Synthesis of covalent DNA–protein conjugates by expressed protein ligation
Marina
Lovrinovic
,
Mark
Spengler
,
Carl
Deutsch
and
Christof M.
Niemeyer
*
Universität Dortmund, Fachbereich Chemie, Biologisch-Chemische Mikrostrukturtechnik, Otto-Hahn Str. 6, D-44227 Dortmund. E-mail: christof.niemeyer@uni-dortmund.de; Fax: +49 (0)231 755 7082; Tel: +49 (0)231 755 7080
First published on 25th April 2005
Abstract
Semisynthetic DNA–protein conjugates are versatile tools for many applications in bioanalytics and nanobiotechnology. We here report a method based on expressed protein ligation (EPL) for the site-specific coupling of cysteine-modified DNA oligomers with recombinant intein-fusion proteins. The latter contain a C-terminal thioester, enabling the mild and highly specific reaction with N-terminal cysteine compounds. To conveniently couple commercially available DNA oligomers with cysteine groups a universal chemical modifier was developed, containing a protected cysteine and an amino-reactive N-hydroxysuccinimide group connected by a hexaethyleneglycol moiety. Using maltose-binding protein (MBP) and green fluorescent protein mutant EYFP as a model systems, we demonstrate the feasibility of this approach, as well as the integrity and functionality of the DNA–protein conjugates synthesized. We anticipate that our concept will enable many applications, such as the generation of large arrays of surface-bound, recombinant proteins assembled by means of DNA-directed immobilization.
Introduction
Protein microarrays are currently explored for a broad range of applications in biomedical diagnostics, to simultaneously determine several parameters in individual samples from a limited amount of material.1–3 A general problem of protein biochips however, concerns their preparation by stepwise, robotic immobilization of multiple proteins at chemically activated surfaces, which is often obstructed by the intrinsic instability of many proteins and, thus, a loss of functionality. To overcome these problems, we have developed the DNA-directed immobilization (DDI) of proteins as a chemically mild procedure for the highly parallel attachment of multiple proteins to a solid support using DNA microarrays as immobilization matrices (Fig. 1).4–8 | ||
| Fig. 1 (A) Schematic drawing of the generation of DNA–protein conjugates 10 and 11 by EPL, using intein-fusion proteins EYFP 8 and MBP 9 and the cysteine–oligonucleotide conjugate 7, generated from amino–oligonucleotide and chemical modifier 1. (B) Schematic drawing of the DNA-directed immobilization of DNA–protein conjugates 10 and 11 at surface-bound capture oligomers 12 (I). Subsequent to immobilization, the conjugates were labeled with a specific antibody (II) and an enzyme-labeled secondary antibody (III). | ||
Despite the many advantages of the DDI method, the chemical conjugation of the DNA with the protein of interest is often cumbersome. In particular, it is rather difficult to control the stoichiometry of coupling and to predict the site of the protein that the nucleic acid is coupled to.4,9 Hence, efficient methodologies are required to allow chemically mild, stoichiometrically controlled and regioselective coupling of nucleic acids with proteins. As an approach to solving these problems, we have recently reported on the expressed protein ligation (EPL)10 of intein-fusion proteins with a conjugate comprising a nucleic acid derivatized with an N-terminal cysteine (Fig. 1A).11 In this initial report however, we had chosen polyamide nucleic acid (PNA) coupled with a short peptide sequence containing a N-terminal cysteine as a model system, due to the convenient synthesis of PNA–cysteine conjugates as opposed to DNA–cysteine conjugates.
We report here on the synthesis of the universal chemical modifier 1 which allows for the convenient modification of commercially available amino-modified DNA oligomers with cysteine groups.12 Using maltose-binding protein (MBP) and green fluorescent protein mutant EYFP as model systems, we demonstrate the use of this modifier to readily synthesize DNA–protein conjugates, which are integer and fully functional with respect to both the DNA and the protein component.
Results and discussion
To effectively take advantage of the expressed protein ligation (EPL) of recombinant proteins with DNA oligonucleotides, the modification of DNA oligomers with a cysteine group is required. Although the incorporation of cysteine during solid-phase oligonucleotide synthesis has already been described,13 the direct coupling to commercially available amino-modified oligomers would offer a more convenient and time-saving preparation at low costs. To this end, we synthesized the chemical modifier 1 comprising an N-hydroxysuccinimide (NHS) ester for the derivatization of amino-modified DNA oligomers with cysteine (Fig. 2). Hexaethyleneglycol was used as spacer between the two reactive moieties. | ||
| Fig. 2 Synthesis of the chemical modifier 1. (a) NaH, THF, tert-butyl bromoacetate (79%); (b) PPh3, H2O, THF (84%); (c) Fmoc–Cys(tButhio)–OPf5, THF (77%); (d) TFA, DCM (100%); (e) NHS, DCC, THF, (93%); (f) DNA oligomer 5, PBS buffer (pH 7.3), DMF; (g) aq. ammonia, 37 °C. | ||
The synthesis was based on commercially available hexaethylenglycol, that was converted to the acidoalcohol 2 using a previously published protocol.14 Ether synthesis and subsequent Staudinger reduction led to compound 3 in 84% yield (steps a and b in Fig. 2). The aminoester 3 was coupled with activated Fmoc-protected cysteine pentafluorophenyl ester (step c in Fig. 2). Subsequent acidolysis cleaved a tert-butylester without affecting the cysteine protection groups (step d in Fig. 2) and the reaction with DCC and NHS under standard conditions led to the universal modifier 1 (step e in Fig. 2).
Following to the establishment of an efficient synthesis of modifier 1, we investigated its functionality in the solution-phase coupling with commercially available amino-modified oligonucleotides by reacting 1 with amino-modified oligonucleotide 5 in aq. buffer solution (for details see Experimental). RP-HPLC and MALDI-TOF MS analysis of the resulting products revealed the formation of cysteine–oligonucleotide conjugate 6 with only small amounts of side-products. A similar outcome was observed for the cleavage of the Fmoc protection group that led to the formation of 7 (Fig. 3). To confirm that the product formation was due to the coupling between modifier 1 and the terminal alkylamino group of oligomer 5, a control experiment was carried out using an unmodified oligonucleotide lacking the terminal alkylamino group. RP-HPLC and MALDI-TOF MS showed neither products nor side products (data not shown), indicating that the reaction between 1 and 5, indeed, occurred as proposed.
 | ||
| Fig. 3 (A) Reversed-phase HPLC analysis of the crude products of the reaction between modifier 1 and alkylamino–DNA 5. Shown are the absorbances at 260 nm (black) and 280 nm (grey). The peak with an asterisk is the cysteine-modified oligonucleotide 7. (B) MALDI-TOF mass spectrum of the desalted reaction mixture shown in (A). The labeled signal at m/z 4989.2 represents 7. | ||
To experimentally investigate the synthesis of DNA–protein conjugates by EPL, we overexpressed two recombinant fusion proteins comprised of MBP (8) or EYFP (9) and intein fused to the chitin binding domain (CBD). CBD enabled the affinity purification of the intein-fusion proteins using a chitin matrix. Thiolysis of the self-cleavable intein-tag using MESNA afforded the activated proteins containing a thioester at their C-termini (Fig. 1A).
Since the S-tert-butylsulfenyl group in 1 was cleaved in the ligation buffer containing MESNA and benzylmercaptan, no further deprotection of cysteine modified oligonucleotide 7 was necessary and 7 was directly coupled with the thioester-activated proteins 8 and 9. The conjugate synthesis was monitored by anion-exchange HPLC15 and native PAGE (Fig. 4). The DNA moiety attached to the conjugate led to an increased electrophoretic mobility, as compared to the native protein.4 Hence, band a in lane 1 and band b in lane 3 of Fig. 4 indicated the formation of EYFP–DNA 10 and MBP–DNA 11 conjugates, respectively. The formation of the two stained bands for the EYFP (c and d in lanes 1 and 2 in Fig. 4), in contrast to MBP (lane 4 in Fig. 4), most likely occurred due to the hydrophobic character of EYFP leading to the formation of non-covalently bound EYFP dimers, which are directly stained by the fluorescent dye SybrGold. DNA–EYFP conjugate 10 however, revealed no such dimer formation (lane 1 in Fig. 4) suggesting that the conjugation of the negatively charged DNA to the C-terminus decreased the EYFP's capabilities for dimerization. These observations are in agreement with earlier studies on the cross-linking of EYFP with DNA oligomers using conventional techniques.9 Grayscale analysis of the stained gels indicated that the ligation had proceeded with yields of approximately 20% for MBP (11) and 30% for EYFP (12).
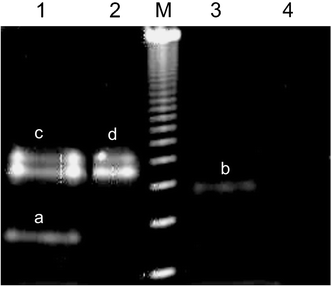 | ||
| Fig. 4 Electrophoretic analysis of DNA–protein conjugates 10 and 11. Shown is an 8.5% non-denaturing PAGE stained with SybrGold. Lane 1: crude reaction mixture of DNA–EYFP conjugate 10; lane 2: native EYFP; lane 3: crude reaction mixture of DNA–MBP; lane 4: native MBP; M: 123 bp DNA ladder; a: EYFP–DNA conjugate 10; b: MBP–DNA conjugate 11; c and d: EYFP and EYFP-dimers. | ||
The biological functionality of DNA–protein conjugates 10 and 11 was investigated by DNA-directed immobilization (DDI) in streptavidin-coated microtiter plates, containing the complementary biotinylated DNA 12 as the capture probe (I in Fig. 1B). The conjugates bound to the solid support were subsequently labeled with a specific antibody (II, mouse anti-EYFP and mouse anti-MBP, respectively) and an enzyme-labeled secondary antibody (III, goat anti-mouse–alkaline phosphatase conjugate). Enzymatic reaction using the fluorogenic substrate Attophos showed signal intensities which clearly indicated the biological functionality of both the DNA and the protein moiety of 10 and 11 (Fig. 5). Moreover, control reactions with non-complementary capture oligomers 13 or lacking any DNA–protein conjugate (nc1 and nc2 in Fig. 5) indicated that, indeed, the immobilization exclusively occurred due to specific Watson–Crick base-paring.
 | ||
| Fig. 5 DNA-directed immobilization of conjugates 10 and 11 in STV-coated microplates, functionalized with either biotinylated complementary capture oligomer 12 or with a non-complementary sequence 13. Nc1 and nc2 represent negative controls, which lack any protein–DNA conjugate. The height of the histograms represents fluorescence signals obtained in the enzymatic reaction of alkaline phosphatase bound to conjugates 10 and 11 by specific antibodies, as illustrated in Fig. 1B. | ||
Conclusions
We have reported here a mild and efficient strategy for the synthesis of DNA–protein conjugates by expressed protein ligation (EPL). With the aid of universal chemical modifier 1, commercially available alkylamino–DNA oligomers were effectively derivatized with a cysteine group in solution. The resulting DNA–cysteine conjugate was coupled with thioester-activated proteins, such as EYFP and MBP, conveniently accessible by overexpression of recombinant intein-fusion proteins. The resulting covalent DNA–protein conjugates retain their full biological activity, as demonstrated here using a DNA-directed immobilization immunoassay.It is particularly noteworthy that the covalent DNA–protein conjugates are well defined with respect to stoichiometric composition and regiospecific linkage. Hence, EPL has clear advantages over conventional coupling techniques. We anticipate that the EPL- and DDI-based immobilization strategy described here will be useful for a variety of applications. These include the synthesis of libraries of recombinant DNA–protein conjugates for protein microarray technologies to study complex biomolecular systems, as well as the fabrication of oligofunctional protein nanostructures for biomedical diagnostics and the arising field of nanobiotechnology.8,9
Experimental
Synthesis of modifier 1
The cysteine-containing modifier 1 was synthesized in seven steps, using hexaethylenglycol as the starting material (Fig. 2). Initially, azidoalcohol 2 was prepared from hexaethylenglycol according to the method of Svedhem et al.142 was treated with NaH in THF, followed by subsequent reaction with tert-butyl bromoacetate to give a tert-butylester azide. Staudinger reduction of the azide group yielded 84% of amino-compound 3. A cysteine moiety necessary for the EPL reaction was incorporated using the pentafluorophenyl ester of N-α-Fmoc-S-tert-butylsulfenylcysteine. After stirring for 3 h at 70 °C, the crude product was purified by flash chromatography (EtOAc–MeOH 10 : 1) to give 77% of the compound 4. 1H NMR (500 MHz, CDCl3): δ = 7.75 (d, 2H), 7.60 (d, 2H), 7.39 (t, 2H), 7.30 (dt, 2H), 4.45 (t, 2H), 4.35 (t, 1H), 4.22 (t, 1H), 3.99 (s, 2H), 3.66 (bm, 22H), 3.47 (t, 3J(H,H) = 6,5 Hz, 1H), 3.09 (d, 3J(H,H) = 6.5 Hz, 2H), 1.46 (s, 9H), 1.33 (s, 9H); 13C NMR (125 MHz, CDCl3) δ = 169.5, 143.7, 141.2, 127.7, 127.1, 125.1, 120.0, 81.4, 72.5, 70.49, 70.35, 70.33, 70.30, 70.0, 68.8, 41.3, 39.5, 37.3, 29.8, 28.1; FAB-HRMS (m/z): calcd for C40H60O11N2S2 808.3639 (M+), found 809.3701.The tert-butylester moiety in 4 was removed by treatment with trifluoroacetic acid (TFA). The resulting acid was activated with dicyclohexyl carbodiimide (DCC) and reacted with N-hydroxysuccinimide (NHS) to yield 93% of 1 as a yellow oil. 1H NMR (500 MHz, CDCl3) δ = 9.52 (sb, 1H), 7.77 (d, 2H), 7.60 (d, 2H), 7.39 (t, 2H), 7.31 (dt, 2H), 4.46 (s, 2H), 4.39 (t, 2H), 4.22 (t, 1H), 4.17 (s, 2H), 3.65 (bm, 22H), 3.47 (t, 3J(H,H) = 6.5 Hz, 1H), 3.06 (d, 3J(H,H) = 6.5 Hz, 2H), 2.73 (s, 4H), 1.33 (s, 9H); 13C NMR (125 MHz, CDCl3) δ = 172.3, 170.6, 168.9, 156.0, 143.6, 141.3, 127.8, 120.0, 70.8–67.8, 41.9, 39.9, 29.7, 24.7; FAB-HRMS (m/z): calcd for C40H55O13N3S2 850.3176 ([M + H]+) and 872.3176 ([M + Na]+), found 850.3294 and 872.3027, respectively.
Conjugate synthesis and purification
A solution of 100 µl of modifier 1 (10 mM in DMF) was added to a solution of the amino-modified DNA 5 (5′-NH2–CCTGTGTGAAATTG-3′, Thermo Electron, Germany) in 100 µl PBS buffer (100 mM, pH 7.3). The reaction mixture was incubated at room temperature for 12 h. The solvent was removed in vacuo and the resulting residue was dissolved in water. After purification by gel filtration chromatography (NAP 5 and NAP 10 columns, Pharmacia) the product 6 was characterized by MALDI-TOF mass spectrometry. MALDI-TOF MS (matrix: 3-hydroxypicolinic acid): calcd 5211.0 [M+H]+, found 5211.1.The amino protection group Fmoc was removed by treatment with aq. ammonia at 37 °C for 6 h. The crude product was purified by gel filtration chromatography (NAP 5) and reversed-phase HPLC (Nucleodur Gravity C 18, Macherey-Nagel) using a gradient of 5–95% acetonitrile in 1 M triethylammonium acetate over 40 min at a flow rate of 1 ml min−1 (Fig. 3A). The conjugate 7 obtained from HPLC was characterized by MALDI-TOF MS (Fig. 3B) (matrix: 3-hydroxypicolinic acid): calcd 4989.8 [M + H]+, found 4989.2.
Expression and purification of fusion proteins
Maltose binding protein (MBP) 8 was expressed as an intein-fusion protein in E.coli using the control plasmid provided in the IMPACT™-CN kit (New England Biolabs), according to manufacturers instructions.The EYFP gene pEYFP-C1 (Invitrogen) was digested with Nco I and Sal I (New England Biolabs), purified with a spin column kit (Quiagen) and cloned into commercially available pTYB1 vector (New England Biolabs) to yield the EYFP–intein construct. The EYFP 9 was expressed in E.coli as fusion protein with the intein affinity tag at its C-terminus. Upon harvest and lysis, the recombinant protein was purified by affinity chromatography using chitin beads and the intein tag was cleaved using 2-mercaptoethanesulfonic acid (MESNA).
Expressed protein ligation of protein thioesters with DNA
Cys-modifier 7 (80 µM) was mixed with the thioester-containing proteins 8 and 9 (20 µM) in ligation buffer (20 mM Tris–HCl, 200 mM NaCl, 4% MESNA, 4% benzylmercaptan, pH 8.0) and the mixture was incubated for 48 h at 4 °C. The resulting DNA–protein conjugates, 10 and 11, respectively, were purified by dialysis and characterized by gel electrophoresis using 8.5% acrylamide gels at the constant voltage of 150 V (Fig. 4) and anion-exchange HPLC.15 Gels were stained with SybrGold (Molecular Probes) and silver developing kit (Bio-Rad) according to the manufacturer's instructions. Documentation was performed on a UV–vis gel documentation system (AlphImager, Biozym) using appropriate filters. The yield was determined by the grayscale analysis using the AlphaEaseFC software (Biozym).DNA-directed immobilization (DDI)
The DNA–protein conjugate's functionality was proven by site-specific DNA-directed immobilization (DDI) using complementary DNA capture oligomers immobilized in microplates. DDI was carried out as previously described.5 In brief, the DNA–protein conjugates were immobilized in STV-coated microplates containing complementary oligomers 12 (5′-CAATTTCACACAGG-3) or, as a negative control, non-complementary oligomers 13 (5′-TGATAGGGTGCTTGC-3′) as capture strands. The hybridized conjugates 10 and 11 were detected using either an anti-MBP or anti-GFP antibody, respectively, and an alkaline phosphatase-labeled secondary antibody (Fig. 1B). Attophos (Roche) was used as the fluorogenic substrate for alkaline phosphatase. Fluorescence signals were detected using a microplate reader (Synergy HT, Bio-Tek).Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG) through grant Ni399/6-1. We thank Andreas Arndt for help with the overexpression of the intein fusion proteins.References
- M. F. Templin, D. Stoll, M. Schrenk, P. C. Traub, C. F. Vohringer and T. O. Joos, Trends Biotechnol., 2002, 20, 160–166 CrossRef CAS.
- B. Schweitzer and S. F. Kingsmore, Curr. Opin. Biotechnol., 2002, 13, 14–19 CrossRef CAS.
- W. Kusnezow and J. D. Hoheisel, BioTechniques, 2002, 33(suppl), 14–23.
- C. M. Niemeyer, T. Sano, C. L. Smith and C. R. Cantor, Nucleic Acids Res., 1994, 22, 5530–5539 CrossRef CAS.
- C. M. Niemeyer, L. Boldt, B. Ceyhan and D. Blohm, Anal. Biochem., 1999, 268, 54–63 CrossRef CAS.
- R. Wacker and C. M. Niemeyer, ChemBioChem, 2004, 5, 453–459 CrossRef CAS.
- R. Wacker, H. Schroeder and C. M. Niemeyer, Anal. Biochem., 2004, 330, 281–287 CrossRef CAS.
- C. M. Niemeyer, in NanoBiotechnology: Concepts, Methods and Applications, ed. C. M. Niemeyer and C. A. Mirkin, Wiley-VCH, Weinheim, 2004, pp. 227–243 Search PubMed.
- F. Kukolka and C. M. Niemeyer, Org. Biomol. Chem., 2004, 2, 2203–2206 RSC.
- R. S. Goody, K. Alexandrov and M. Engelhard, ChemBioChem, 2002, 3, 399–403 CrossRef CAS.
- M. Lovrinovic, R. Seidel, R. Wacker, H. Schroeder, O. Seitz, M. Engelhard, R. Goody and C. M. Niemeyer, Chem. Commun., 2003, 822–823 RSC.
- Universal cysteine modifiers, such as 1, for the functionalization of DNA oligomers and their use in EPL: C.M. Niemeyer, M. Lovrinovic, C. Deutsch, German patent application, priority date, October 30th, 2003. During the preparation of this manuscript, Takeda and coworkers reported on the conjugation of DNA oligonucleotides to the C-terminus of a green fluorescent protein variant, ECFP, by EPL using a cysteine-derivatized isonipecotic acid–NHS ester ( S. Takeda, S. Tsukiji and T. Nagamune, Bioorg. Med. Chem. Lett., 2004, 14, 2407–2410 Search PubMed ). However, no functionality of the conjugated protein was demonstrated. Very recently, dsDNA–protein conjugates were prepared by EPL and used in a PCR-based immunoassay: I. Burbulis, K. Yamaguchi, A. Gordon, R. Carlson and R. Brent, Nat. Methods, 2005, 2, 31–37 CrossRef CAS . For related immunoassays see: C. M. Niemeyer, M. Adler and R. Wacker, Trends Biotechnol., 2005, 23, 208–216 Search PubMed.
- D. A. Stetsenko and M. J. Gait, J. Org. Chem., 2000, 65, 4900–4908 CrossRef CAS.
- S. Svedhem, C. A. Hollander, J. Shi, P. Konradsson, B. Liedberg and S. C. Svensson, J. Org. Chem., 2001, 66, 4494–4503 CrossRef CAS.
- The DNA–EYFP conjugates were purified by anion-exchange HPLC (Nucleosil 4000-7 PEI, Macherey-Nagel) by gradually increasing the NaCl concentration from 0 to 0.7 M. The fluorescence of the conjugate was detected using a 515 nm excitation and a 527 nm emission filter, thereby clearly indicating the biological activity of the protein component.
| This journal is © The Royal Society of Chemistry 2005 |