Crystal structures and physical properties of single-component molecular conductors consisting of nickel and gold complexes with bis(trifluoromethyl)tetrathiafulvalenedithiolate ligands
Masaaki
Sasa
a,
Emiko
Fujiwara
a,
Akiko
Kobayashi
*a,
Shoji
Ishibashi
b,
Kiyoyuki
Terakura
c,
Yoshinori
Okano
d,
Hideki
Fujiwara
d and
Hayao
Kobayashi
d
aResearch Centre for Spectrochemistry, Graduate School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: akiko@chem.s.u-tokyo.ac.jp
bResearch Institute for Computational Sciences, AIST, Tsukuba, Ibaraki 305-8568, Japan
cCenter for Advanced Science and Technology Creative Research Initiative, “Sousei” Division of Frontier Research, Hokkaido University, Kita 8 Nishi 5, Sapporo 060-0808, Japan
dInstitute for Molecular Science and CREST, JST, Myodaiji, Okazaki 444-8585, Japan
First published on 29th November 2004
Abstract
The neutral nickel and gold complexes with bis(trifluoromethyl)tetrathiafulvalenedithiolate ligands, [M(hfdt)2] (M = Ni, Au) were prepared in order to examine the possibility of the development of single-component molecular conductors soluble in organic solvents. However, in contrast to the previous report, the crystals did not show any solubility in the usual organic solvents. On the other hand, the crystal structure analyses showed unique two-dimensional layered structures, despite that the single-component molecular conductors usually tend to take a compact three-dimensional molecular arrangement. Each layer is separated by the terminal CF3 groups to form the “CF3 bilayer structure”. The shortest intermolecular F⋯F distance (3.018 Å for [Ni(hfdt)2] and 2.862 Å for [Au(hfdt)2]) is significantly longer than the van der Waals F⋯F distance (2.70 Å) and the distribution of the frontier electrons is almost zero around the CF3 bilayer region. This is due to the strong F⋯F segregation effect, which will provide a useful way to control the molecular aggregation in the single-component molecular conductors. Extended-Hückel tight-binding band structure calculations and the ab initio local density approximation (LDA) band structure calculations were made for [Ni(hfdt)2], which explains the semiconducting and non-magnetic properties of the system. Extended-Hückel tight-binding band structure calculations were also made for [Au(hfdt)2]. The calculated band structure is consistent with the semiconducting and almost non-magnetic properties of [Au(hfdt)2].
Introduction
It has been long believed that metal electrons cannot exist in molecular crystals consisting of single-component molecules. However, we have recently succeeded in preparing a crystal of a neutral transition metal complex with extended-TTF (tetrathiafulvalene) dithiolate ligands, [Ni(tmdt)2] (tmdt = trimethylenetetrathiafulvalenedithiolate) exhibiting metallic behavior down to 0.6 K.1,2 Very recently, direct experimental evidence for the Fermi surface was obtained by torque magnetometry measurements using a very tiny single crystal of [Ni(tmdt)2] and a sensitive microcantilever at low temperatures at high magnetic fields.3 The quantum oscillations of magnetization, the de Haas–van Alphen (dHvA) effect, observed for all the directions of magnetic field, showed the presence of three-dimensional electron and hole Fermi surfaces.2c,3Unlike traditional molecular conductors composed of π molecules forming a conduction band and the counter-ions producing conduction electrons by accepting or donating electrons from π molecules, crystals of single-component molecular conductors definitely consist of identical neutral molecules. One of the new possibilities arising from the realization of single-component molecular metals is the possibility of developing molecular metals with high magnetic transition temperatures. [Ni(tmdt)2] can be changed into a magnetic molecule by replacing the central Ni2+ atom with magnetic atoms such as Cu2+ and Co2+, where the magnetic interaction between 3d magnetic moments of central transition metal atoms can be mediated by the surrounding π conduction electrons.4,5 The Curie temperature of the ferromagnetic crystal composed of single-component organic molecules with N–O˙ radicals is very low,6 but the magnetic transition temperature is expected to become much higher if the intermolecular magnetic interaction is mediated by conduction electrons.
Soluble metals or conductors might be another new possibility. It is expected that the solubility will be enhanced by introducing long alkyl-chain substituents or highly polar substituents. If a metallic (or highly conducting) crystal soluble in an organic solvent could be obtained, they could provide a new method of electrical wiring. In 1979, Engler et al. submitted an attractive paper on the synthesis of a neutral nickel complex with bis(trifluoromethyl)tetrathiafulvalenedithiolate ligands: the reaction of disodium bis(trifluoromethyl)tetrathiafulvalenedithiolate with nickel acetate followed by treatment with O2, and purification of the product by Soxhlet extraction (using PhCl) gave green-black platelets.7 We took notice of this description and tried to synthesize the neutral metal complex [bis(trifluoromethyl)tetrathiafulvalenedithiolato]nickel, [Ni(hfdt)2], in order to obtain a conducting crystal soluble in organic solvents.
Recently, the strong segregation patterns resulting from the formation of a fluorous bilayer structure in highly fluorinated molecules with CF2 groups on a TTF core aroused a good deal of interest.8 The overlap interaction of molecules in the crystal with the nonbonded interactions of the fluorinated moieties produced a layered structure with full segregation of the aromatic and fluorinated moieties despite the limited number of CF2 groups. Such segregation effects can be thought to be one of the tools for controlling the aggregation patterns of the molecules. If we can prepare single-component molecular conductors that are easily dissolved or sublimed, they will provide a new way to obtain molecular conducting films.
Here, we report the preparation, crystal structure determination, physical properties and band structure calculations of neutral [bis(trifluoromethyl)tetrathiafulvalenedithiolato]nickel and gold complexes [M(hfdt)2] (M = Ni, Au; hfdt = bis(trifluoromethyl)tetrathiafulvalenedithiolate) with fluorinated moieties on the TTF core (Chart 1). In terms of valence electrons, the bis(dithiolato)gold complex is isoelectronic with the planar monoanionic bis(dithiolato)nickel complex with one unpaired electron per molecule, which makes the electromagnetic properties of the neutral gold complex very attractive.9,10 These unpaired electrons or holes will produce various possible electrical properties. The system can exhibit a half-filled metallic band, a spin-paired insulating state with a dimeric structure and a Mott insulating state with strongly correlated conduction electrons. According to this idea, bis(dithiolato)gold complexes were prepared and their physical properties were examined.
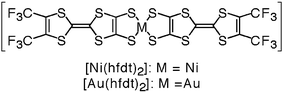 | ||
| Chart 1 | ||
Experimental
Synthesis of neutral [Ni(hfdt)2]
The syntheses of the extended TTF ligand and the corresponding metal complex were performed according to the literature methods2a,11,12 under a strictly inert atmosphere using the Schlenk technique. The synthetic procedure is shown in Scheme 1. A 25 wt% solution of tetramethylammonium hydroxide (0.3 ml, 0.6 mmol) in MeOH was added to 4,5-bis(2′-cyanoethylthio)-4,5-bis(trifluoromethyl)tetrathiafulvalene (72 mg, 0.14 mmol) in dry MeOH (2 ml), and the reaction mixture was stirred at room temperature for 1 h, followed by the addition of nickel(II) dichloride hexahydrate (16 mg, 0.067 mmol) in dry MeOH (2 ml) at −78 °C. The solution was then stirred overnight and the reaction mixture was warmed gradually to room temperature. The resultant precipitate was filtered off, washed with MeOH and dried in vacuo to give the tetramethylammonium salt of the metal complex as a dark reddish solid.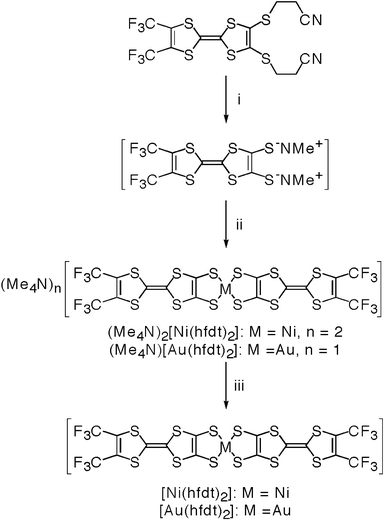 | ||
| Scheme 1 Reagents and conditions: i, 25 wt% Me4NOH/MeOH (4.0 equiv.), dry MeOH, rt, 1 h; ii, NiCl2·6H2O/dry MeOH (0.5 equiv.) or HAuCl4·4H2O/dry MeOH (0.5 equiv.), −78 °C to rt, overnight; iii, electrochemical oxidation (constant current of 2.0 µA for 2 weeks at room temperature). | ||
A mixture of this metal complex and tetra-n-butylammonium hexafluorophosphate (400 mg) as a supporting electrolyte were dissolved in PhCl, and the solution was poured into H-shaped glass cells with Pt electrodes. Electrochemical oxidation was performed by the use of these glass cells under a constant current of ca. 2.0 µA for 2 weeks at room temperature. The black plate single crystals of the neutral nickel complex were obtained on the surface of the platinum electrodes. EPMA (electron probe microanalysis) was performed, indicating the ratio of Ni and S to be about 1 : 11.2. No traces of other elements were detected.
Synthesis of neutral [Au(hfdt)2]
The synthesis of neutral [Au(hfdt)2] was almost the same as that of [Ni(hfdt)2], as shown in Scheme 1. A 25 wt% solution of tetramethylammonium hydroxide (0.15 ml, 0.3 mmol) in MeOH was added to 4,5-bis(2′-cyanoethylthio)-4,5-bis(trifluoromethyl)tetrathiafulvalene (36 mg, 0.075 mmol) in dry MeOH (1 ml), and the reaction mixture was stirred at room temperature for 1 h, followed by the addition of hydrogen tetrachloroaurate(III) tetrahydrate (12 mg, 0.035 mmol) in dry MeOH (1 ml) at −78 °C. The solution was then stirred overnight and the reaction mixture was warmed gradually to room temperature. The resultant precipitate was filtered off, washed with MeOH and dried in vacuo to give the tetramethylammonium salt of the metal complex as a dark greenish solid. The conditions of electrochemical oxidation were almost the same as those of nickel complex described above. EPMA (electron probe microanalysis) was performed, indicating the ratio of Au and S to be about 1 : 12.9. No traces of other elements were detected.Crystal structure determination
Crystal structure determinations were carried out for [M(hfdt)2] (M = Ni, Au). Data collection for a black plate-shaped crystal of [Ni(hfdt)2] was performed on a Rigaku AFC7R instrument with graphite-monochromated Mo Kα radiation (λ = 0.7107 Å) at room temperature. Crystal dimensions 0.4 × 0.2 × 0.02 mm3. Crystal data: C16F12NiS12, M = 863.63, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a
= 7.964(1)
Å, b
= 16.701(2)
Å, c
= 5.0608(9)
Å, α
=90.29(1)°, β
= 92.54(1)°, γ
= 98.40(1)°, V
= 665.2(2)
Å3, Z
= 1, dcalcd
= 2.156 g cm−3, μ
= 17.67 cm−1, F000
= 424.0. Intensity data were collected to a maximum 2θ value of 60.1° by the ω-scan technique. The number of collected reflections was 4141. The structure was solved by a direct method (SIR97).13 The atomic scattering factors were taken from the International Tables for X-ray Crystallography.14 The non-hydrogen atoms were anisotropically refined by the full-matrix least-squares method, based on 2215 observed reflections [I > 4.0σ(I)] and 188 variable parameters. The structure was refined to R
= 0.067, Rw
= 0.119, GOF = 2.97. All the calculations were performed using the teXsan crystallographic software package of the Molecular Structure Corporation.15 CCDC reference number 249813. See http://www.rsc.org/suppdata/jm/b4/b413597k/ for crystallographic data in .cif or other electronic format.
(no. 2), a
= 7.964(1)
Å, b
= 16.701(2)
Å, c
= 5.0608(9)
Å, α
=90.29(1)°, β
= 92.54(1)°, γ
= 98.40(1)°, V
= 665.2(2)
Å3, Z
= 1, dcalcd
= 2.156 g cm−3, μ
= 17.67 cm−1, F000
= 424.0. Intensity data were collected to a maximum 2θ value of 60.1° by the ω-scan technique. The number of collected reflections was 4141. The structure was solved by a direct method (SIR97).13 The atomic scattering factors were taken from the International Tables for X-ray Crystallography.14 The non-hydrogen atoms were anisotropically refined by the full-matrix least-squares method, based on 2215 observed reflections [I > 4.0σ(I)] and 188 variable parameters. The structure was refined to R
= 0.067, Rw
= 0.119, GOF = 2.97. All the calculations were performed using the teXsan crystallographic software package of the Molecular Structure Corporation.15 CCDC reference number 249813. See http://www.rsc.org/suppdata/jm/b4/b413597k/ for crystallographic data in .cif or other electronic format.
Data collection for tiny black block-shaped crystals of the [Au(hfdt)2] was performed on a Rigaku/MSC Mercury CCD system with graphite-monochromated Mo Kα radiation (λ
= 0.7107 Å) and confocal mirror at room temperature. Crystal dimensions 0.10 × 0.07 × 0.01 mm3. Crystal data: C16AuF12S12, M
= 1001.84, triclinic, space group P
(no. 2), a
= 8.019(3)
Å, b
= 8.752(3)
Å, c
= 19.552(6)
Å, α
= 84.26(2)°, β
= 84.30(2)°, γ
= 81.59(2)°, V
= 1345.7(8)
Å3, Z
= 2, dcalcd
= 2.472 g cm−3, μ
= 65.11 cm−1, F000
= 950.0. The number of collected reflections was 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 043. The structure was solved by a direct method (SIR92).16 The atomic scattering factors were taken from the International Tables for X-ray Crystallography.14 The non-hydrogen atoms were anisotropically refined by the full-matrix least-squares method, based on 4725 observed reflections [I > 3.0σ(I)] and 370 variable parameters. The structure was refined to R
= 0.070, Rw
= 0.086, GOF = 2.237. All the calculations were performed using the CrystalStructure17 crystallographic software package. CCDC reference number 249812. See http://www.rsc.org/suppdata/jm/b4/b413597k/ for crystallographic data in .cif or other electronic format.
043. The structure was solved by a direct method (SIR92).16 The atomic scattering factors were taken from the International Tables for X-ray Crystallography.14 The non-hydrogen atoms were anisotropically refined by the full-matrix least-squares method, based on 4725 observed reflections [I > 3.0σ(I)] and 370 variable parameters. The structure was refined to R
= 0.070, Rw
= 0.086, GOF = 2.237. All the calculations were performed using the CrystalStructure17 crystallographic software package. CCDC reference number 249812. See http://www.rsc.org/suppdata/jm/b4/b413597k/ for crystallographic data in .cif or other electronic format.
Physical measurements
Temperature dependencies of the electrical resistivities were measured by using the conventional four-probe method by use of Huso Electro Chemical System HECS 994C-1 Multi-channel four-terminal conductometer. Electrode contacts were made by using gold wire (15 µm diameter) with gold paste.Static magnetic susceptibility (χ–T measurements of [Ni(hfdt)2] (4.57 mg, 0.0054 mmol) and [Au(hfdt)2] (1.3 mg, 0.0013 mmol) were performed by a Quantum Design MPMS-2 (for [Ni(hfdt)2]) or MPMS-7XL (for [Au(hfdt)2]) SQUID (superconducting quantum interference device) magnetometer in the temperature range 2.0–300 K. The applied magnetic fields were 30 kOe for [Ni(hfdt)2] and 20 kOe for [Au(hfdt)2]. The samples were wrapped with clean aluminum foil whose magnetic susceptibilities were separately measured and subtracted. The diamagnetic contribution was calculated from Pascal's constants (χdia = −3.3 × 10−4 emu mol−1 for [Ni(hfdt)2] and χdia = −3.5 × 10−4 emu mol−1 for [Au(hfdt)2]).
The visible and IR spectra of [Ni(hfdt)2] were measured on crystalline powder samples using a JASCO-MSV-370 spectrometer (40000–5000 cm−1) and a JASCO-FT/IR-420 + IRT30 spectrometer (7800–1000 cm−1).
Results and discussion
Syntheses
The neutral complexes [M(hfdt)2] (M = Ni, Au) were prepared by electrochemical oxidation of the anionic complexes (Me4N)2[Ni(hfdt)2] and (Me4N)[Au(hfdt)2] (Scheme 1) in the presence of tetra-n-butylammonium hexafluorophosphate as the supporting electrolyte in dry PhCl under a constant current of 2.0 µA at room temperature. Air-stable plate-shaped black single crystals of neutral [M(hfdt)2] (M = Ni, Au) grew on the electrode within about 2 weeks.The crystal of [Ni(hfdt)2] was found to be insoluble in the usual organic solvents (such as MeCN, THF, PhCl, MeOH, DMF, DMSO, CH2Cl2, C6H14, CS2, etc.), contrary to the report of Engler et al., who tried to synthesize the neutral nickel complex [Ni(hfdt)2] from Na2[Ni(hfdt)2] by O2 oxidation.7 The neutral compound they obtained was purified by Soxhlet extraction (using PhCl) and a peak was observed in the electronic absorption spectrum at 1.35 µm using DMSO as solvent. However, unlike their reports,7 [Ni(hfdt)2] was found to be insoluble in the usual organic solvents. On the other hand, the dianionic complex (Me4N)2[Ni(hfdt)2] has high solubility in the usual organic solvents.
Crystal structures
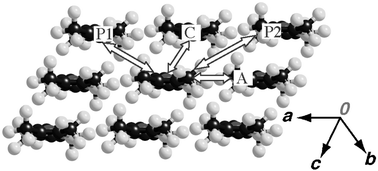
Similar to other single-component molecular metals such as M(tmdt)2 (M = Ni, Au), and Ni(dmdt)2,1,2 neutral [Ni(hfdt)2] molecules crystallize into a very simple triclinic structure. The unit cell contains only one molecule. The central Ni atoms are on the lattice points and half of the molecule is crystallographically independent. Fig. 1 shows the molecular and crystal structures of [Ni(hfdt)2]. The [Ni(hfdt)2] unit is almost planar except for the terminal C–CF3 group, and regularly arranged along the c axis. The nickel atom has a square planar coordination with an average Ni–S distance of 2.156(1) Å and an S(1)–Ni–S(2) angle of 93.15 (8)° (see Fig. 1(a)). These values well correspond to those of neutral bis(dithiolato)nickel complex [Ni(tmdt)2] (Ni–S 2.177(7) Å, S–Ni–S 92.29(4)° at 123 K).1,2 A comparison of the average C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–S bond lengths in neutral [Ni(hfdt)2], [Ni(tmdt)2] and [Ni(ptdt)2]18
(ptdt = propylenedithiotetrathiafulvalenedithiolate) is shown in Table 1. No significant differences in the bond distances were observed between these molecules. The average C–CF3 and C–F distances are 1.495(7)
Å and 1.293(8)
Å, respectively, which are slightly shorter than the corresponding lengths of the sum of the atomic covalent radii (1.54 Å
(C–C), 1.41 Å
(C–F)). The molecules form a one-dimensional column structure along the c axis with an interplanar distance of 3.52 Å and a slip distance along the long axis of the molecule of 1.61 Å
(Fig. 1(c)–(d)). The shortest Ni⋯Ni distance along the c direction is 5.061 Å. The overlapping mode of [Ni(hfdt)2] molecules is shown in Fig. 1(d).
C and C–S bond lengths in neutral [Ni(hfdt)2], [Ni(tmdt)2] and [Ni(ptdt)2]18
(ptdt = propylenedithiotetrathiafulvalenedithiolate) is shown in Table 1. No significant differences in the bond distances were observed between these molecules. The average C–CF3 and C–F distances are 1.495(7)
Å and 1.293(8)
Å, respectively, which are slightly shorter than the corresponding lengths of the sum of the atomic covalent radii (1.54 Å
(C–C), 1.41 Å
(C–F)). The molecules form a one-dimensional column structure along the c axis with an interplanar distance of 3.52 Å and a slip distance along the long axis of the molecule of 1.61 Å
(Fig. 1(c)–(d)). The shortest Ni⋯Ni distance along the c direction is 5.061 Å. The overlapping mode of [Ni(hfdt)2] molecules is shown in Fig. 1(d).
![(a) Molecular structures of [Ni(hfdt)2] with atomic numbering schemes. (b) Crystal structure of [Ni(hfdt)2] projected approximately onto the molecular plane. The intermolecular S⋯S distances (3.549 Å) less than the van der Waals contact (3.70 Å) are shown as dotted lines (see Table 2). (c) Side view of the molecular arrangement of [Ni(hfdt)2]. Molecules form a one-dimensional column structure along the c axis with interplanar distance of 3.52 Å
(A). The shortest intermolecular F⋯F distances (3.018 Å) are shown as dotted lines. (d) Intermolecular overlapping mode of neutral molecules of [Ni(hfdt)2]. Slip distances are 1.61 Å
(i) and 3.26 Å
(ii) along the short and long molecular axes, respectively.](/image/article/2005/JM/b413597k/b413597k-f1.gif) | ||
| Fig. 1 (a) Molecular structures of [Ni(hfdt)2] with atomic numbering schemes. (b) Crystal structure of [Ni(hfdt)2] projected approximately onto the molecular plane. The intermolecular S⋯S distances (3.549 Å) less than the van der Waals contact (3.70 Å) are shown as dotted lines (see Table 2). (c) Side view of the molecular arrangement of [Ni(hfdt)2]. Molecules form a one-dimensional column structure along the c axis with interplanar distance of 3.52 Å (A). The shortest intermolecular F⋯F distances (3.018 Å) are shown as dotted lines. (d) Intermolecular overlapping mode of neutral molecules of [Ni(hfdt)2]. Slip distances are 1.61 Å (i) and 3.26 Å (ii) along the short and long molecular axes, respectively. | ||
|
|
|
|
|
|---|---|---|---|
| a At 123 K. | |||
| a | 2.183(1) | 2.177(1) | 2.156(1) |
| b | 1.726(4) | 1.717(4) | 1.695(6) |
| c | 1.384(8) | 1.352(6) | 1.380(7) |
| d | 1.736(4) | 1.750(4) | 1.737(6) |
| e | 1.743(3) | 1.742(4) | 1.752(6) |
| f | 1.380(7) | 1.362(5) | 1.360(7) |
| g | 1.752(3) | 1.751(4) | 1.746(7) |
| h | 1.765(4) | 1.739(4) | 1.749(7) |
| i | 1.384(8) | 1.333(6) | 1.310(7) |
The molecules are arranged to form two-dimensional layers parallel to the ac plane. The adjacent layers are separated by the CF3 group along the b direction. The shortest intermolecular F⋯F distance is 3.018 Å, which is remarkably longer than the F⋯F van der Waals contact (2.70 Å)
(see Fig. 1(c)). These strong segregation patterns of the molecular arrangements are often observed in the crystals of highly fluorinated molecules.8 Intermolecular S⋯S contacts less than the sum of the van der Waals radii (3.70 Å) were not found within the column. The shortest S⋯S short contact (3.549 Å) was observed along the [10] direction (see Fig. 1(b) and Table 2).
|
|
||||
|---|---|---|---|---|
| H–H | H–L | L–L | L–H | |
| a The short S⋯S distance is found for the intermolecular contact: P1 (3.549 Å). | ||||
| A | 0.6 | 0.1 | 0.1 | −0.1 |
| C | −0.7 | −0.5 | −0.8 | 0.5 |
| P1 | −3.2 | −2.0 | 1.3 | 2.0 |
| P2 | −3.2 | 2.0 | 1.3 | −2.9 |
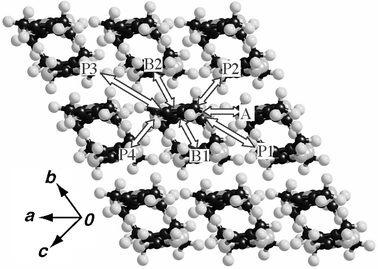
The crystal structure of [Au(hfdt)2] was investigated. Neutral [Au(hfdt)2] molecules crystallize into a simple triclinic structure. Fig. 2 shows the molecular and crystal structure of [Au(hfdt)2]. Unlike [Ni(hfdt)2], [Au(hfdt)2] is on the general position and the unit cell contains two molecules connected by inversion symmetry. The [Au(hfdt)2] molecule is also almost planar. The gold atom has a square planar coordination with average Au–S distance of 2.323(3)
Å and S–Au–S angle of 91.45(9)°. These values correspond to those of neutral bis(dithiolato)gold complex [Au(tmdt)2]
(Au–S 2.296(2)
Å, S–Au–S 89.9(1)°).9 The average C–CF3 and C–F distances are 1.504(9)
Å and 1.288(6)
Å, respectively. [Au(hfdt)2] forms a dimeric structure along the b axis with an interplanar distance of 3.60 Å and a slip distance of 0.67 Å. This dimeric structure is rather similar to that of Pd(dmit)2n− or Pt(dmit)2n− complexes, where the overlap mode of Pd complexes within the dimer have an eclipsed form.19 The Au⋯Au distances are 3.86 Å within each dimer and 5.40 Å between dimers. [Ni(hfdt)2] is almost planar except the terminal C–CF3 group. However, [Au(hfdf)2] has a bent structure at the positions of sulfur atoms of the TTF part, with a dihedral angle of 13.7° and a chair conformation (see Fig. 2(a)). [Au(hfdf)2] molecules are arranged to form a dimeric column along the b axis but there are many intercolumnar S⋯S short contacts less than the sum of the van der Waals radii, especially along [10]. The large number of the short intermolecular S⋯S contacts in the ab conduction layer is consistent with the relatively high conductivity of [Au(hfdt)2], as described below, but the terminal CF3 groups separate the [Au(hfdt)2] layers along the c direction (Fig. 2(c)). Similar to [Ni(hfdt)2], the crystal structure of [Au(hfdt)2] showed a strong segregation pattern of the fluorinated groups. The shortest F⋯F distance of 2.862 Å is much longer than the F⋯F van der Waals distance (2.70 Å). Figs. 2(d) and 2(e) show the overlapping mode within a dimer and between dimers.
![(a) Molecular structures of [Au(hfdt)2] with atomic numbering schemes. (b) Crystal structures of [Au(hfdt)2] projected approximately onto the molecular plane. The short intermolecular S⋯S contacts less than the sum of the van der Waals radius (3.70 Å) are shown as dotted lines (see Table 3). (c) Side view of the molecular arrangement of [Au(hfdt)2]. Molecules form a dimeric structure along the b axis. The distance within a dimer (A) is 3.60 Å and the distance between dimers (B) is 3.61 Å. The shortest intermolecular F⋯F distances (2.862 Å) are shown as dotted lines. (d) Overlapping mode within a dimer of [Au(hfdt)2] . Slipping distances are 1.37 Å
(i) and 0.67 Å
(ii) along the short and long molecular axes, respectively. (e) Overlapping mode between dimers of [Au(hfdt)2]. Slip distances are 1.68 Å
(iii) and 3.64 Å
(iv) along the short and long molecular axes, respectively.](/image/article/2005/JM/b413597k/b413597k-f2.gif) | ||
| Fig. 2 (a) Molecular structures of [Au(hfdt)2] with atomic numbering schemes. (b) Crystal structures of [Au(hfdt)2] projected approximately onto the molecular plane. The short intermolecular S⋯S contacts less than the sum of the van der Waals radius (3.70 Å) are shown as dotted lines (see Table 3). (c) Side view of the molecular arrangement of [Au(hfdt)2]. Molecules form a dimeric structure along the b axis. The distance within a dimer (A) is 3.60 Å and the distance between dimers (B) is 3.61 Å. The shortest intermolecular F⋯F distances (2.862 Å) are shown as dotted lines. (d) Overlapping mode within a dimer of [Au(hfdt)2] . Slipping distances are 1.37 Å (i) and 0.67 Å (ii) along the short and long molecular axes, respectively. (e) Overlapping mode between dimers of [Au(hfdt)2]. Slip distances are 1.68 Å (iii) and 3.64 Å (iv) along the short and long molecular axes, respectively. | ||
Electrical conductivities
The electrical conductivity of single crystals for [Ni(hfdt)2] along the direction approximately parallel to the c axis showed that [Ni(hfdt)2] is semiconducting (Fig. 3(a)). The room temperature conductivity was 1.6 × 10−3 S cm−1 and the activation energy (Ea) was 0.14 eV. The electrical conductivity of a compressed pellet of [Au(hfdt)2] also exhibited semiconducting behavior, with a room temperature conductivity of 3.2 × 10−3 S cm−1 and an activation energy of 0.12 eV (Fig. 3(b)). Considering that the measurements were made on a compressed pellet, crystalline [Au(hfdt)2] should be a fairly good conductor.![Temperature dependences of resistivities of (a) a single crystal of [Ni(hfdt)2] approximately along the c axis and (b) a compacted pellet of [Au(hfdt)2]. The room temperature resistivities are 6.3 × 102
Ω cm for [Ni(hfdt)2] and 3.1 × 102
Ω cm for [Au(hfdt)2].](/image/article/2005/JM/b413597k/b413597k-f3.gif) | ||
| Fig. 3 Temperature dependences of resistivities of (a) a single crystal of [Ni(hfdt)2] approximately along the c axis and (b) a compacted pellet of [Au(hfdt)2]. The room temperature resistivities are 6.3 × 102 Ω cm for [Ni(hfdt)2] and 3.1 × 102 Ω cm for [Au(hfdt)2]. | ||
Magnetic susceptibilities
The temperature-dependence of magnetic susceptibility of [Ni(hfdt)2] corrected for the Pascal diamagnetic term (−3.3 × 10−4 emu mol−1) is shown in Fig. 4. The room temperature susceptibility was 2.4 × 10−5 emu mol−1 and the temperature dependence was well explained by the Curie(–Weiss) behavior, which indicated that the paramagnetic impurities correspond to 1.9% of S = ½ spin and a very small Weiss temperature of −0.14 K. Thus, the susceptibility measurements suggest that the system is essentially non-magnetic, which is consistent with the ab initio band calculation indicating a small gap between the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) bands, as mentioned below.![Temperature dependence of magnetic susceptibility of [Ni(hfdt)2].](/image/article/2005/JM/b413597k/b413597k-f4.gif) | ||
| Fig. 4 Temperature dependence of magnetic susceptibility of [Ni(hfdt)2]. | ||
The result of SQUID susceptibility measurements on [Au(hfdt)2] was very noisy, but indicated a very weak paramagnetic susceptibility of [Au(hfdt)2] (<0.8 × 10−4 emu mol−1), indicating the essentially non-magnetic property of the system, which is consistent with the result of ESR experiments exhibiting no significant signal.
Visible and IR spectra measurement
The single-component molecular metals such as [Ni(tmdt)2] and [Ni(dmdt)2]2a,2c have an electronic absorption in the infrared region which indicates a very small HOMO–LUMO gap in these complexes.20 As reported before, the absorption maxima of the metallic crystals such as [Ni(tmdt)2] and [Ni(dmdt)2] appears around 2200 cm−1 and the absorption maxima of the crystalline powder samples of analogous single-component molecular conductors shows a blue shift with decreasing the room temperature conductivity. The absorption maximum of semiconducting [Ni(ptdt)2],16 with a room temperature conductivity of 7 S cm−1, appears around 4700 cm−1. The electronic absorption spectra of the crystals of [Ni(hfdt)2] measured on crystalline powder samples showed broad electronic absorption peaks around 6090 cm−1 (Fig. 5), which is consistent with the semiconducting properties of this system.![The visible and IR spectra of (A)
[Ni(tmdt)2] and (C)
[Ni(hfdt)2], and the IR spectrum of (B)
[Ni(ptdt)2], measured on powder samples using a JASCO-MSV-370 spectrometer (40000–5000 cm−1) and a JASCO-FT/IR-420 + IRT30 spectrometer (7800–1000 cm−1).](/image/article/2005/JM/b413597k/b413597k-f5.gif) | ||
| Fig. 5 The visible and IR spectra of (A) [Ni(tmdt)2] and (C) [Ni(hfdt)2], and the IR spectrum of (B) [Ni(ptdt)2], measured on powder samples using a JASCO-MSV-370 spectrometer (40000–5000 cm−1) and a JASCO-FT/IR-420 + IRT30 spectrometer (7800–1000 cm−1). | ||
Band structure calculation
The extended-Hückel molecular orbital and tight-binding band structure calculations of [Ni(hfdt)2] were performed by using the following parameters sets of atomic orbitals:21 the valence shell ionization potential Hii (eV) and the exponent ζi of the Slater-type atomic orbital χi are −20.0 and 1.817 for S 3s, −13.3 and 1.817 for S 3p, −21.4 and 1.625 for C 2s, −11.4 and 1.625 for C 2p, −13.6 and 1.0 for H 1s, −10.95 and 2.1 for Ni 4s, −3.74 and 2.1 for Ni 4p, respectively. The d orbitals of Ni are represented by a linear combination of two Slater-type orbitals. Hii, ζi, χi, ζii and χii values are −10.58, 5.75, 0.5681, 2.00 and 0.6294 for Ni 3d. The HOMO and LUMO are shown in Fig. 6. As reported before,2 these frontier orbitals are expressed as: ϕHOMO = ϕ1 + ϕ2 and ϕLUMO = ϕ1 − ϕ2 + cMϕd, where ϕ1 and ϕ2 are right and left ligand orbitals and cMϕd, is a small contribution from the 3d orbital of the central Ni atom. ϕ1 and ϕ2 have symmetry similar to that of the HOMO of the TTF-like π donor molecule. The intermolecular overlap integrals are given in Table 2. Because the HOMO–LUMO interactions are comparable to the HOMO–HOMO and the LUMO–LUMO interactions, the system tends to have an energy gap between the HOMO and LUMO bands. Since it is very difficult to estimate a reliable value for the HOMO–LUMO gap (ΔEHL), the tight-binding band structure was calculated by changing ΔEHL. The semiconducting band structure of [Ni(hfdt)2] was calculated for ΔEHL < 0.05 eV and ΔEHL > 0.1 eV. It is interesting that, due to the HOMO–LUMO interaction, very small band gap was obtained even when ΔEHL ≈ 0.0 eV. The local density approximation (LDA) band calculation of [Ni(hfdt)2] was also performed on the basis of the ab initio plane-wave norm-conserved pseudopotential method with Troullier–Martins potentials.22 Needless to say, the band of [Ni(hfdt)2] has almost no energy dispersion along the b* direction but exhibits a considerable dispersion along the a* and c* directions, indicating the two-dimensional nature of the system (Fig. 7(a)). The band widths of the HOMO bands and LUMO bands are 0.45 eV and 0.20 eV, respectively. There is no overlap between the HOMO and LUMO bands, which is consistent with the observed semiconducting property of [Ni(hfdt)2], but the calculated band gap of 0.12 eV is a little less than half of the observed band gap (0.28 eV (= 2Ea)), as is usual with LDA. The density of state of [Ni(hfdt)2] obtained by LDA band structure calculation is shown in Fig. 7(b). The extended-Hückel tight-binding band structure calculation gave an almost similar density of states, but the bandwidth obtained by extended-Hückel tight-binding band structure calculation was less than half of the band width of the LDA calculation. In order to reproduce the band gap of the LDA band structure by the extended-Hückel calculation, ΔEHL must be assumed to be 0.14–0.22 eV, which is consistent with our previous observation of electronic absorption spectra of single-component molecular conducting systems indicating ΔEHL to be less than about 0.2 eV.20![Schematic drawings of the HOMO and LUMO of neutral molecule [Ni(hfdt)2].](/image/article/2005/JM/b413597k/b413597k-f6.gif) | ||
| Fig. 6 Schematic drawings of the HOMO and LUMO of neutral molecule [Ni(hfdt)2]. | ||
![(a) Band energy dispersion curve of [Ni(hfdt)2] obtained by ab initio LDA band calculations. The symbols Γ, X, Y, Z, S, T and U represent the following positions in the reciprocal space: Γ
(0,0,0), X (½,0,0), Y (0,½,0), Z (0,0,½), S (½,−½,0), T (0,½,−½) and U (−½,0,½). (b) The density of states of [Ni(hfdt)2] obtained by ab initio LDA band calculation. The gray line indicates the vacant LUMO band and the black lines indicate occupied HOMO and next-HOMO bands.](/image/article/2005/JM/b413597k/b413597k-f7.gif) | ||
| Fig. 7 (a) Band energy dispersion curve of [Ni(hfdt)2] obtained by ab initio LDA band calculations. The symbols Γ, X, Y, Z, S, T and U represent the following positions in the reciprocal space: Γ (0,0,0), X (½,0,0), Y (0,½,0), Z (0,0,½), S (½,−½,0), T (0,½,−½) and U (−½,0,½). (b) The density of states of [Ni(hfdt)2] obtained by ab initio LDA band calculation. The gray line indicates the vacant LUMO band and the black lines indicate occupied HOMO and next-HOMO bands. | ||
Fig. 8 shows the electron distribution of the HOMO band based on the LDA band calculation. The conduction electrons are not distributed around the region of the CF3 group. A similar depression of the electron density around the terminal CF3 region was also observed for LUMO band electrons. These results are consistent with the fact that the band of [Ni(hfdt)2] has almost no energy dispersion along the b* direction. Thus the crystal and electronic structures of [Ni(hfdt)2] strongly suggest the idea of the F⋯F segregation effect in the molecular aggregation to be valid also in the single-component molecular systems.
![The distribution of electrons in HOMO and LUMO bands projected to the bc plane. The electron density was calculated on the basis of the wavefunction of ab initio LDA calculations. The electron density is scaled by electrons/a02
(a0
= Bohr radius (0.529 Å)). Red plus symbols (+) indicate the position of S atoms. The black region indicates the very low density of frontier electrons in the CF3-bilayer region due to the strong F⋯F segregation effect in this system. Such a low-density region of frontier electrons could not be found in the similar calculation for [Ni(tmdt)2] without a terminal CF3
(or CF2) group.](/image/article/2005/JM/b413597k/b413597k-f8.gif) | ||
| Fig. 8 The distribution of electrons in HOMO and LUMO bands projected to the bc plane. The electron density was calculated on the basis of the wavefunction of ab initio LDA calculations. The electron density is scaled by electrons/a02 (a0 = Bohr radius (0.529 Å)). Red plus symbols (+) indicate the position of S atoms. The black region indicates the very low density of frontier electrons in the CF3-bilayer region due to the strong F⋯F segregation effect in this system. Such a low-density region of frontier electrons could not be found in the similar calculation for [Ni(tmdt)2] without a terminal CF3 (or CF2) group. | ||
The extended-Hückel tight-binding band structure calculation was made also for [Au(hfdt)2]. The intermolecular overlap integrals are given in Table 3. The valence shell ionization potential Hii (eV) and the exponent ζi of the Slater-type atomic orbital χi of the Au atom are −10.92 and 2.602 for 6s and −5.55 and 2.584 for 6p, respectively.21 The d orbitals are represented by a linear combination of two Slater-type orbitals. Hii, ζi, χi, ζii and χii values are −15.07, 6.163, 0.6851, 2.794 and 0.5696 for 5d.21 The energy dispersion curves and the density of states are shown in Fig. 9(a) and (b), respectively. Due to the dimeric structure, both HOMO and LUMO bands are split into two bands, similar to the band structure of [Pd(dmit)2] conductors with strongly dimeric columns. The highest vacant band mainly originates from the antibonding combination of LUMOs of the [Au(hfdt)2] dimer, and the highest occupied band mainly comes from the antibonding combination of HOMOs of the [Au(hfdt)2] dimer. The resultant band gap was very small (≈0.02 eV), which is consistent with the small activation energy of the resistivity of the compressed pellet sample (Fig. 3 and Fig. 9).
![(a) Band energy dispersion curve of [Au(hfdt)2] calculated by extended-Hückel tight-binding band calculation. (b) The density of states calculated by extended-Hückel tight-binding band calculation. The black and gray lines indicate the density of states of occupied and unoccupied bands, respectively.](/image/article/2005/JM/b413597k/b413597k-f9.gif) | ||
| Fig. 9 (a) Band energy dispersion curve of [Au(hfdt)2] calculated by extended-Hückel tight-binding band calculation. (b) The density of states calculated by extended-Hückel tight-binding band calculation. The black and gray lines indicate the density of states of occupied and unoccupied bands, respectively. | ||
|
|
||||
|---|---|---|---|---|
| H–H | H–L | L–L | L–H | |
| a The short S⋯S distances are found in the intermolecular contacts: A1, P2 and P4: 3.614 Å (A), 3.502 Å (P2), 3.530 Å (P2), 3.675 Å (P2), 3.615 Å (P4), 3.633 Å (P4) and 3.400 Å (P4). | ||||
| A | 0.2 | 0.2 | 0.2 | 0.2 |
| B1 | 2.2 | 0 | −1.1 | 0 |
| B2 | 5.0 | 0 | −4.1 | 0 |
| P1 | 0 | 0.1 | 0 | 0 |
| P2 | 3.9 | 0 | −0.8 | 0 |
| P3 | 0 | 0 | 0 | 0 |
| P4 | 0.9 | 0 | 7.9 | 0.1 |
Conclusion
If molecules consisting of single-component molecular conductors can be sublimed or dissolved, then conductng films of single-component molecular conductors will be obtained. However, due to the strong three-dimensional intermolecular interactions, this seems not to be so easy. On the other hand, the F⋯F segragation effect in the molecular aggregation might provide a way to control the molecular assembly of single-component molecular conducting systems. As expected, the crystal structures of [M(hfdt)2] (M = Ni, Au) with extended-TTF dithiolate ligands having CF3 groups demonstrate the F⋯F segragation effect. Unlike typical single-component molecular metals such as the first single-component molecular metal [Ni(tmdt)2] and analogous [Au(tmdt)2], with three-dimensional tight intermolecular interactions, the crystals of [M(hfdt)2] (M = Ni, Au) are composed of two-dimensional [M(hfdt)2] (M = Ni, Au) layers separated by terminal CF3 groups. The intermolecular F⋯F distances are significantly longer than the sum of the van der Waals radii. Thermal studies will be made in the near future. The resistivity measurements on single crystals of [Ni(hfdt)2] and compressed pellet samples of [Au(hfdt)2] showed the semiconducting but fairly high-conducting properties, which is consistent with the extended-Hückel tight-binding and also the LDA band structure calculations giving semiconducting bands with a small band gap. The observed non-magnetic susceptibility behavior of [M(hfdt)2] (M = Ni, Au) is consistent with the band structure calculations.Acknowledgements
This work is financially supported by a Grant-in-Aid for Scientific Research (S) (No. 14103005) and by the 21st Century COE Program for Frontiers in Fundamental Chemistry from the Ministry of Education, Culture, Sports, Science and Technology. This work is also supported by CREST [Core Research for Evolutional Science and Technology of JST (Japan Science and Technology Agency)].References
- H. Tanaka, Y. Okano, H. Kobayashi, W. Suzuki and A. Kobayashi, Science, 2001, 291, 285 CrossRef CAS.
- (a) A. Kobayashi, H. Tanaka and H. Kobayashi, J. Mater. Chem., 2001, 11, 2078 RSC; (b) A. Kobayashi, W. Suzuki, E. Fujiwara, T. Otsuka, H. Tanaka, Y. Okano and H. Kobayashi, Mol. Cryst. Liq. Cryst., 2002, 379, 19 CAS; (c) A. Kobayashi, E. Fujiwara and H. Kobayashi, Chem. Rev., 2004, 104, 5243 CrossRef CAS.
- H. Tanaka, M. Tokumoto, S. Ishibashi, D. Graf, E. S. Choi, J. S. Brooks, S. Yasuzuka, Y. Okano, H. Kobayashi and A. Kobayashi, J. Am. Chem. Soc., 2004, 126, 10518 CrossRef CAS.
- H. Tanaka, H. Kobayashi and A. Kobayashi, J. Am. Chem. Soc., 2002, 124, 10002 CrossRef CAS.
- A. Kobayashi, W. Suzuki, M. Sasa, E. Fujiwara, Y. Fujishiro, E. Nishibori, M. Takata, M. Sakata, Y. Okano and H. Kobayashi, J. Phys. IV, 2004, 114, 419 Search PubMed.
- For example: (a) M. Tamura, Y. Nakazawa, D. Shiomi, K. Nozawa, Y. Hosokoshi, M. Ishikawa, M. Takahashi and M. Kinoshita, Chem. Phys. Lett., 1991, 186, 401 CrossRef CAS; (b) R. Chiarell, M. A. Novak, A. Rassat and J. L. Tholence, Nature, 1993, 363, 147 CrossRef CAS.
- N. M. Rivera and E. M. Engler, J. Chem. Soc., Chem. Commun., 1979, 184 RSC.
- (a) O. J. Dautel and M. Fourmigue, Inorg. Chem., 2001, 40, 2083 CrossRef CAS; (b) O. J. Dautel, M. Fourmigue, E. Canadell and P. Auban-Senzier, Adv. Funct. Mater., 2002, 12, 693 CrossRef CAS.
- W. Suzuki, E. Fujiwara, A. Kobayashi, Y. Fujishiro, E. Nishibori, M. Takata, M. Sakata, H. Fujiwara and H. Kobayashi, J. Am. Chem. Soc., 2003, 125, 1486 CrossRef CAS.
- (a) D. Belo, H. Alves, E. B. Lopes, M. T. Duarte, V. Gama, R. T. Henriques, M. Almeida, A. Pérez-Benítez, C. Rovira and J. Veciana, Chem. Eur. J., 2001, 7, 511 CrossRef CAS; (b) N. C. Schiødt, T. Bjørnholm, K. Bechgaard, J. J. Neumeier, C. Allgeier, C. S. Jacobsen and N. Thorup, Phys. Rev. B: Condens. Matter, 1996, 53, 1773 CrossRef; (c) A. J. Schultz, H. H. Wang, L. C. Soderholm, T. L. Sifter, J. M. Williams, K. Bechgaard and M.-H. Whangbo, Inorg. Chem., 1986, 26, 3757.
- (a) L. Binet, J. M. Fabre, C. Montginoul, K. B. Simonsen and J. Becher, J. Chem. Soc., Perkin Trans. 1, 1996, 783 RSC; (b) L. Binet, C. Montginoul, J. M. Fabre, L. Ouahab, S. Golhen and J. Becher, Synth. Met., 1997, 86, 1825 CrossRef CAS.
- M. Kumasaki, H. Tanaka and A. Kobayashi, J. Mater. Chem., 1998, 8, 301 RSC.
- SIR97: A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Cryst., 1999, 32, 115 Search PubMed.
- D. T. Cromer and J. T. Waber, in International Tables for X-ray Crystallography, vol. IV, The Kynoch Press, Birmingham, England, 1974 Search PubMed, Table 2.2 A, .
- teXsan: Crystal Structure Analysis Package, Molecular Structure Corporation, The Woodlands, TX, USA, 1985 and 1999.
- SIR92: A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi and G. Polidori, J. Appl. Cryst., 1994, 27, 435 Search PubMed.
- CrystalStructure 3.0: Crystal Structure Analysis Package, Rigaku/MSC and Rigaku Corporation, The Woodlands, TX, USA, 2000–2002.
- A. Kobayashi, H. Tanaka, M. Kumasaki, H. Torii, B. Narymbetov and T. Adachi, J. Am. Chem. Soc., 1999, 121, 10763 CrossRef CAS.
- (a) A. Kobayashi, A. Miyamoto, H. Kobayashi, A. Clark and A. E. Underhill, J. Mater. Chem., 1991, 1, 827 RSC; (b) P. Cassoux, L. Valade, H. Kobayashi, A. Kobayashi, R. A. Clark and A. E. Underhill, Coord. Chem. Rev., 1191, 110, 115 Search PubMed; (c) A. Kobayashi, H. Kim, Y. Sasaki, K. Murata, R. Kato and H. Kobayashi, J. Chem. Soc., Faraday Trans., 1990, 86, 361 RSC.
- A. Kobayashi, M. Sasa, W. Suzuki, E. Fujiwara, H. Tanaka, M. Tokumoto, Y. Okano, H. Fujiwara and H. Kobayashi, J. Am. Chem. Soc., 2004, 126, 426 CrossRef CAS.
- S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1976, 198, 7255 CrossRef.
- (a) D. R. Hamann, M. Schlüter and C. Chiang, Phys. Rev. Lett., 1979, 43, 1494 CrossRef CAS; (b) D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS; (c) J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter, 1981, 23, 5048 CrossRef CAS; (d) N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter, 1991, 43, 1993 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |