-H)(CO)9{μ3-η1:η3:η1-(OCHCHCCCCH3)}] and [Os3(μ-H)(CO)9{μ3-η1:η3:η1-Ph(C)(C9H6)}] produced by similar decarbonylation reactions.24,34 However, there are subtle differences between these products and complex 3.
![The molecular structure of [Os3(μ-H)(CO)9{μ3-η1:η2:η1-(C5H4N)–C–(C8H5N)}]
3.](/image/article/2005/NJ/b412578a/b412578a-f3.gif) | ||
| Fig. 3 The molecular structure of [Os3(μ-H)(CO)9{μ3-η1:η2:η1-(C5H4N)–C–(C8H5N)}] 3. | ||
In particular, the Os(2)–C(49) distance [2.670(10)
Å] is considerably longer than the Os(2)–C(47)
[2.229(10)
Å] and Os(2)–C(48)
[2.311(11)
Å] distances, and the bonding mode may be better described as μ3-η1:η2:η1 coordination of the C(47)C(48) double bond to Os(2) rather than μ3-η1:η3:η1 coordination of an allylic C(47)–C(49) unit. This is supported by the C–C distances (Table 2). Whilst the C(47)–C(48) distance increases on coordination to Os(2), the C(48)–C(49) distance is not significantly altered. The C(47)–C(49) vector is twisted relative to the Os(2)–Os(3) edge in 3, whereas in 1 they are approximately parallel.
This behaviour contrasts with that of [Os3(μ-H)(CO)9{μ3-η1:η3:η1-CH3CC–C(H)C(H)–NPh}],24 which contains a pyrrole rather than indolizine ring system, in which C(49) is clearly bonding to Os(2)
(Table 2), although the C(49)–Os(2) distance is significantly longer [2.359(4)
Å] than the C(47)–Os(2) distance [2.281(4)
Å], whereas the Os(2)–C(47) and Os(2)–C(49) distances are not significantly different in [Os3(μ-H)(CO)9{μ3-η1:η3:η1-Ph(C)(C9H6)}]
(Table 2). The bond distance changes in the pyrrole complex suggest that some rearrangement of electron density in the pyrrole ring may facilitate π-donation from the C(48)–C(49) bond, whereas no statistically significant changes in the distances in the indolizine ring are apparent when complex 1 is decarbonylated to generate 3. There are, however, significant changes in the Os–Os bond distances, the Os(1)–Os(3) distance increasing and the Os(1)–Os(2) and Os(2)–Os(3) distances decreasing, as in the decarbonylation of [Os3(μ-H)(CO)9{μ-η1:η1-CH3CC–C(H)C(H)–NPh}] to generate [Os3(μ-H)(CO)9{μ3-η1:η3:η1-CH3CC–C(H)C(H)-NPh}].24
The UV/Vis absorption spectrum of 3 indicates the inter-valence charge transfer is now a higher energy process, the absorption being at 496 nm, whilst the inter-ligand charge transfer occurs at 349 nm. This is rather unexpected as the increased interaction between the bridging ligand and the triosmium core could be anticipated to lower the energy associated with the inter-valence charge transfer.
Reaction of 1,4-dipyridylbutadiyne with [Os3(CO)10(MeCN)2]
The room-temperature reaction of 1,4-dipyridylbutadiyne with [Os3(CO)10(MeCN)2] in dichloromethane generates three products, the yellow cluster [Os3(μ-H)(CO)10{μ-η2-(NC5H3)–C2C2–(C5H5N)}] 4 in 50% yield, a second yellow product [{Os3(μ-H)(CO)10}2{μ-η2-(NC5H3)–C2–}2] 5 in 20% yield and a navy-blue minor product [{Os3(CO)10}2{μ3-η1-η1-C2–(NC5H4)}2] 6 in 10% yield (Scheme 3). | ||
| Scheme 3 | ||
The 1H NMR spectrum of 4 revealed aromatic resonances consistent with a structure containing a single orthometallated pyridyl ring along with a single hydride resonance at δ
−14.65 ppm. The 13C NMR spectrum revealed the expected resonance in the carbonyl region, as well as those for the uncoordinated alkynes. As with 3, the expected resonances associated with the quaternary carbons were not observed, due in part to the long relaxation time for these centres in the cluster. The FAB-MS spectrum exhibited the [M − H]+ ion at m/z 1056. The IR spectrum of 4 was characterised by a series of absorptions for terminal carbonyls, however the absorptions characteristic of bridging carbonyl ligands were not observed. The free alkyne moieties gave rise to a weak ν(CC) absorption band at 2103 cm−1.
The 1H NMR spectrum of 5 is very similar to the portion of the 1H NMR spectrum of 4 which involves the ligand coordinated to the cluster core, leading to the conclusion that 5 is the bis-ortho-metallated adduct of 4. The 1H NMR spectrum also displays a single hydride resonance present at δ
−14.56 ppm. The FAB-MS spectrum exhibited the [M − CO]+ ion at m/z 1878, while the IR spectrum was characterised again by a series of absorptions for terminal carbonyls while the free alkyne moieties gave rise to a weak ν(CC) signal unchanged to that observed in 4. Low solubility of 5 precluded the acquisition of satisfactory 13C NMR data. Although no solid state molecular structure was obtained for 5, other clusters comprising two Os3 units linked by other orthometallated bis(pyridyl) ligands have recently been reported,43,44 and it is likely that the molecular structure of 5 resembles these clusters.
The 1H NMR spectrum of 6 displays resonances consistent with the presence of C(C5H4N) as a bridging unit in the cluster, with no hydride proton detected. The FAB-MS spectrum did not exhibit the molecular ion, however, a fragment ion at m/z 1878 arising from the loss of one carbonyl group from the parent cluster was observed. The IR spectrum was characterised by seven absorptions for terminal carbonyls, and an absorption at 1843 cm−1 arising from a bridging carbonyl group. The free alkyne moieties gave rise to a weak ν(CC) absorption band at 2101 cm−1. Low solubility again precluded the acquisition of satisfactory 13C NMR data.
The solid state molecular structure of a solvent free 6 as well as a dichloromethane solvated cluster 6·2[CH2Cl2] was determined by single-crystal X-ray diffraction (Fig. 4, Table 2). The molecular structure of 6 illustrates that the C(C5H4N) unit bridges two Os3(CO)10 units, but in this case there is no evidence that orthometallation has occurred and the coordination is via a μ2-carbene and a σ-N interaction, similar to that found for 2, where the organic ligand acts as a 4-electron donor to each Os3 cluster.
![The molecular structure of [{Os3(CO)10}2{μ3-η1-η1-C2–(NC5H4)}2]
6.](/image/article/2005/NJ/b412578a/b412578a-f4.gif) | ||
| Fig. 4 The molecular structure of [{Os3(CO)10}2{μ3-η1-η1-C2–(NC5H4)}2] 6. | ||
The metal–metal, C–N, Os–N and Os–C distances in the μ3-η1:η1:η1-RC–C(R)NR unit in 6 are comparable with those in 2. The carbene-bridged edge Os(2)–Os(3) is bridged by a carbonyl ligand. The C(15)–C(16) and C(16)–C(17) distances are in the single bond range, whilst the central C(17)–C(17A) bond length [1.19(2)
Å] is characteristic of a triple bond. Thus the diyne is coordinated as a buta-2-yne-1,1,4,4-tetrayl ligand in 6 and the pyridyl rings are essentially coplanar with respect to the triosmium planes. In 6·2[CH2Cl2] the complex is located on an inversion centre in the crystal, whereas in 6 the two crystallographically independent molecules occupy general positions. Both the independent molecules in 6 and the single molecule in 6·2[CH2Cl2] have closely similar geometries.
It is perhaps surprising that no analogues are produced in this reaction of structural types [Os3(μ3-η2-RC2CCR′)(μ-CO)(CO)9] and [Os3(μ3-η4-RC2C2R′)(CO)9], as are observed in the corresponding reactions with other diynes.30 It would appear that there is a considerable driving force for the orthometallation of the pyridyl rings, associated with the formation of strong N–Os bonds. Complexes 4 and 5 probably result from nucleophilic attack of the pyridyl nitrogen on the activated Os cluster, whilst 6 may result from initial coordination of the alkyne followed by nucleophilic attack of the pyridyl nitrogen on the remaining MeCN-coordinated Os site, activating the ligand to coordinate to a further Os3 unit in a similar manner. This is consistent with the observation that no mixed coordination mode product was obtained, i.e. one with both a μ3-η1:η1:η1 and μ-η1:η1 coordinated Os3 unit.
Reaction of 4 with [Os3(μ-H)2(CO)10]
Reaction of a molar equivalent of 4 with [Os3(μ-H)2(CO)10] led to the isolation of a deep purple product (Scheme 4) identified as [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}] 7via spectroscopic and single-crystal X-ray diffraction methods (Table 1). | ||
| Scheme 4 | ||
The 1H NMR spectrum of 7 revealed the presence of two discrete hydride resonances at δ −13.71 and δ −14.33 ppm, consistent with chemically inequivalent Os3(μ-H)(CO)10 units. The 13C NMR spectrum revealed eight resonances in the aromatic region, while the remaining expected resonances were not observed. The FAB-MS spectrum exhibited the [M + H]+ ion at m/z 1909. The IR spectrum of 7 was characterised by a series of absorptions for terminal carbonyl ligands while the absorptions characteristic of bridging carbonyl ligands were not observed.
The solid state molecular structure of 7 consists of two osmium triangles linked by a pyridylindolizin-2-ylidene ligand (Fig. 5, Table 2). The structure also reveals that one triosmium unit is coordinated in the same manner as in 1 while the other is coordinated in a manner proposed for 4. This result suggests that the reaction of complex 4 with [Os3(μ-H)2(CO)10] proceeds via a similar mechanism to that for 1,4-dipyridylbutadiyne itself. The orthometallated coordination of one of the pyridyl ligands to another Os3 unit does significantly deactivate the diyne unit which is preserved in 4. However, it does prevent a transformation of the kind which complex 1 undergoes to generate 2, since the remaining pyridyl group is already coordinated to another Os3 unit in 4.
![The molecular structure of [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}]
7.](/image/article/2005/NJ/b412578a/b412578a-f5.gif) | ||
| Fig. 5 The molecular structure of [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}] 7. | ||
The triosmium unit attached to the pyridyl ligand adopts a pseudo-octahedral environment, with Os(5) bound to four approximately linear CO ligands, and Os(4) and Os(6) bound to three similar carbonyl groups. The associated hydride was located by potential energy calculations that place the hydrogen atom below the triangular metal plane, bridging Os(4) and Os(6). The indolizinyl unit bridges Os(1)–Os(3), with the hydride also bridging this metal vertex. As with 1, the C (49)–C(50) interaction is significantly shorter than the adjacent bonds and a similar bonding applies to 7.
Conclusions
The current study shows that triosmium clusters can act as templates for the rearrangement of 1,4-dipyridylbuta-1,3-diyne, leading to the formation of fused ring organic products, in a manner similar to that previously reported for other 1,3-diyne compounds. However, there are subtle differences in the reaction products that are a formulation of the original diyne. For example, the reaction of [Os3(μ-H)2(CO)10] with 1,4-dipyridylbuta-1,3-diyne produces the substituted indolizine ring system, while the reaction of [Os3(μ-H)2(CO)10] with 1,4-diphenylbutadiyne34 produces a substituted indane ring system via similar cyclisation of the diyne. In the latter reaction, the isomeric products are 50-electron open triangles which exhibit an allylic η1:η3:η1 coordination mode involving the indane ring system rather than the η1:η1:η1 manner occurring for 1.In addition, the relatively slow kinetics of the osmium centres means that it is possible to identify both kinetic and thermodynamic products within the reaction sequence, e.g., when a dichloromethane solution of [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}] 1 is subjected to a nitrogen purge for 48 h, cluster [Os3(μ-H)(CO)10-{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}] 2 is produced in 40% yield. This suggests that 2 is the thermodynamic product, while 1 is the kinetic product; the bridging carbene cluster 2 being stabilized by the nitrogen donor bond.
In the reactions of 1,4-dipyridylbuta-1,3-diyne with [Os3(CO)10(NCMe)2] the production of the linked cluster [{Os3(CO)10}2{μ3-η1:η1:η1-C2–(NC5H4)}2] 6, and the reaction of [Os3(μ-H)(CO)10{μ-η1:η1-(NC5H3)–C2C2–(C5H5N)}] 4 with further [Os3(μ-H)2(CO)10] affording [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}] 7, both highlight the possibility of synthesising longer chain oligomeric molecules by further modification of the cluster complexes. Further work in this area is in progress.
Experimental
All manipulations were performed under an atmosphere of dry, oxygen-free nitrogen using standard Schlenk and vacuum-line techniques, at room temperature, unless otherwise stated. Solvents were distilled over appropriate drying agents, under an inert nitrogen atmosphere, prior to use. Routine separation of products was performed by thin layer chromatography (TLC), using commercially prepared glass plates, precoated to 0.25 mm thickness with Merck Kieselgel 60 F254, as supplied by Merck; or using laboratory prepared glass plates, coated to 1 mm thickness with Merck Kieselgel 60 F254. Infrared spectra were recorded as solution spectra on a Perkin-Elmer 1710 Fourier Transform infra-red spectrophotometer using 0.5 mm NaCl or CaF2 cells. NMR spectra were recorded on a Bruker WM250, WM400 or WM500 FT-NMR spectrometer. FAB mass spectra were recorded on a Kratos AEI MS 902 instrument. Solution UV/Vis absorption spectra were recorded on a Perkin-Elmer Lambda 35 spectrometer.The reagents [Os3H2(CO)10]45,46 and [Os3(CO)10(MeCN)2]47,48 were prepared and purified according to literature methods. The compounds 1,4-dipyridylbutadiyne and Me3NO were used as purchased without further purification.
Preparation of [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}] and [Os3(μ-H)(CO)10{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}] (1 and 2)
The compound [Os3H2(CO)10] (0.20 g, 0.211 mmol) was stirred in CH2Cl2 (40 cm3) and 1,4-dipyridylbutadiyne (45 mg, 0.22 mmol) was added as a CH2Cl2 solution (10 cm3) over a period of 5 min. The solution colour changed immediately to deep red. Stirring for 3 h and subsequent purification by preparative TLC using CH2Cl2–hexane (3 ∶ 7) as eluant resulted in the isolation of a deep red coloured band (low Rf) and a blue-black minor band (high Rf). The red product [Os3(μ-H)(CO)10{μ-η1:η1-(C8H5N)–C–(C5H4N)}] 1 was isolated in 90% yield (0.21 g). Evaporation of a CH2Cl2–hexane solution of 1 provided dark red crystalline plates. The blue-black product [Os3(μ-H)(CO)10{μ3-η1:η1:η1-(C5H4N)–C–C(C8H6N)}] 2 was isolated in 8% yield (18.5 mg) and black crystals were grown from a CH2Cl2–hexane solution at −20 °C.Preparation of [Os3(μ-H)(CO)9{μ3-η1:η2:η1-(C5H4N)–C–(C8H5N)}] (3)
Complex 1 (0.07 g, 0.066 mmol) was dissolved in heptane and was heated under reflux for 4 h at 100 °C. A rose-red solid was precipitated on cooling the crude mixture to room temperature which was isolated as a red powder on removal of solvent. The crude product was purified by preparative TLC plates using hexane as eluant, affording a single rose coloured product [Os3(μ-H)(CO)9{μ3-η1:η2:η1-(C5H4N)–C–(C8H5N)}] 3 (95%, 0.065 g). Red crystals were grown from a CH2Cl2–hexane solution at −20 °C.Preparation of [Os3(μ-H)(CO)10{μ-η2-(NC5H3)–C2C2–(C5H4N)}], [{Os3(μ-H)(CO)10}2{μ-η2-(NC5H3)–C2–}2] and [{Os3(CO)10}2{μ3-η1:η1:η1-C2–(NC5H4)}2] (4–6)
The compound [Os3(CO)10(MeCN)2] (0.20 g, 0.214 mmol) was dissolved in CH2Cl2 (40 cm3) and 1,4-dipyridylbutadiyne (44 mg, 0.215 mmol) was added as a CH2Cl2 solution at room temperature. After removal of solvent, the crude product was purified by preparative TLC using hexane as eluant. The major yellow product (low Rf) [Os3(μ-H)(CO)10{μ-η2-(NC5H3)–C2C2–(C5H4N)}] 4 was isolated in 50% yield (0.115 g) as a microcrystalline powder, the minor yellow product (high Rf) [{Os3(μ-H)(CO)10}2{μ-η2-(NC5H3)–C2–}2] 5 in 20% yield (0.08 g) and minor blue product (high Rf) [{Os3(CO)10}2{μ3-η1:η1:η1-C2–(NC5H4)}2] 6 in 10% yield (0.04 g) which was recrystallised from a CH2Cl2–hexane solution, affording crystals of both 6 and 6·2[CH2Cl2].Preparation of [{Os3(μ-H)(CO)10}2{μ-η1:η1,μ-η1:η1-(C8H5N)–C–(C5H3N)}] (7)
A molar equivalent CH2Cl2 solution (25 cm3) of [Os3H2(CO)10] (30 mg, 0.035 mmol) was stirred with complex 4 (38 mg, 0.035 mmol) for 6 h at room temperature, resulting in a deep red solution. After solvent removal, the products were separated on TLC plates using hexane as eluant. The deep purple product [{Os3(μ-H)(CO)10}2{μ-η1:η1;μ-η1:η1-(C8H5N)–C–(C5H3N)}] 7 was isolated in 50% yield. Single crystals of 7 were grown from a CH2Cl2 solution.Crystallography
Details of crystal data, data collection and structure refinement are summarised in Table 3. Structures were solved via direct methods49 and refined by full-matrix least squares on F2.50,51 In all cases the Os atoms were refined anisotropically, while the lighter atoms were refined anisotropically if treating them anisotropically improved the refinement and gave sensible displacement parameters. Hydrogen atoms were included in geometrically idealised positions. The bridging hydride ligands were located by a potential-energy minimization method (HYDEX52). Crystallographic data for 1, 2, 3, 6·2[CH2Cl2], 6 and 7 have been deposited in CIF format at the Cambridge Crystallographic Data Centre.‡ These data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK CB2 1EZ; Fax: +44 1223 336033; or data_request@ccdc.cam.ac.uk).| Complex | 1 | 2 | 3 | 6·2[CH2Cl2] | 6 | 7 |
|---|---|---|---|---|---|---|
| a Data in common. b R1 = Σ|Fo| − |Fc|/Σ|Fo|, wR2 = [Σw(Fo2 − Fc2)2/ΣwFo4]1/2, w = 1/[σ2(Fo)2 + (xP)2 + yP], P = (Fo2 + 2Fc2)/3, where x and y are parameters adjusted by the program; Goodness-of-fit = [Σ[w(Fo2 − Fc2)2]/(n − p)]2, where n is the number of reflections and p the number of parameters. | ||||||
| Molecular formula | C24H10N2O10Os3 | C24H10N2O10Os3 | C23H10N2O9Os3 | C34H8N2O20 Os6, 2(CH2Cl2) | C34H8N2O20Os6 | C34H10N2O20Os6 |
| M | 1056.94 | 1056.94 | 1028.93 | 2075.48 | 1905.62 | 1907.64 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| a/Å | 16.374(5) | 21.296(2) | 10.250(5) | 11.258(2) | 8.7840(6) | 11.211(4) |
| b/Å | 17.289(5) | 13.576(2) | 14.515(4) | 12.994(3) | 28.201(3) | 19.277(4) |
| c/Å | 9.688(2) | 18.931(2) | 8.843(2) | 8.381(2) | 16.7970(9) | 19.435(3) |
| α/° | 98.63(2) | 90 | 107.27(2) | 104.69(3) | 90 | 90 |
| β/° | 106.33(2) | 107.150(10) | 102.64(4) | 103.26(3) | 97.085(5) | 105.13(2) |
| γ/° | 74.81(2) | 90 | 85.36(3) | 85.71(2) | 90 | 90 |
| U/Å3 | 2531.3(12) | 5229.9(11) | 1225.7(7) | 1154.2(4) | 4129.1(5) | 4054.6(18) |
| Space group | P-1 | P21/c | P-1 | P-1 | P21 | P21/n |
| Z | 4 | 8 | 2 | 1 | 4 | 4 |
| D c/Mg m3 | 2.773 | 2.685 | 2.788 | 2.986 | 3.065 | 3.125 |
| Crystal size/mm | 0.20 0.12 0.10 | 0.40 0.27 0.15 | 0.18 0.10 0.08 | 0.30 0.20 0.20 | 0.12 0.05 0.02 | 0.15 0.10 0.10 |
| Crystal habit | Red block | Dark red block | Red prism | Orange block | Black prism | Red block |
| F(000) | 1904 | 3808 | 924 | 926 | 3368 | 3376 |
| Radiation | MoKα | MoKα | MoKα | MoKα | MoKα | MoKα |
| Wavelength/Å | 0.710 73 | 0.710 73 | 0.710 73 | 0.710 73 | 0.710 73 | 0.710 73 |
| μ/mm−1 | 15.082 | 14.600 | 15.567 | 16.759 | 18.472 | 18.812 |
| Transmission | 0.120–0.221 | 0.059–0.150 | 0.171–0.288 | 0.026–0.040 | 0.565–0.691 | 0.124–0.152 |
| Temperature/K | 150 | 293 | 293 | 150 | 180 | 150 |
| Diffractometer | Rigaku AFC7R | Stoe-Siemens | Rigaku AFC5R | Rigaku AFC5R | Nonius Kappa CCD | Rigaku AFC5R |
| Scan type | ω/2θ scans | ω/2θ scans | ω/2θ scans | ω/2θ scans | ω/2θ scans | |
| Data collection range/° | 2.50–22.50 | 3.52–22.50 | 2.53–27.51 | 2.57–27.49 | 3.66–25.01 | 2.63–22.54 |
| Index ranges | 0 17 | −17 22 | 0 13 | −12 14 | −9 10 | 0 12 |
| −17 18 | −14 6 | −18 18 | −16 16 | −31 33 | 0 20 | |
| −10 10 | −20 20 | −11 11 | −10 10 | −19 19 | −20 20 | |
| Reflections measured | 6865 | 7025 | 7491 | 7402 | 24123 | 5609 |
| Independent reflections | 6595 (Rint = 0.068) | 6808 (Rint = 0.087) | 5627 (Rint = 0.031) | 5297 (Rint = 0.051) | 12818 (Rint = 0.086) | 5294 (Rint = 0.115) |
| Parameters, restraints | 443, 0 | 503, 0 | 324, 0 | 307, 0 | 1117, 631 | 279, 15 |
| wR2(all data)b | 0.105 | 0.096 | 0.107 | 0.148 | 0.112 | 0.200 |
| x, yb | 0.060, 1.89 | 0.047, 16.12 | 0.041, 1.31 | 0.041, 0.00 | 0.043 | 0.056, 44.78 |
| R1[I > 2σ(I)]b | 0.040 | 0.045 | 0.041 | 0.038 | 0.050 | 0.064 |
| Observed reflections | 5503 | 4790 | 3738 | 3942 | 10371 | 2831 |
| Goodness-of-fit on F2 (all data)b | 1.024 | 1.051 | 1.012 | 1.037 | 0.986 | 1.023 |
| Maximum shift/σ | 0.001 | 0.001 | 0.014 | <0.001 | 0.001 | < 0.000 |
| Peak, hole/eÅ−3 | 1.51, −2.35 | 1.32, −1.00 | 1.21, −1.87 | 1.918, −2.198 | 1.96, −2.30 | 2.31, −2.26 |
Acknowledgements
We are grateful for financial support from the EPSRC (LPC, OFK, GPS) and the Cambridge Crystallographic Data Centre (JED, GPS) and Johnson Matthey PLC for the generous loan of Os salts.References
- (a) N. E. Schore, Chem. Rev., 1988, 88, 1081 CrossRef CAS; (b) M. I. Bruce and P. J. Low, Adv. Organomet. Chem., 2004, 50, 179 CrossRef CAS.
- P. L. Pauson and I. U. Khand, Ann. N. Y. Acad. Sci., 1977, 295, 2 CAS.
- P. L. Pauson, Tetrahedron, 1985, 41, 5855 CrossRef CAS.
- P. L. Pauson, in Organometallics in Organic Synthesis, Aspects of a Modern Interdisciplinary Field, eds. A. de Heijere and H. Tom Dieck, Springer-Verlag, Berlin, 1988 Search PubMed.
- B. F. G. Johnson, J. Organomet. Chem., 1994, 475, 31 CrossRef CAS.
- B. F. G. Johnson, J. Lewis, C. E. Housecroft, M. A. Gallop, M. Martinelli, D. Braga and F. Grepioni, J. Mol. Catal., 1992, 76, 61 CrossRef CAS.
- P. J. Low and M. I. Bruce, Adv. Organomet. Chem., 2002, 48, 71 CrossRef.
- M. Tachikawa, J. R. Shapley and C. J. Pierpont, J. Am. Chem. Soc., 1975, 977, 172.
- A. J. Deeming, S. Hasso and M. Underhill, J. Chem. Soc., Dalton Trans., 1975, 1614 RSC.
- B. F. G. Johnson, R. Khattar, J. Lewis and P. R. Raithby, J. Organomet. Chem., 1987, 335, C17 CrossRef CAS.
- B. F. G. Johnson, R. Khattar, F. J. Lahoz, J. Lewis and P. R. Raithby, J. Organomet. Chem., 1987, 319, C51 CrossRef CAS.
- G. A. Vaglio, O. Gambino, R. P. Farrari and G. Cetini, Inorg. Chim. Acta, 1973, 7, 193 CrossRef CAS.
- G. A. Vaglio, O. Gambino, R. P. Farrari and M. Chinone, Inorg. Chim. Acta, 1975, 12, 155 CrossRef CAS.
- G. A. Vaglio, O. Gambino, R. P. Farrari and G. Cetini, J. Organomet. Chem., 1971, 30, 381 CrossRef CAS.
- G. A. Vaglio, O. Gambino, R. P. Farrari, M. Valle and G. Cetini, J. Chem. Soc., Dalton Trans., 1972, 1998 RSC.
- B. F. G. Johnson, R. Khattar, J. Lewis, P. R. Raithby and D. N. Smit, J. Chem. Soc., Dalton Trans., 1988, 1421 RSC.
- M. G. Karpov, S. P. Tunik, V. R. Denisov, G. L. Starova, A. B. Nikd'skii, F. M. Dolgushin, A. I. Yanovsky and Y. T. Struchkov, J. Organomet. Chem., 1995, 485, 219 CrossRef CAS.
- M. I. Bruce, P. J. Low, A. Werth, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1996, 1551 RSC.
- L. P. Clarke, J. E. Davies, P. R. Raithby, M.-A. Rennie, G. P. Shields and E. E. Sparr, J. Organomet. Chem., 2000, 609, 169 CrossRef CAS.
- A. J. Amoroso, L. P. Clarke, J. E. Davies, J. Lewis, H. R. Powell, P. R. Raithby and G. P. Shields, J. Organomet. Chem., 2001, 635, 119 CrossRef CAS.
- R. D. Adams, B. Qu and M. D. Smith, Organometallics, 2002, 21, 4847 CrossRef CAS.
- A. J. Deeming, M. S. B. Felix, B. A. Bates and M. B. Hursthouse, J. Chem. Soc., Chem. Commun., 1987, 461 RSC.
- A. J. Deeming, M. S. B. Felix and D. Nuel, Inorg. Chim. Acta, 1993, 213, 3 CrossRef CAS.
- S. P. Tunik, V. D. Khripun, I. A. Balova, E. Nordlander and P. R. Raithby, Organometallics, 2001, 20, 3584 CrossRef CAS.
- N. Pirio, D. Touchard, L. Troupet and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1991, 980 RSC.
- A. J. Deeming, M. S. B. Felix and M. B. Hursthouse, Inorg. Chim. Acta, 1993, 213, 3 CrossRef CAS.
- R. D. Adams and B. Qu, Organometallics, 2000, 19, 2411 CrossRef CAS.
- R. D. Adams, B. Qu and M. D. Smith, J. Organomet. Chem., 2001, 637–639, 514 CrossRef CAS.
- C. J. Adams, L. P. Clarke, A. M. Martín-Castro, P. R. Raithby and G. P. Shields, J. Chem. Soc., Dalton Trans., 2000, 4015 RSC.
- R. D. Adams, O.-S. Kwon, B. Qu and M. D. Smith, Organometallics, 2001, 20, 5225 CrossRef CAS.
- R. D. Adams and B. Qu, Organometallics, 2000, 19, 4090 CrossRef CAS.
- R. D. Adams and B. Qu, J. Organomet. Chem., 2000, 619, 271 CrossRef CAS.
- S. P. Tunik, I. A. Balova, M. E. Borivitov, E. Nordlander, M. Haukka and T. A. Pakkanen, J. Chem. Soc., Dalton Trans., 2002, 827 RSC.
- L. P. Clarke, J. E. Davies, D. V. Krupenya, P. R. Raithby, G. P. Shields, G. L. Starova and S. P. Tunik, J. Organomet. Chem., 2003, 683, 313 CrossRef CAS.
- U. Klement, Z. Kristallogr., 1995, 210, 226 CAS.
- O. Au, V. A. Tafeenko and L. A. Aslanov, Rev. Cubana Fis., 1989, 9, 295 Search PubMed.
- G. Suss-Fink and P. R. Raithby, Inorg. Chim. Acta, 1983, 71, 109 CrossRef.
- R. D. Adams and J. P. Selegue, Inorg. Chem., 1980, 19, 1795 CrossRef CAS.
- M. Langenbahn, H. Stoeckli-Evans and G. Suss-Fink, J. Organomet. Chem., 1991, 402, C12 CrossRef CAS.
- M. Day, W. Freeman, K. I. Hardcastle, M. Isomaki, S. E. Kabir, T. McPhillips, E. Rosenberg, L. G. Scott and E. Wolf, Organometallics, 1992, 11, 3376 CrossRef CAS.
- S. E. Kabir, D. S. Kolwaite, E. Rosenberg, L. G. Scott, T. McPhillips, R. Duque, M. Day and K. I. Hardcastle, Organometallics, 1996, 15, 1979 CrossRef CAS.
- M. Akhter, K. A. Azam, S. M. Azad, S. E. Kabir, K. M. A. Malik and R. Mann, Polyhedron, 2003, 22, 355 CrossRef.
- W.-Y. Wong, S. H. Cheung, S.-M. Lee and S.-Y. Leung, J. Organomet. Chem., 2000, 596, 36 CrossRef CAS.
- W.-Y. Wong, S.-H. Cheung, X. Huang and Z. Lin, J. Organomet. Chem., 2002, 655, 39 CAS.
- H. D. Kaesz, S. A. R. Knox, J. W. Keopke and R. B. Saillant, J. Chem. Soc., Chem. Commun., 1971, 477 RSC.
- H. D. Kaesz, Inorg. Synth., 1990, 28, 238 CAS.
- B. F. G. Johnson, J. Lewis and D. A. Pippard, J. Chem. Soc., Dalton Trans., 1981, 407 RSC.
- J. N. Nicholls and M. D. Vargas, Inorg. Synth., 1990, 28, 232 CAS.
- G. M. Sheldrick, SHELXS-86: ‘Program for Crystal Structure Refinement’, University of Göttingen, 1990 Search PubMed.
- G. M. Sheldrick, SHELXS-93: ‘Program for Crystal Structure Refinement’, University of Göttingen, 1993 Search PubMed.
- G. M. Sheldrick, SHELXS-97: ‘Program for Crystal Structure Refinement’, University of Göttingen, 1997 Search PubMed.
- A. G. Orpen, J. Chem. Soc., Dalton Trans., 1980, 1, 2509 RSC.
Footnotes |
| † Present address: CCDC, 12 Union Road, Cambridge, UK CB2 1EZ. |
| ‡ CCDC reference numbers 248097–248102. See http://www.rsc.org/suppdata/nj/b4/b412578a/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |