One-electron oxidation-induced dimerising C–C coupling of a 2,5-diamino-1,4-benzoquinonediimine: a chemical and electrochemical investigation†
Jean-philippe
Taquet
a,
Olivier
Siri
a,
Jean-Paul
Collin
b,
Abdellatif
Messaoudi
a and
Pierre
Braunstein
*a
aLaboratoire de Chimie de Coordination (UMR 7513 CNRS), Université Louis Pasteur, 4, rue Blaise Pascal, 67070, Strasbourg cedex, France. E-mail: braunst@chimie.u-strasbg.fr
bLaboratoire de Chimie Organo-Minérale (UMR 7513 CNRS), Université Louis Pasteur, 4, rue Blaise Pascal, 67070, Strasbourg cedex, France
First published on 9th December 2004
Abstract
The one-electron oxidation of N,N′,N″,N‴-tetraneopentyl-2,5-diamino-1,4-benzoquinonediimine 3 has been carried out using Ag(I) as an oxidising agent and led to the formation of the postulated nitrogen-based radical cation B. This unstable species can evolve by following two competing pathways, either a hydrogen atom abstraction from a THF solvent molecule, which leads to the N,N′,N″,N‴-tetraneopentyl-2,5-diamino-1,4-benzoquinonemonoiminemonoiminium triflate 5, or a dimerisation of two carbon-based radical cations C, which affords the new dimer 4. An X-ray diffraction study of the latter established the presence of a centre of symmetry in the middle of the newly formed Csp3–Csp3 bond. Breaking of this bond upon reduction regenerates 3 under mild conditions. This type of chemically induced formation/breaking of a C–C single bond appears to be unprecedented in quinonoid chemistry. Electrochemical studies performed in THF confirm the proposed mechanism for the one-electron oxidation process.
Introduction
The transfer of one electron from a strong electron donor to a strong electron acceptor molecule has emerged over the last 15 years as a valuable concept for accomplishing novel reactions in organic1 and transition metal chemistry.2 Among them, oxidative dimerisation of radical cations is generating increasing interest, and this process plays a key role in the industrial electropolymerisation of pyrroles and thiophenes.3 The oxidative dimerisation induced by metal ions such as Ag(I), Tl(III) or Sc(III) is also an efficient method to couple aromatic compounds such as benzenoid derivatives4,5 and porphyrins,6–12 allowing the preparation of larger nanoscale arrays. A number of organic one-electron oxidising agents, including triarylaminium salts and tetracyanoolefinic compounds such as tetracyanoethylene (TCNE) and 7,7,8,8-tetracyanodiquinodimethane (TCNQ), are also used efficiently in radical organic synthesis. In particular, Clyburne and coworkers have recently coupled N-heterocyclic carbenes 1 under mild conditions by tetracyanoethylene-induced one-electron oxidation, which afforded the new air-sensitive 1,2-dicationic dimers of type 2 [eqn. (1)]:13 | (1) |
The importance of quinonoid molecules in many areas of chemistry and biology has been well-recognised.14 They represent a large family of electroactive molecules with a well-documented behaviour toward reduction, which leads to the formation of radical anions semiquinones and/or aromatic hydroquinones.14 We recently observed an unprecedented reaction in quinonoid chemistry whereby reaction of a 6π + 6π zwitterion with TCNE or TCNQ resulted in the regioselective, formal insertion of the olefin into a C–H bond of the quinone ring.15 In contrast, very few examples of oxidation reactions of quinonoid compounds have been reported in the literature, such as the direct oxidative dimerisation resulting in symmetrical bisquinones linked by Csp2 atoms.16–21 As part of our interest in the synthesis, electronic structure and coordination properties of new multifunctional quinonoid ligands,22–24 we decided to explore the behaviour of the “potentially antiaromatic”, acidichromic N,N′,N″,N‴-tetraneopentyl-2,5-diamino-1,4-benzoquinonediimine 323a,b in oxidation chemistry. This molecule has been shown to contain the first example of two separated, conjugated, and localized 6π-electron systems that can be tuned by reversible protonation to become delocalised.23b The corresponding N-benzyl derivative was found to be a sensitive H+ chemosensor with large Stokes shifts (>120 nm) by comparison with conventional fluorophores.23c

We describe in this work the chemical and electrochemical one-electron oxidation of 3 and the first regioselective oxidative dimerisation reaction without dehydrogenation of the coupling site in quinonoid chemistry. The chemical reversal of this reaction will also be discussed.
Results and discussion
Treatment of the tetraneopentyl derivative 3 in THF with 0.8 equiv. of AgOTf for 1 week in the absence of light resulted in a colour change from yellow to red and precipitation of the pale-yellow compound 4 in 34% yield (Scheme 1). Its 1H NMR spectrum revealed the presence of an olefinic proton (δ = 6.70) and of a tertiary methine sp3 proton (δ = 4.80). Furthermore, the presence of two different signals for the neopentyl groups clearly indicated a lower molecular symmetry of the dication in 4 compared to 3. We observed only one signal at δ = 8.35 for the N–H protons, which is consistent with the presence of C![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) NH groups.25 The 13C spectrum (DEPT 135 experiment) confirmed the presence of a sp3 carbon bearing only one proton (δC–H = 40.88). Formation of a dicationic dimer was supported by electrospray mass spectrometry on the solid dissolved in CH3CN, which showed in the positive ion mode peaks at 416.3999 and 831.7947 amu corresponding to [4]2+ and [4
NH groups.25 The 13C spectrum (DEPT 135 experiment) confirmed the presence of a sp3 carbon bearing only one proton (δC–H = 40.88). Formation of a dicationic dimer was supported by electrospray mass spectrometry on the solid dissolved in CH3CN, which showed in the positive ion mode peaks at 416.3999 and 831.7947 amu corresponding to [4]2+ and [4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −H]+, respectively. Evaporation of the red THF filtrate, followed by the addition of hexane to remove soluble 3 in excess, led in addition to a red precipitate. Comparison of its 1H and 13C NMR spectra with that of analogues23a,b clearly showed that 5 had formed (Scheme 1). The identity of this compound as the monoprotonated form of 3 was then confirmed by the preparation of an authentic sample of 5via direct protonation of 3 with triflic acid.
−H]+, respectively. Evaporation of the red THF filtrate, followed by the addition of hexane to remove soluble 3 in excess, led in addition to a red precipitate. Comparison of its 1H and 13C NMR spectra with that of analogues23a,b clearly showed that 5 had formed (Scheme 1). The identity of this compound as the monoprotonated form of 3 was then confirmed by the preparation of an authentic sample of 5via direct protonation of 3 with triflic acid.
 | ||
| Scheme 1 The proposed mechanism for the formation of 4 and 5 | ||
Colourless crystals of compound 4·2H2O were obtained by slow evaporation of an acetone–chlorobenzene solution and subjected to analysis by X-ray diffraction. However, owing to the insufficient quality of the crystals, and despite numerous attempts and changes in the crystallisation solvents, only the structure motif could be determined, which, nevertheless, clearly established a dicationic dimeric structure resulting from C–C coupling of two C6 ring moieties without dehydrogenation of the coupling site (Fig. 1). Selected bond lengths and bond angles are reported in Table 1. There is a centre of symmetry in the middle of the newly formed C(1)–C(1′) bond whose length of 1.576(6) Å corresponds to a single bond between the two cycles. Examination of the bond distances within the N(1)–C(3)–C(4)–C(5)–N(2) moiety shows an equalisation of the C–C and C–N bond distances, which is consistent with the delocalisation of the 6π-electron system and the planar geometry of this moiety.23b In contrast, the C–C and C–N distances within the N(3)–C(6)–C(1)–C(2)–N(4) moiety reveal two consecutive C–C single bonds and two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds, consistent with the presence of the sp3 carbon atom C(1). The C(2)–C(3) and C(5)–C(6) distances of 1.504(4) and 1.503(4) Å, respectively, correspond to single bonds and account for the lack of conjugation between the two C6 ring moieties.
N double bonds, consistent with the presence of the sp3 carbon atom C(1). The C(2)–C(3) and C(5)–C(6) distances of 1.504(4) and 1.503(4) Å, respectively, correspond to single bonds and account for the lack of conjugation between the two C6 ring moieties.
 | ||
| Fig. 1 Structure motif of the dicationic part of 4 in 4·2H2O (methyls of t-Bu groups, CH2 protons and triflate counterions have been omitted for clarity). Thermal ellipsoids enclose 50% of the electron density. | ||
| C(1)–C(1′) | 1.576(6) | C(1′)–C(1)–C(2) | 109.9(3) |
| C(1)–C(2) | 1.518(4) | C(1′)–C(1)–C(6) | 110.0(3) |
| C(1)–C(6) | 1.516(4) | C(2)–C(1)–C(6) | 107.9(2) |
| C(2)–C(3) | 1.504(4) | C(1)–C(2)–C(3) | 115.2(3) |
| C(5)–C(6) | 1.503(4) | C(2)–C(3)–C(4) | 120.2(3) |
| C(4)–C(5) | 1.392(4) | N(1)–C(3)–C(4) | 125.7(3) |
| C(2)–N(4) | 1.265(4) | N(4)–C(2)–C(1) | 129.0(3) |
| C(6)–N(3) | 1.263(4) | ||
| C(3)–N(1) | 1.315(4) | ||
| C(5)–N(2) | 1.324(4) |
We observed that AgBF4 was similarly effective for this coupling reaction but with a significant reduction of the reaction time (3 days instead of 1 week), and that the reaction with either AgOTf or AgBF4 was further accelerated by the addition of 1 equiv. of iodine (reaction in 1 day). Note that direct treatment of 3 with only I2 resulted in no reaction after 1 week.
By analogy with related studies,19,26,27 a possible mechanism (Scheme 1) for this reaction could involve first the one-electron oxidation of an amino nitrogen atom of 3, leading to the nitrogen-centred radical cation A. Proton migration to the more basic proximal sp2 nitrogen atom would result in the protonation of the other vinamidine moiety and formation of a cyanine-type structure stabilised by intramolecular electronic delocalisation between the two nitrogen atoms (intermediate B).23b This postulated radical cation B could evolve by following two competing pathways. The first one would involve hydrogen atom abstraction from the solvent THF, a well-known reaction in radical chemistry,28–30 which leads to the benzoquinonemonoiminemonoiminium triflate 5 (the corresponding chloride has been previously characterised).23b The second possibility consists in the delocalisation of the radical onto the central electrophilic carbon atom (intermediate C), which restores two CN double bonds and maintains delocalisation of the positive charge between the NHR groups. Dimerisation of two such radical cations [radical radical cation (RRC) mechanism],1 would then lead to the triflate salt of a dicationic C–C dimer, 4. Interestingly, related σ-dimeric structures have been isolated as intermediates in the oxidative dimerisation of aromatic compounds (aminobenzenes).4 Similar pathways involving hydrogen atom abstraction or radical cations dimerisation have also been discussed for the reactions of N-heterocyclic carbenes 1, but in this case only hydrogen atom abstraction or only dimerisation of two radical cations occurred, selectively depending on the one-electron oxidant used.13
We were unable to spectroscopically confirm the formation of a radical cation [3]˙+ (i.e., in A, B or C) so we turned to computational studies in order to obtain some information on its structure. Qualitative computations were performed on [3]˙+ using the extended Hückel theory (EHT)31 with the CACAO program (computer aided composition of atomic orbitals).32 Details on the calculated radical cation obtained from 3 are given in the ESI and the most interesting result is the high spin density concentrated on the central olefinic carbon atoms. This is also consistent with the calculated net charge on these carbon atoms, which increases from −0.296 e in the neutral molecule 3 to −0.049 e in the radical cation [3]˙+. The shape of the singly occupied molecular orbital (SOMO) (Fig. 2) clearly suggests that dimerisation of the radical cation will occur through C–C bond formation.
![Representation of the frontier orbital SOMO for the radical cation [3]˙+.](/image/article/2005/NJ/b408762c/b408762c-f2.gif) | ||
| Fig. 2 Representation of the frontier orbital SOMO for the radical cation [3]˙+. | ||
The yields obtained for 4 and 5, slightly in favour of 5, could be explained by the relatively slow kinetics of the dimerisation reaction. Indeed, the rate-determining step of the overall reaction is presumably the formation of the radical cation from 3 and, under our reaction conditions, at any given time only a small amount of radical cations is generated, which limits the dimerisation process. Thus, alternative reaction pathways become possible and abstraction of a hydrogen atom from the solvent leads to 5. Nevertheless, the yield of the C–C dimer 4 is comparable to those observed in the oxidative coupling reactions of Zn(II) porphyrins.11,12,33
The proposed mechanism for the formation of 4 and 5 suggests that electrochemical oxidation could also lead to the formation of the dicationic dimer present in 4, as also observed for the formation of meso,meso-coupled porphyrin arrays.11 Thus, exploration of the electrochemical oxidation of 3 was performed using cyclic voltammetry and controlled potential electrolysis in anhydrous THF containing N(n-Bu)4PF6 as supporting electrolyte, with a platinum net as working electrode. The voltammogram of 3 (Fig. 3) shows an irreversible oxidation wave at 1.08 V vs. SCE, resulting from a one-electron oxidation process, and consequently leading to the formation of a radical cation. The irreversibility of the electrochemical process is consistent with an irreversible formation of the radical cation in B by rapid proton migration in the radical cation precursor A. Increasing the scan rate to 8 V s−1 did not allow to reach reversibility. A controlled potential electrolysis during 2 h in anhydrous THF was realised at 1.3 V vs. SCE in order to see whether the electrochemical oxidation leads to the same ratio of products 4 and 5 as the chemical oxidation. Unfortunately, no precipitation of 4 was observed and the colour of the solution, which turned from yellow before electrolysis to red, obviously indicated the formation of 5 by hydrogen atom abstraction from THF, consistent with the formation at the electrode of only a small amount of radical cation, which disfavours its dimerisation in solution.
![Cyclic voltammogram of 3 in anhydrous THF [0.1 M N(n-Bu)4PF6] at a scan rate of 100 mV s−1.](/image/article/2005/NJ/b408762c/b408762c-f3.gif) | ||
| Fig. 3 Cyclic voltammogram of 3 in anhydrous THF [0.1 M N(n-Bu)4PF6] at a scan rate of 100 mV s−1. | ||
Deprotonation of 4 was also attempted, in particular in order to prepare a new neutral bis(aminotriimine) ligand of interest in coordination chemistry. Thus, 4 was reacted in CH2Cl2 with NEt3 and after 5 min, the reaction mixture was quenched with water, the organic phase was separated and the solvent evaporated to dryness. The solid residue was taken up in hexane and the yellow filtrate was slowly evaporated, leading to the formation of pure yellow prisms that could be separated and analysed by 1H NMR spectroscopy. Unexpectedly, the benzoquinonediimine 3 was recovered in 50% yield. Formation of a white, still unidentified, compound in irreproducible yields was sometimes observed (its 1H and 13C NMR data suggest a hydrolysis product of 4). The clean chemical reduction of the dimer 4 was achieved by redox titration with 2 equiv. of benzophenoneketyl radical (−1.88 V vs. SCE),34 previously generated by reaction of excess sodium with benzophenone in THF, and 3 was obtained in quantitative yield. This reaction, which consumes two electrons, clearly shows that the formation of the C–C σ-bond that led to 4 can be reversed upon reduction. Cyclic voltammetry of 4 in anhydrous THF or CH2Cl2 showed only an irreversible reduction wave at −0.72 V vs. SCE. Controlled potential electrolysis for 0.5 h at −1.35 V vs. SCE of 4 in anhydrous THF or CH2Cl2 allowed the observation of an oxidation wave of the product corresponding to that of 3. Although new in quinonoid chemistry, this type of chemically induced formation/breaking of a C–C single bond is not unprecedented and Berke and coworkers have recently observed this process in dimeric manganese carbyne complexes.35 There is also an interesting analogy between the behaviour of 4 in basic medium and the slow decomposition in CHCl3 of σ-dimeric 5,15-dioxoporphodimethenes, obtained by oxidation of Zn(II) octaethylporphyrin with Tl(III), into the monomeric dioxoporphodimethene and an unknown compound.33
Conclusion
The AgI promoted oxidative coupling reaction of N,N′,N″,N‴-tetraneopentyl-2,5-diamino-1,4-benzoquinonediimine 3, which is itself the two-electron oxidation product of the aromatic compound 6, resulted in the formation of a radical cation, which evolves either by hydrogen atom abstraction from the solvent or by a dimerisation reaction, the latter being the first non-dehydrogenative and reversible C–C coupling in quinonoid chemistry.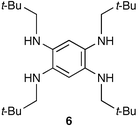
The extension of this reaction to other benzoquinonediimines is in progress and should provide an easy entry into a new class of aminoimine type ligands. Further studies on the deprotonation of the C–C coupling product 4 are also in progress in order to fully understand the mechanism of the C–C bond breaking induced by deprotonation, as well as studies of the influence of the nature of the one-electron oxidising agent used on the reactivity of 3.
Experimental
General details
All solvents were dried and distilled using common techniques unless otherwise stated. All manipulations were performed using standard Schlenk techniques under dry nitrogen atmosphere. 1H NMR (300.13 or 500.13 MHz) and 13C NMR (75.5 or 125.77 MHz) spectra were recorded on a Bruker AC-300 or AMX-500 instrument. ESI mass spectra were recorded on an Autospec HF mass spectrometer. Elemental analyses were performed by the “Service de Microanalyse”, Université Louis Pasteur (Strasbourg, France). The N,N′,N″,N‴-tetraneopentylamino-1,4-benzoquinonediimine 3 was prepared according to the literature.23a,bSyntheses of 4 and 5
A mixture of 3 (1.00 g, 2.40 mmol) and AgOTf (0.49 g, 1.92 mmol) in dry THF (150 ml) was stirred at room temperature, in the absence of light, for 1 week under nitrogen. Formation of a silver mirror and a precipitate was observed. After filtration, the solid was extracted with CH2Cl2 and flash evaporation of the solvent and drying under vacuum afforded 4 as a cream powder. The red filtrate of the reaction mixture was taken to dryness. Addition of hexane removed excess 3 and afforded 5 as a red powder.
4 (0.37 g, 34% yield): 1H NMR (500.13 MHz, CDCl3) δ: 1.00 (s, 36 H, CH3), 1.03 (s, 36 H, CH3), 3.30 (s, 8 H, CH2–N), 3.58–3.64 (m, 8 H, CH2–NH), 4.80 (s, 2 H, NC–C–H), 6.70 (s, 2 H, NCC–H), 8.35 (br s, 4 H, N–H); 13C{1H} NMR (125.77 MHz, CDCl3) δ: 27.48 (CMe3), 27.69 (CMe3), 32.60 (CMe3), 33.01 (CMe3), 40.88 (NC–C–H), 54.82 (CH2–NH), 63.76 (CH2–N), 93.08 (NCC–H), 151.85 (CN), 156.25 (CN); HRMS (ESI) calcd. for C52H96N8: m/z 416.3879 [M]2+, found: 416.3999; 831.7679 [M−H]+, found: 831.7947; 981.7278 [M + OTf]+, found: 981.7595; anal. calcd for C54H96F6N8O6S2: C, 57.32; H, 8.55; N, 9.90; found: C, 57.00; H, 8.47; N, 10.02; UV-vis (CH2Cl2) λmax: 368(br) nm (log ε = 4.64).
5 (0.48 g, 44% yield): 1H NMR (300.13 MHz, CDCl3) δ: 1.03 (s, 18 H, CH3), 1.04 (s, 18 H, CH3), 3.13 (s, 4 H, CH2–N), 3.21 (d, 3JHH = 6.06 Hz, 4 H, CH2–NH), 5.46 (s, 2 H, HCsp2), 7.69 (br t, 2 H, NH), 8.29 (br s, 1 H, NH); 13C{1H} NMR (75.5 MHz, CDCl3) δ: 27.70 (CH3), 27.90 (CH3), 32.32 (CMe3), 33.89 (CMe3), 55.50 (CH2N), 58.59 (CH2N), 87.24 (HCsp2), 150.74 (Csp2N), 152.47 (Csp2N). The NMR data of 5 are consistent with a rapid equilibrium between two tautomers, generating an average structure of higher symmetry in solution.23b
Reaction of 5 with the benzophenoneketyl radical
A blue solution of benzophenoneketyl radical in 5 ml THF, previously generated by reaction of benzophenone (0.07 mg, 0.4 mmol) with excess solid Na in 5 ml of dry THF, was added dropwise to a suspension of 4 (0.23 g, 0.20 mmol) in dry THF (150 ml) at room temperature. The yellow solution was taken to dryness and 3 was recovered quantitatively as a yellow powder. It was characterised by comparison of its 1H NMR data with those of an authentic sample.23bElectrochemical measurements
Electrochemical experiments were performed with a three-electrodes system consisting of a platinum working electrode, a platinum wire counter electrode, and a standard reference saturated calomel electrode (SCE), versus which all potentials are reported. All measurements were carried out under Ar, in degassed THF (previously distilled from Na/benzophenone under N2) or CH2Cl2 (distilled from CaH2 under N2), using 0.1 M N(n-Bu)4PF6 solutions as the supporting electrolyte. An EG&G Princeton Applied Research Model 273A potentiostat connected to a computer (Programme Research Electrochemistry Software) and a Brucker EI 30M potentiostat connected to a printing table were used for the cyclic voltammetry measurements.X-Ray data
Diffraction data were collected on a Kappa CCD diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The relevant data are summarised in Table 2. Data were collected using phi scans and the structures were solved by direct methods using the SHELX 97 software;36,37 the refinement was by full-matrix least squares on F2. No absorption correction was used. All non-hydrogen atoms were refined anisotropically with H atoms introduced as fixed contributors (dC–H = 0.95 Å, U11 = 0.04). The insufficient quality of the crystals precluded a completely satisfactory refinement of the structure, which was therefore not deposited at the CCDC.| Formula | C54H96N8F6O6S2·2H2O |
| Formula wt/g mol−1 | 1167.54 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 12.3030(3) |
| b/Å | 17.0200(4) |
| c/Å | 16.8770(6) |
| β/° | 96.6510(11) |
| U/Å3 | 3510.21(17) |
| Z | 2 |
| ρ calc/g cm−3 | 1.105 |
| μ(MoKα)/mm−1 | 0.141 |
| F(000) | 1260 |
| T/K | 173 |
| Total reflect. | 12183 |
| Unique reflect. | 12182 |
| R int | 0.1451 |
Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique and the Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche (Ph.D. grant for J.-p. T.). We are also grateful to Prof. R. Welter (ULP Strasbourg) for the crystal structure analysis, to Dr. G. Royal (Université Joseph Fourier Grenoble) for preliminary electrochemical studies on 3 and to Dr. M. Bénard (ULP Strasbourg) for discussions on the theoretical analysis.References
- M. Schmittel and A. Burghart, Angew. Chem., Int. Ed., 1997, 36, 2551 CAS.
- D. Astruc, Electron Transfer and Radical Processes in Transition-Metal Chemistry, Wiley–VCH, Weinheim, 1995 Search PubMed.
- A. F. Diaz, in Organic Electrochemistry, eds. H. Lund and M. M. Baizer, Wiley, New York, 1991 Search PubMed.
- F. Effenberger, K. E. Mack, R. Niess, F. Reisinger, A. Steinbach, W. D. Stohrer, J. J. Stezowski, I. Rommel and A. Maier, J. Org. Chem., 1988, 53, 4379 CrossRef CAS.
- A. McKillop, A. G. Turrell, D. W. Young and E. C. Taylor, J. Am. Chem. Soc., 1980, 102, 6504 CrossRef CAS.
- X. Peng, Y. Nakamura, N. Aratani, D. Kim and A. Osuka, Tetrahedron Lett., 2004, 45, 4981 CrossRef CAS.
- (a) A. Nakano, N. Aratani, H. Furuta and A. Osuka, Chem. Commun., 2001, 1920 RSC; (b) A. Tsuda, Y. Nakamura and A. Osuka, Chem. Commun., 2003, 1096 RSC.
- N. Aratani, A. Osuka, Y. H. Kim, D. H. Jeong and D. Kim, Angew. Chem., Int. Ed., 2000, 39, 1458 CrossRef CAS.
- T. Ogawa, Y. Nishimoto, N. Yoshida, N. Ono and A. Osuka, Angew. Chem., Int. Ed., 1999, 38, 176 CrossRef CAS.
- N. Yoshida, H. Shimidzu and A. Osuka, Chem. Lett., 1998, 55 CrossRef CAS.
- T. Ogawa, Y. Nishimoto, N. Yoshida, N. Ono and A. Osuka, Chem. Commun., 1997, 337 Search PubMed.
- H. Shimidzu and A. Osuka, Angew. Chem., Int. Ed., 1997, 36, 135 CrossRef CAS.
- T. Ramnial, I. McKenzie, B. Gorodetsky, E. M. W. Tsang and J. A. C. Clyburne, Chem. Commun., 2004, 1054 RSC.
- S. Pataï and Z. Rappoport, The Chemistry of the Quinonoid Compounds, Wiley, Chichester, 1988 Search PubMed.
- J.-p. Taquet, Q.-Z. Yang, O. Siri and P. Braunstein, unpublished results.
- L. S. Liebeskind and S. W. Riesinger, Tetrahedron Lett., 1991, 32, 5681 CrossRef CAS.
- J. H. P. Schutte, J. A. K. du Plessis, G. Lachmann, A. M. Viljoen and H. D. Nelson, J. Chem. Soc., Chem. Commun., 1989, 1290 RSC.
- P. Joseph-Nathan, M. E. Garibay and R. L. Santillan, J. Org. Chem., 1987, 52, 759 CrossRef CAS.
- A. R. Forrester, A. S. Ingram, I. L. John and R. H. Thomson, J. Chem. Soc., Perkin Trans. 1, 1975, 12, 1115 RSC.
- K. Chandrasenan and R. H. Thomson, Tetrahedron, 1971, 27, 2529 CrossRef CAS.
- J. F. Corbett, J. Chem. Soc. (C), 1970, 1912 RSC.
- (a) P. Braunstein and O. Siri, Chem. Commun., 2002, 208 RSC; (b) P. Braunstein, O. Siri, J.-p. Taquet, M.-M. Rohmer, M. Bénard and R. Welter, J. Am. Chem. Soc., 2003, 125, 12246 CrossRef CAS; (c) J.-p. Taquet, O. Siri, P. Braunstein and R. Welter, Inorg. Chem., 2004, 43, 6944–6953 CrossRef CAS.
- (a) O. Siri and P. Braunstein, Chem. Commun., 2000, 2223 RSC; (b) O. Siri, P. Braunstein, M.-M. Rohmer, M. Bénard and R. Welter, J. Am. Chem. Soc., 2003, 125, 13793 CrossRef CAS; (c) M. Elhabiri, O. Siri, A. Sornosa-Tent, A.-M. Albrecht-Gary and P. Braunstein, Chem. Eur. J., 2004, 10, 134 CrossRef CAS; (d) P. Braunstein, A. Demessence, O. Siri and J.-p. Taquet, C. R. Chim., 2004, 7, 909–913 Search PubMed.
- P. Braunstein, O. Siri, J.-p. Taquet and Q.-Z. Yang, Chem. Eur. J., 2004, 10, 3817 CrossRef CAS.
- C. Rabiller, G. Ricolleau, M. L. Martin and G. J. Martin, New J. Chem., 1980, 4, 35 Search PubMed.
- S. P. Rath, R. Koerner, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 11798 CrossRef CAS.
- A. L. Balch, B. C. Noll, S. M. Reid and E. P. Zovinka, J. Am. Chem. Soc., 1993, 115, 2531 CrossRef CAS.
- P. Renaud, F. Beaufils, L. Feray and K. Schenk, Angew. Chem., Int. Ed., 2003, 42, 4230 CrossRef CAS.
- D. Crich, X. Huang and M. Newcomb, Org. Lett., 1999, 1, 225 CrossRef CAS.
- S. Nakahama, S. Hino and N. Yamazaki, Polymer, 1971, 2, 56 CAS.
- (a) R. Hoffmann and W. N. Lipscomb, J. Chem. Phys., 1962, 36, 2179 CrossRef CAS; (b) R. Hoffmann and W. N. Lipscomb, J. Chem. Phys., 1962, 37, 2872 CrossRef CAS; (c) R. Hoffmann, J. Chem. Phys., 1963, 39, 1397 CrossRef CAS . The parameters used and the Mulliken orbital populations obtained for the metal atoms are available in the Electronic supplementary information (ESI, Tables S1 and S2).
- C. Mealli and D. M. Proserpio, J. Chem. Educ., 1990, 67, 399 CrossRef CAS.
- J. H. Fuhrhop, E. Baumgartner and H. Bauer, J. Am. Chem. Soc., 1981, 103, 5854 CrossRef CAS.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 1980 Search PubMed; (b) M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407 CrossRef CAS.
- K. Venkatesan, O. Blacque, T. Fox, M. Alfonso, H. W. Schmalle and H. Berke, Organometallics, 2004, 23, 1183 CrossRef CAS.
- Kappa CCD Operation Manual, Nonius B. V., Delft, The Netherlands, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: computational details and cif file for compound 4·2H2O. See http://www.rsc.org/suppdata/nj/b4/b408762c/ |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |