Effects of flow and diffusion on chemotaxis studies in a microfabricated gradient generator
Glenn M.
Walker†‡
ab,
Jiqing
Sai‡
c,
Ann
Richmond
cd,
Mark
Stremler
e,
Chang Y.
Chung
f and
John P.
Wikswo
*abgh
aVanderbilt Institute for Integrative Biosystems Research and Education, Vanderbilt University, Nashville, TN 37235, USA
bDepartment of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
cDepartment of Cancer Biology, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
dVeterans Affairs Medical Center, Nashville, TN 37212, USA
eDepartment of Mechanical Engineering, Vanderbilt University, Nashville, TN 37235, USA
fDepartment of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
gDepartment of Physics and Astronomy, Vanderbilt University, Nashville, TN 37235, USA
hDepartment of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
First published on 27th April 2005
Abstract
An understanding of chemotaxis at the level of cell–molecule interactions is important because of its relevance in cancer, immunology, and microbiology, just to name a few. This study quantifies the effects of flow on cell migration during chemotaxis in a microfluidic device. The chemotaxis gradient within the device was modeled and compared to experimental results. Chemotaxis experiments were performed using the chemokine CXCL8 under different flow rates with human HL60 promyelocytic leukemia cells expressing a transfected CXCR2 chemokine receptor. Cell trajectories were separated into x and y axis components. When the microchannel flow rates were increased, cell trajectories along the x axis were found to be significantly affected (p < 0.05). Total migration distances were not affected. These results should be considered when using similar microfluidic devices for chemotaxis studies so that flow bias can be minimized. It may be possible to use this effect to estimate the total tractile force exerted by a cell during chemotaxis, which would be particularly valuable for cells whose tractile forces are below the level of detection with standard techniques of traction–force microscopy.
Introduction
Many cell types exhibit the ability to sense certain chemicals (chemokines) and move, or taxis, toward them. The study of chemotaxis is fundamentally important in many areas of biology, including microbiology1,2 and immunology.3 The most studied branch of chemotaxis in microbiology is with the bacterium Escherichia coli, which can sense chemokines and alter its course to swim up the chemokine gradient toward the source by using flagellar motors. Leukocytes, cells involved in healing damaged tissue within the body, respond to chemokines released from damaged cells. The chemokines enter the bloodstream, providing a signal for the leukocytes to move toward, which they do by cytoskeletal reorganization and lamellipodial motion.Chemotaxis studies require a way to deliver chemicals to cells in a controlled gradient because cells need to be able to sense an increase in concentration of chemokine to direct their motion. Several methods have been developed to create gradients of chemotactic reagents. The earliest methods of leukocyte chemotaxis studies used agarose4,5 and collagen6,7 gels. Newer devices do not rely on gels and include the Zigmond chamber,8 the Dunn chamber,9 and more recently, an optical chemotaxis assay system.10 Other methods for studying chemotaxis have been reviewed as well.11
The Zigmond and Dunn chambers rely on diffusion over a short distance between two fluid-filled reservoirs. The agarose and collagen gel methods rely on diffusion of chemokine through the gel. The cells migrate toward the chemokine by crawling under and through the agarose and collagen gels, respectively.
Each of these techniques has advantages, but they all have the same three disadvantages: (1) Only linear gradients can be formed. One can imagine a situation in vivo where the gradient might increase, then decrease, for instance, as a function of distance. The ability to create arbitrary gradient shapes in vitro would therefore be useful. (2) Changing chemokines within the gradient is not feasible mid-experiment. After the gradient has been established, changing chemokines would require a significant amount of time before the new gradient becomes established. A more accessible gradient generation method would allow the chemokine to be changed in a matter of minutes instead of hours. (3) The slope of the gradient changes over time. Creating and maintaining a stable concentration gradient is desirable because it allows for a more controlled experimental environment.
Microfluidic devices are ideal tools for creating and controlling chemotactic gradients. Microfabrication methods allow arbitrary microchannel designs to be created through which cells can crawl. Microchannel dimensions can be created small enough that diffusion occurs in minutes to seconds, thus reducing the waiting time for a gradient to become established.
A device that overcomes the three disadvantages of prior techniques has recently been demonstrated by Jeon et al.12–14 This microfluidic device provides a versatile method of generating gradients and can be used to create a variety of cross sectional profiles (i.e., linear, ramp, parabolic, etc.).15 The device uses laminar flow and diffusional mixing to create a concentration gradient. However, the flow used to maintain the gradient creates a small drag force on the cells (<0.2 dyn cm−2). Also, diffusion between streams causes the chemokine gradient linearity to vary down the length of the microchannel—gradient linearity is a function of distance down the channel length. In this paper, these two problems are explored by characterizing the linearity of the gradient down the microchannel and the effect of drag on cell migration during chemotaxis.
Operational aspects of the gradient generator
The concentration gradient within the device was visualized and quantified with the fluorescent molecule FITC-dextran (FITC = fluorescein isothiocyanate). The gradient device takes two input buffer streams, one containing FITC-dextran at a concentration of C0 and the other containing no FITC-dextran. By repeatedly dividing and mixing the two input streams in different proportions, an arbitrary number of streams can be created with varying concentrations of FITC-dextran. All experiments discussed here use the following design: the two input streams are divided and mixed until five streams are created, which are directed into the main microchannel (Figs. 1a and b). When each stream enters the main microchannel it is about 100 μm wide and its concentration is one of the following multiples of C0: 0, 0.25, 0.5, 0.75, or 1 (Fig. 1c). When first entering the main microchannel, the five streams are easily distinguishable and their cross sectional profile is step-shaped. Further down the microchannel, as the streams mix by diffusion, the five streams become more difficult to identify (Fig. 1d) and the cross sectional profile becomes more linear. A linear cross sectional profile now exists that ranges from C0 to 0. Still further down the microchannel, the five streams are no longer distinctly identifiable (Fig. 1e).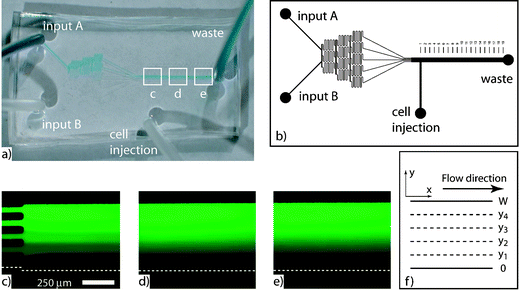 | ||
| Fig. 1 (a) A microfluidic gradient generator was fabricated from a previously published design. (b) Two inputs are divided and mixed until five streams are created. FITC-dextran was imaged at channel lengths (c) 1 mm, (d) 4 mm, and (e) 8 mm. (f) A mathematical model based on the channel geometry was used to confirm the FITC-dextran observations. | ||
Constant fluid flow is required within the microchannel for the concentration gradient to maintain its shape. A fixed arbitrary point within the microchannel will continuously experience the same concentration only if flow is present. Cessation of flow would cause the gradient to continuously change shape until diffusional mixing had homogenized the contents of the microchannel. Faster flows cause each cross sectional geometry of the gradient to persist for a longer portion of the microchannel length than do slower flow rates. In the limiting case of very fast flow, the step-profile would persist down the entire length of the microchannel. At the other extreme, no flow would cause the streams to mix by diffusion and the cross sectional profile to become flat. Figs. 1c–e are representative of a slow flow rate (they portray a situation near the extreme of no flow).
Because flow is required to establish and maintain the gradient, shear forces are always present within the microchannel. Shear forces deserve special consideration at the microscale, where microchannel dimensions can be of the same order of magnitude as cellular dimensions. A recent paper by Gaver and Kute predicted that shear forces on adherent cells within microchannels can be greater than what is predicted by calculating wall shear.16 In the experiments presented here, the ratio of cell height to channel height is less than 0.1. Therefore, wall shear stress is used as an approximation of actual shear forces on the cells.
The amount of shear experienced by a microchannel wall in laminar flow can be calculated from Newton's law of viscosity
 | (1) |
 | (2) |
Some cell types, such as endothelial cells, require shear stress to develop properly.18 Other cell types have shear responses but do not have altered phenotypes.19 Therefore, the aim of this article is to quantify the effect of flow on cellular chemotaxis studies conducted in the gradient devices developed by Jeon et al.12–14
Materials and methods
Microfluidic devices
We utilized our class 100 clean rooms to fabricate microfluidic devices that consist of three tiers of “divide and mix” microchannels that empty into the main microchannel (Fig. 1b). The purpose of these preconditioning microchannels is to take the two input streams, divide and mix them until five streams of varying concentration are generated, and introduce the five streams into the main microchannel flowing next to each other. In the models and experiments presented here, the main microchannel dimensions are 500 μm wide, ∼100 μm tall, and 1 cm long.Polydimethylsiloxane (PDMS) was chosen as the construction material because it allows devices to be fabricated rapidly and is optically transparent, facilitating the concentration gradient observation. Microfluidic devices were made by molding microchannels in the PDMS (Sylgard 184, Dow Corning, Midland, MI) and then bonding the mold to glass coverslips, as detailed elsewhere.20 Briefly, curing agent and pre-polymer were mixed together (1 ∶ 10 wt/wt) and placed in a desiccator for 45 min. The mixture was then poured over a SU-8-2100 (Microchem Corp., Newton, MA) master in a tissue culture dish which contained the positive relief of the microchannel design. The PDMS mold was cured for 2.5 h at 80 °C on a programmable hotplate (Barnstead/Thermolyne International, Dubuque, IA). Once the mold had cooled, it was peeled off the master, cut to size, and access holes were punched for tubing. A blunt 16 gauge needle (Becton Dickinson, Franklin Lakes, NJ) with an outer diameter (od) of 1.65 mm and an inner diameter (id) of 1.19 mm was used as the punching tool. Each PDMS mold was then treated with a plasma cleaner (Harrick Scientific Corporation, Ossining, NY) for 20 s and placed on a no. 1 24 × 50 mm glass coverslip (VWR International, West Chester, PA). The bonded device was treated again with the plasma cleaner for 15 s to facilitate filling with liquid. Tygon tubing (Cole Parmer, Vernon Hills, IL) with an od of 1.52 mm and an id of 0.508 mm, cut into four 30.5 cm lengths, was inserted into each access hole. Sterilized DI water was injected into the device via the waste line until the device and the remaining three tubes were filled with water.
Bubbles were removed by pinching the three lines closed and applying pressure to the waste line syringe. The increased fluid pressure within the device caused the air in trapped bubbles to diffuse through the PDMS.
Modeling
The diffusive transport of particles between multiple streams in laminar flow can be modeled with the two-dimensional transport equation21 | (3) |
| c(y,0) = 0 for 0 ≤ y ≤ y1 |
| c(y,0) = 0.25C0 for y1 < y ≤ y2 |
| c(y,0) = 0.50C0 for y2 < y ≤ y3 |
| c(y,0) = 0.75C0 for y3 < y ≤ y4 | (4) |
| c(y,0) = C0 for y4 < y ≤ W |
| ∂c/∂y = 0 for y = 0 and W at all x |
MATLAB was used to solve eqn. (3)via a previously published algorithm for calculating a two-dimensional concentration profile within a channel.24 Concentrations were calculated in a mesh of 40 × 43 nodes (length × width).
Numerical simulation of different flow rates
The concentration profile within the main microchannel was calculated for five different flow rates: 1, 2, 3, 6, and 20 μl min−1. These flow rates were chosen because they represented a range of velocities from what has been used previously (v = 333 μm s−1)12 to a physiologically unrealistic velocity (v = 6667 μm s−1).The linearity of all forty calculated cross sections was determined for each flow rate by using the least-squares error. The calculated cross sections of the gradient were experimentally confirmed with FITC-dextran (8 kDa, D = 8 × 10−7 cm2 s−1)25 dissolved in Hank's balanced salt solution (HBSS). FITC-dextran was used because it has a diffusion coefficient similar to the chemokine CXCL8 (8 kDa, D = 2.5 × 10−6 cm2 s−1),12 which was used in the chemotaxis experiments.
Imaging and cell movement
Epifluorescence and cell tracking experiments were performed on a Zeiss Axiovert 200M inverted microscope (Zeiss, Thornwood, NY). A monochrome, cooled CoolSNAP HQ CCD camera (Roper Scientific, Trenton, NJ) was used for both DIC imaging and epifluorescence. Openlab software (Improvision Inc., Lexington, MA) was used to analyze the FITC-dextran profiles. MetaMorph software (Universal Imaging Corp., Downingtown, PA) was used to analyze cell movement. Cell movement for each flow rate was quantified by analyzing the change in position of seven cells on a frame-by-frame basis. Cell starting positions were manually entered in MetaMorph and then each cell's position was subsequently tracked. Images were captured every 10 s over 25 min and 150 frames were analyzed for each migration experiment. Statistical analysis of migration distances was performed using single factor analysis of variance (ANOVA) and Student's t-test.Chemotaxis assay
Stable HL-60 cells transfected to express the chemokine receptor CXCR2 were differentiated to neutrophil-like cells by culturing them in 1.3% DMSO for 7 days at 37 °C and 5% CO2. Differentiated cells were washed with serum-free RPMI1640 medium, re-suspended in modified HBSS at 2 × 106 cells ml−1 and injected into the microfluidic device, which had been pre-coated with fibronectin (100 μg ml−1) for 1 h at room temperature. The fibronectin was injected through the cell injection line (Fig. 1b) with inputs A and B blocked. Cells were incubated for 20 min at 37 °C and 5% CO2 which allowed them to settle and attach to the microchannel floor. Cells were uniformly distributed on the microchannel floor prior to exposing them to the chemokine. Chemokine (CXCL8) was diluted so that the gradient concentrations ranged from 0 to 50 ng ml−1 inside the device. A Harvard Apparatus Pump 11 Pico Plus syringe pump (Harvard Apparatus, Holliston, MA) was used to provide flow to the A and B inputs. The cell injection line was closed during the chemotaxis experiments.Results and discussion
Device characterization
The gradient evolves from a step-like cross section at the entrance to a linear cross section farther down the microchannel. The cross section that is most linear represents the position down the microchannel (along the x axis) that provides the most linear, and therefore most desirable, chemokine gradient.Fig. 2a shows the calculated concentration of FITC-dextran throughout the main microchannel for a flow rate of 1 μl min−1. Figs. 2b–d show that the cross sectional profile of the gradient is a function of position along the x axis. Therefore, cells attached to the bottom of the microchannel experience different linear gradients depending on their x axis location. Cells attached 1 mm down the microchannel (Fig. 2b) experience a linear gradient across the entire microchannel width, even though the gradient is still “bumpy” from lack of diffusional mixing. Cells attached in the microchannel at length 4 mm (Fig. 2c) experience a linear gradient only between widths 100 μm and 400 μm. Cells outside of this range experience a less steep gradient, and depending on the slope might not migrate toward a higher concentration. Similarly, cells attached to the microchannel at length 8 mm (Fig. 2d) only experience a linear gradient between widths 150 μm and 350 μm.
 | ||
| Fig. 2 (a) The concentration gradient was modeled in MATLAB assuming a flow rate of 1 μl min−1. Cross sectional profiles were validated experimentally at (b) 1 mm, (c) 4 mm, and (d) 8 mm down the microchannel. Circles represent experimental observations, solid lines represent the model, and dashed lines represent the linear fit. A flow rate of 1 μl min−1 was used in the experiment. | ||
The changing cross sectional gradient linearity as a function of microchannel length implies that there is a cross section down the length of the microchannel that is the most linear. To find this region, the concentration gradient was simulated for five flow rates: 1, 2, 3, 6, and 20 μl min−1. Each simulation comprised forty cross sectional gradient profiles (three of which are shown in Figs. 2b–d for the 1 μl min−1 flow rate). A line was fit to each cross section and the summed squares error was calculated. All summed squares errors for each flow rate were plotted as a function of distance down the microchannel (Fig. 3).
 | ||
| Fig. 3 Lower values for squared errors indicate a more linear gradient. As flow rate increases, the ideal linear gradient region occupies a larger portion of microchannel length. For clarity, only every thirtieth data point is marked with a symbol. | ||
The summed squares error for each flow rate initially declines along the channel. Slower flow rates reach a local minimum in error and then increase for the duration of the microchannel length (Fig. 3). The summed squares errors trace out the evolution of the gradient within the microchannel. When the five input streams first enter the main microchannel, their cross sectional profile is step-like. As these five streams mix by diffusion, they form a sigmoidal cross sectional profile, which is represented by the local minima in Fig. 3. As the sigmoidal profile continues to evolve via diffusion, it becomes less linear due to the flat concentration profile regions near the microchannel wall (Figs. 2c and d). In a very long microchannel, or at very slow flow rates, the gradient would become uniformly mixed by diffusion (i.e., the cross sectional profile would be horizontal) and the summed squares error would be zero. None of the flow rates observed or simulated here were slow enough to achieve a horizontal profile by the end of the microchannel—a flow rate slower than 0.03 μl min−1 would be required.
As shown in Fig. 3, changing the flow rate has the effect of changing the position and duration (or length) of the optimal linear concentration gradient within the microchannel. In general, slow flow rates cause the gradient to transition from a step cross sectional profile to a linear profile in a short distance down the length of the microchannel because the residence time between neighboring streams is long. Diffusion has time to equilibrate the concentrations between streams. The result is that slower flow rates cause the optimal linear region to occupy a small fraction of the microchannel length. As the flow rate is increased, the error plot is essentially “stretched out” along the horizontal axis in the plot (Fig. 3). An increasing flow rate causes the optimal linear region to occupy an ever-longer portion of the microchannel length. Faster flow rates are more desirable because they make the gradient more linear for a longer fraction of microchannel length. The drawback is that they cause increased drag on the attached cells, which can bias migration patterns during chemotaxis.
Shear effects
Attached cells are assumed to be flat and to not interfere with fluid flow. The microchannel heights are ∼100 μm. DIC measurements showed attached cells to be 1–4 μm tall and their morphology unaffected by flow rate. Because the cells are essentially flat, they can be considered a region of the wall. Using eqn. (2), the wall shear stress can be calculated for different flow rates. Cells used in this study experienced shear stresses ranging from 0.229 to 4.58 dyn cm−2 for flow rates of 1 μl min−1 and 20 μl min−1, respectively. For reference, the shear experienced by endothelial cells in the vascular system of humans is approximately 10 dyn cm−2. The wall shear stresses and forces on cells for all flow rates used in this study are shown in Table 1.| Flow rates/μl min−1 | Shear stress/dyn cm−2 | Force/pN |
|---|---|---|
| 1 | 0.229 | 7.19 |
| 2 | 0.458 | 14.4 |
| 3 | 0.687 | 21.6 |
| 6 | 1.37 | 43.1 |
| 20 | 4.58 | 144 |
The shear stress that an attached cell will experience is linearly related to the flow rate. As mentioned previously, some cells require shear for proper development and others are adversely affected by it even though their phenotype may not change. In the microfluidic gradient generator a higher flow rate is desirable because of more favorable gradient conditions but the higher flow rate also causes more shear stress on the cells. The shear stress, and therefore flow rate, influences motion by biasing the cell's direction, as shown below.
Chemotaxis assay
Cell chemotaxis was observed with five different flow rates: 1, 2, 3, 6, and 20 μl min−1. Cell motion was analyzed in terms of cumulative distance along perpendicular axes, in this case the x and y axis (Fig. 4a). The movement vector for each selected cell in each frame of the image sequence was broken down into its x and y components. All x components were summed over the entire image sequence (150 frames) to obtain the total x displacement. Similarly, all y components were summed to obtain the total y displacement. The cumulative distance a cell crawled across the channel (i.e., up the chemotaxis gradient) was measured along the y axis while the x axis cumulative distance was used to keep track of cell migration down the channel, along the same axis as flow. The total path length, or total length of cell migration, was used to determine the relationship between total migration distance and flow. | ||
| Fig. 4 (a) Schematic for analyzing cell migration within the microchannel. Note the polarity of the x and y axes with respect to the flow direction. (b) Representative results showing cell migration trajectories in a chemokine gradient with a flow rate of 3 μl min−1 over 20 min. Cell positions at the beginning (c) and end of the experiment (d). Images flipped for coordinate system consistency. The opposite microchannel wall (top of image) is just beyond the viewable area. | ||
Fig. 4b is representative of cell migration patterns in the chemotaxis experiments. Cell migration up the chemokine gradient (from bottom to top) was biased by the 3 μl min−1 flow rate, which caused the cells to move downstream (left to right). The initial and final cell positions are shown in Figs. 4c and d, respectively.
Linear fits to the cumulative average x and y distances were calculated and are shown in Figs. 5a and b, respectively. In Fig. 5a the slope of the linear fit to x axis distance for 1 μl min−1 is greater than the slope for 2 μl min−1. The same is true for the slopes of 3 μl min−1 and 6 μl min−1. However, the linear fits in Fig. 5a show a general trend of increasing linear fit slope as flow rate increases. In Fig. 5b the slope of the 20 μl min−1 linear fit is greater than the 6 μl min−1 linear fit. The general trend though shows decreasing linear fit slopes for y axis distances as flow rate increases.
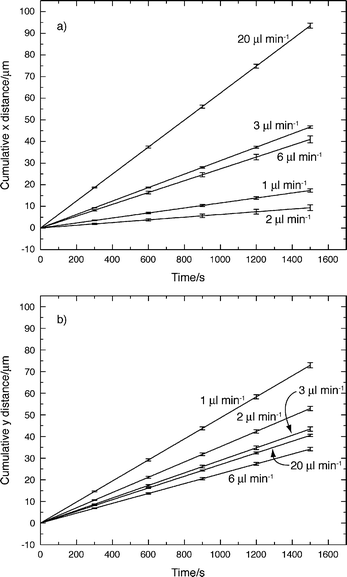 | ||
| Fig. 5 Linear fits to the average cumulative x axis (a) and y axis (b) migration distances for each flow rate. | ||
Table 2 summarizes the effect of flow rate on cell migration for all five flow rates. The final x, y, and total distances were averaged among the seven tracked cells in each of the five flow rates. As flow rate increased, cells on average migrated shorter distances across the microchannel. Conversely, at higher flow rates, cells migrated further downstream. The total path length did not vary significantly as the flow rate was increased.
| 1 μl min−1 | 2 μl min−1 | 3 μl min−1 | 6 μl min−1 | 20 μl min−1 | |
|---|---|---|---|---|---|
| Total x axis distance | 19.0 ± 36.8 | 20.0 ± 38.9 | 48.3 ± 34.0 | 55.2 ± 30.8 | 83.1 ± 44.3 |
| Total y axis distance | 73.3 ± 20.3 | 60.9 ± 43.6 | 47.9 ± 22.1 | 32.2 ± 25.5 | 36.3 ± 38.6 |
| Total distance | 131 ± 25.4 | 144 ± 25.4 | 123 ± 16.5 | 141 ± 32.4 | 168 ± 55.8 |
The results in Table 2 demonstrate the effect of flow rate on cell migration. Even though the same range of chemokine concentration was present within the microchannel for all five flow rates, the average distance cells migrated up the gradient (along the y axis) decreased from 73.3 ± 20.3 μm at 1 μl min−1 to 36.3 ± 38.6 μm at 20 μl min−1. As flow rates increased, average cell migration distances down the microchannel increased from 19.0 ± 36.8 μm at 1 μl min−1 to 83.1 ± 44.3 μm at 20 μl min−1 (p < 0.05). The average x axis migration distance also varied significantly (p < 0.05) between flow rates of 2 μl min−1 and 20 μl min−1 (20 ± 38.9 μm and 83.1 ± 44.3 μm, respectively). The total average total distance cells migrated did not significantly change with respect to the flow rate. These results indicate that flow used to generate microfluidic gradients can bias cell migration during chemotaxis.
Fig. 6a summarizes the findings of the present study. Cell migration distances along the x axis increase with increasing flow rate while migration distances along the y axis decrease. Total migration distance remains relatively constant over the five flow rates. Fig. 6b summarizes the data in a different light by converting the linear fit of the cell migration into the θ angle with respect to the horizontal. Larger values of θ indicate average cell migration across the width of the microchannel (up the chemokine gradient). Smaller values of θ indicate average cell migration down the length of the channel due to forces on the cell by the fluid.
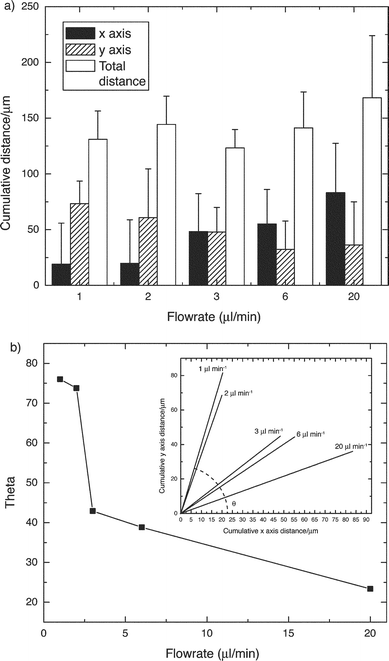 | ||
| Fig. 6 (a) Cell migration distances were affected by the flow rate within the microfluidic gradient generator. Cells migrated across the microchannel at slower flow rates and down the length of the channel at higher flow rates. (b) The linear fit to cell migration at different flow rates was calculated (inset). The slope of each linear fit was converted to an angle to get a convenient representation of the effect of flow rate on cell migration. | ||
Conclusions
Microfluidics provides a convenient and effective way to create a chemokine gradient for studies of cell chemotaxis. In the microdevice design investigated here, continuous flow is required within the device to create and maintain the chemokine gradient. An undesirable consequence of the flow is its effect on cell migration. Two characteristics of the gradient device have been characterized in this paper.First, the cross sectional linearity of the chemokine gradient depends on the position down the length of the microchannel. Cells attached near the beginning of the microchannel will experience a step-like cross sectional profile while cells attached farther down the microchannel experience a more linear cross section. The width of the linear portion of the cross section decreases down the microchannel (i.e., linearity is a function of position in the x direction). Therefore, cells should ideally be observed only in the middle of the microchannel (in the y direction) and far enough downstream so that the step cross sectional profile has disappeared. Changing the flow rate modulates how far down the microchannel each cross sectional gradient profile exists. Higher flow rates cause the gradient to “stretch out”, allowing each cross section to exist for a longer portion of the microchannel length. Slower flow rates have the opposite effect, causing the gradient to go from a step cross sectional profile to a flat profile in a short distance down the microchannel.
Second, while higher flow rates are more desirable from a gradient viewpoint, the higher flow rate affects cell movement. Higher flow rates guide the cells down the microchannel, in the direction of flow, thus biasing their chemotactic response. Certain cell lines are known to be shear sensitive while others require shear for proper development. The concern when using these gradient microdevices is not cell damage from shear, but rather the possibility of biased chemotactic migration due to fluid flow. Therefore, the influence of the chemokine gradient must be separated from flow (shear) effects when analyzing cell trajectories in continuous flow microfluidic concentration gradients.
The data we present suggest that it may be possible to use a combination of chemokine gradients and shear forces to measure the tractile force exerted by a cell. Additional research, beyond the scope of this paper, will be required to explore the complexities of the hydrodynamic interactions between the cell and the flowing fluid, and how it may affect the relative contributions of the motile and the hydrodynamic forces. Possible effects include distortion of the chemokine gradient by the fluid flowing past the cell (Eberhard Bodenschatz, personal communication), or reorientation of the cell axis in response to the shear forces.
The gradient device demonstrated by Jeon et al.12–14 is a novel and valuable tool for chemotaxis research. However, the device characteristics presented in this article should be considered when using the device to conduct chemotaxis experiments and interpreting the results. The possibility of using this approach to estimate cell chemotactic force is worthy of more detailed investigation.
Acknowledgements
The authors wish to thank S. Gruver for his assistance in using the cell tracking software and for helpful discussions and L. McCawley for helpful suggestions. This work was supported in part by the Vanderbilt University Academic Venture Capital Fund, the Vanderbilt Institute for Integrative Biosystems Research and Education (G. W., J. W.), the Vanderbilt University Medical Center (G. W.), the Department of Veterans Affairs (A. R.), and NIH grant CA-34590 (A. R., J. S.).References
- J. Adler, Annu. Rev. Biochem., 1975, 44, 341–356 CrossRef CAS.
- H. C. Berg, Annu. Rev. Biophys. Bioeng., 1975, 4, 119–136 Search PubMed.
- B. Moser, M. Wolf, A. Walz and P. Loetscher, Trends Immunol., 2004, 25(2), 75–84 Search PubMed.
- T. J. John and O. F. Sieber, Jr., Life Sci., 1976, 18(2), 177–181 CrossRef CAS.
- R. D. Nelson, P. G. Quie and R. L. Simmons, J. Immunol., 1975, 115(6), 1650–1656 CAS.
- J. L. Haddox, I. W. Knowles, C. I. Sommers and R. R. Pfister, J. Immunol. Methods, 1994, 171(1), 1–14 CrossRef CAS.
- J. L. Haddox, R. R. Pfister and C. I. Sommers, J. Immunol. Methods, 1991, 141(1), 41–52 CrossRef CAS.
- S. H. Zigmond, J. Cell Biol., 1977, 75(2 pt. 1), 606–616 CAS.
- D. Zicha, G. A. Dunn and A. F. Brown, J. Cell Sci., 1991, 99(pt. 4), 769–775 Search PubMed.
- S. Kanegasaki, Y. Nomura, N. Nitta, S. Akiyama, T. Tamatani, Y. Goshoh, T. Yoshida, T. Sato and Y. Kikuchi, J. Immunol. Methods, 2003, 282(1–2), 1–11 CrossRef CAS.
- L. P. Bignold, J. Immunol. Methods, 1988, 108(1–2), 1–18 CrossRef CAS.
- N. L. Jeon, H. Baskaran, S. K. W. Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nat. Biotechnol., 2002, 20(8), 826–830 CAS.
- F. Lin, C. M. C. Nguyen, S. J. Wang, W. Saadi, S. P. Gross and N. L. Jeon, Biochem. Biophys. Res. Commun., 2004, 319(2), 576–581 CrossRef CAS.
- F. Lin, W. Saadi, S. W. Rhee, S. J. Wang, S. Mittal and N. L. Jeon, Lab Chip, 2004, 4(3), 164–167 RSC.
- S. Dertinger, D. Chiu, N. Jeon and G. Whitesides, Anal. Chem., 2001, 73(6), 1240–1246 CrossRef CAS.
- D. Gaver and S. Kute, Biophys. J., 1998, 75, 721–733 CrossRef CAS.
- F. M. White, Fluid mechanics, McGraw-Hill, New York, 1986 Search PubMed.
- R. Nerem, R. Alexander, D. Chappell, R. Medford, S. Varner and W. Taylor, Am. J. Med. Sci., 1998, 316(3), 169–175 Search PubMed.
- J. Keane, D. Ryan and P. Gray, Biotechnol. Bioeng., 2003, 81(2), 211–220 CrossRef CAS.
- D. Duffy, J. McDonald, O. Schueller and G. Whitesides, Anal. Chem., 1998, 70(23), 4974–4984 CrossRef CAS.
- R. Bird, W. Stewart and E. Lightfoot, Transport Phenomena, John Wiley and Sons, New York, 1960 Search PubMed.
- R. Ismagilov, A. Stroock, P. Kenis, G. Whitesides and H. Stone, Appl. Phys. Lett., 2000, 76(17), 2376–2378 CrossRef CAS.
- A. Kamholz, E. Schilling and P. Yager, Biophys. J., 2001, 80, 1967–1972 CrossRef CAS.
- P. Williams, S. Levin, T. Lenczycki and J. Giddings, Ind. Eng. Chem. Res., 1992, 31, 2172–2181 CrossRef CAS.
- S. Jayaraman, N. S. Joo, B. Reitz, J. J. Wine and A. S. Verkman, Proc. Natl. Acad. Sci. USA, 2001, 98(14), 8119–8123 CrossRef CAS.
Footnotes |
| † Present address: Joint Department of Biomedical Engineering, North Carolina State University, Raleigh, NC 27695, USA and University of North Carolina, Chapel Hill, NC 27599, USA. |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2005 |