In situ Sr-isotope analysis of carbonates by LA-MC-ICP-MS: interference corrections, high spatial resolution and an example from otolith studies
Jon
Woodhead
*a,
Stephen
Swearer
b,
Janet
Hergt
a and
Roland
Maas
a
aSchool of Earth Sciences, The University of Melbourne, Melbourne, VIC 3010, Australia. E-mail: jdwood@unimelb.edu.au
bDepartment of Zoology, The University of Melbourne, Melbourne, VIC 3010, Australia
First published on 30th November 2004
Abstract
In situ Sr-isotope analysis by laser ablation multi-collector ICP-MS is a potentially powerful tracer technique with widespread application to many fields of study. The usefulness of the method, however, depends very strongly upon the quality of data that can be obtained (compared with conventional ‘solution-based’ analyses), and the spatial resolution, particularly in samples with strong compositional zonation or fine-scale growth banding. In this contribution we show that highly accurate (∼50 ppm) and precise (external precision ∼125 ppm) analyses of carbonate materials can be obtained in situ and further demonstrate that, by utilising the aperture-imaging optics of an excimer laser system with appropriate time-resolved software, isotopic variations on the scale of tens of micrometres can be resolved. An example is shown using relatively small (∼500 μm diameter) otoliths from a diadromous fish species, Galaxias maculatus.
Introduction
Although the field of laser ablation MC-ICP-MS, which allows in situ isotopic analysis, is a relatively youthful one, a number of research groups have been quick to realise the potential of the technique for both geochronological and tracer applications. As an isotopic tracer, the 87Sr/86Sr ratio is particularly interesting due to the strong contrast between different reservoirs. For example, Christensen et al.1 first demonstrated the feasibility of in situ Sr-isotope analysis using a Nd-YAG laser (Fisons Laser Probe) coupled to a P54 (Fisons VG) MC-ICP-MS, obtaining results comparable to TIMS data (albeit with lesser precision) on both carbonate and feldspar samples. In the latter they obtained a variety of isotope ratios spanning a range beyond analytical error and providing the first hint of the potential power of the technique. Subsequently, Davidson et al.2 undertook a far more detailed study of Sr-isotope variations in a single feldspar, proving conclusively that abrupt changes in isotopic composition can be correlated with distinct petrographic features. Most recently, Bizzarro et al.3 used LA-MC-ICP-MS to demonstrate significant Sr-isotopic heterogeneity between co-existing magmatic minerals.Although all of the aforementioned studies have stressed the importance of interference corrections in producing high-quality data, they concentrate largely on the influence of Kr and Rb isobaric interferences, with no detailed investigation of the role played by other potential interferences derived from the carrier gas. In this study we demonstrate the necessity to correct for Ca argide/dimer species, in addition to Kr and Rb, when analysing carbonate materials. In this way extremely accurate and precise data can be obtained using relatively short analysis times, and importantly data quality can be continuously monitored using the 84Sr/86Sr ratio, which is invariant in nature, as is the norm in TIMS analysis. Without this facility there is no easy way to determine whether 87Sr/86Sr ratios, measured on unknowns, have been compromised by matrix and/or interference effects.
In addition, all previous studies have employed simple spot analyses with ablation pits generally larger than 100 μm in diameter and often up to 300 μm. The size and/or geometry of many analytical materials, however, precludes the use of such conditions. Herein we explore a potential alternative analytical approach, namely ablation employing a slit which can be traversed across the sample at an appropriate rate to produce a time-resolved signal with ∼10 μm resolution. The power of this technique is demonstrated by analysis of a relatively small otolith from the early juvenile-stage of a diadromous fish species, Galaxias maculatus (Galaxiidae) at very high resolution.
Experimental
Instrumentation
Our analytical system consists of a “Nu Plasma” MC-ICP-MS (Nu Instruments, Wrexham, UK), coupled to a HelEx (Laurin Technic, Canberra, Australia, and the Australian National University) laser ablation system constructed around a Compex 110 (Lambda Physik, Gottingen, Germany) excimer laser operating at 193 nm (see Table 1 for summary of key operational parameters).| Nu Plasma MC-ICP-MS | |
| Forward power | 1350 W |
| Reflected power | <2 W |
| Accelerating voltage | 4000 V |
| Analyser pressure | 4 × 10−9 mbar |
| Cones | Ni |
| Plasma gas | 13 l min−1 |
| Auxiliary gas | 0.90 l min−1 |
| Nebuliser gas | 0.85 l min−1 |
| Helex laser ablation system | |
| Lambda Physik Compex 110 ArF excimer | 193 nm |
| Laser fluence | ∼10 J cm−2 |
| Spot size | 105 μm |
| Repetition rate | 5 Hz |
| He gas to cell | 200 cm3 min−1 |
Eight of the twelve available Faraday cups of the Nu Plasma are employed to collect ion currents at masses 82, 83, 84, 85, 86, 87, 88 and 89 (see Table 2). Mass bias is corrected by reference to an 86Sr/88Sr ratio of 0.1194, after appropriate interference corrections (see later section), and using an exponential mass bias law. Note that although the Nu Plasma detector array also incorporates three ion counters, analyses of this type are best performed on Faraday cups due to the large number of simultaneous detectors required (jumping routines are difficult to implement in time-resolved analyses) and the relative ease of accurately cross-calibrating amplifier responses for these detectors. Fortunately, Sr contents are generally very high (thousands of ppm) in most carbonates and thus analysis using Faraday cups is straightforward for reasonable spot sizes/ablation rates.
| Collectora | H6 | H5 | H4 | H3 | H2 | H1 | Ax | L1 | L2 | IC0 | L3 | IC1 | L4 | IC2 | L5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a H1–H6, Ax, L1 to L5 are Faraday cups, IC0 to IC2 are ion counters. Note gaps in the array between H4, H5 and H6. | |||||||||||||||
| Analyte isotopes | 89Y | 88Sr | 87Sr | — | 86Sr | — | — | — | 84Sr | — | — | — | — | — | |
| Gas-related interferences | — | 86Kr | — | — | — | 84Kr | — | 83Kr | — | 82Kr | — | — | |||
| Sample-related interferences | 48Ca40Ar 48Ca40Ca | 87Rb | — | 46Ca40Ar 46Ca40Ca | — | 85Rb | — | 44Ca40Ar 44Ca40Ca | — | 43Ca40Ar 43Ca40Ca | — | 42Ca40Ar 42Ca40Ca | — | — | |
Design characteristics and applications of the HelEx system have been discussed previously,4,5 and a detailed description of the system will be the subject of a separate publication by the ANU group. For the purposes of the present study, however, it is sufficient to note that ablation is performed under pure He to minimise the re-deposition of ablated material4,6 and the sample is then rapidly entrained into the Ar carrier gas flow. Although the cell has the capacity to take samples up to 10 cm × 10 cm in size, the effective volume ‘seen’ by the ablated sample is very small (∼2 cm3), providing rapid signal response. In addition, for high resolution profiling, the signal-smoothing device, which is used for routine analysis, is removed, providing an almost instantaneous response time. A detailed discussion of the advantages of the HelEx system in terms of signal response and washout can be found in Eggins and Shelley,7 while a thorough investigation of the more general consequences of cell design and gas flows on aerosol transport efficiency can be found in Bleiner and Gunther.8 The laser system itself can be operated in two modes. Routine ‘spot’ analyses are conducted using a variety of circular apertures providing a range of ablation spot sizes from 10 to 350 μm in diameter. Alternatively, for high-resolution line scans, the system utilises a variable aperture formed from two micrometer-driven knife-edges combined with an iris. In this way it is possible to ablate a variety of rectangular shapes of variable aspect ratio as an alternative to conventional circular spots. This feature will be exploited later in this study.
For static spot analyses the laser is operated in constant energy mode with fluence at the target of ∼10 J cm−2, and a repetition rate of ∼5 Hz, depending upon the Sr content of the materials being ablated (we aim for Sr signals between 5 and 10 V). For slit line scan analyses the repetition rate is increased to 15–20 Hz to provide similar signal intensity.
Sample preparation
Fragments of modern sea-water carbonates were embedded in epoxy resin blocks and the surface polished to enhance visual observation of any zonation. Prior to analysis, the surface of the sample blocks was cleaned by wiping with a methanol-soaked lens tissue. We collected juvenile common galaxiid (Galaxias maculatus) using seine nets in the Otway region of Victoria, Australia. G. maculatus is a diadromous fish species, spending the first 3–6 months of life developing out at sea and then migrating into freshwater as young juveniles. Collected juveniles were euthanized using clove oil mixed in freshwater and preserved by freezing at −4 °C. Otoliths (both sagittae) were removed from thawed fish and cleaned of adhering tissue. Once dry, we embedded the otoliths in thermoplastic cement (Crystalbond) on glass slides and ground them in the sagittal plane using diamond lapping films to expose all daily growth rings from the time of hatching to capture. Polished otoliths were sonicated in 18 MΩ water for 5 min, dried in a laminar flow bench, and stored in plastic bags until analysis.Results and discussion
The importance of interference corrections
In carbonate materials, the analysis of Sr isotope variations in situ is potentially compromised by a variety of isobaric interferences (see Table 2). Of these, perhaps the most important is that from Kr, which is usually present as a trace impurity in the Ar and He carrier gases used in ICP-MS. On our instrument we typically observe total Kr signals of the order of 20 mV. Although relatively small, such signals are sufficient to produce a ca. 0.6% shift in a 87Sr/86Sr ratio measured at 5 V total Sr beam, and thus represent a significant impediment to accurate isotope ratio determination (such a shift is ca. 600 × within-run precision on a typical 87Sr/86Sr ratio determination). Although some authors have advocated peak-stripping Kr interferences by reference to a 83Kr monitor,1,9 our own experience with solution-based MC-ICPMS Sr-isotope analysis has demonstrated that the most effective method of dealing with Kr interference for high-accuracy Sr-isotope analysis is the measurement of zeros ‘on peak’ (essentially a gas blank) prior to each analysis. This technique avoids the difficulties associated with performing a mass bias correction on Kr signals which may only represent a few millivolts measured on Faraday cups, a strategy which can result in considerable loss of accuracy since any errors propagate into the bias correction for Sr itself. We have experimented with a variety of Ar gas sources and delivery options (liquid Ar boil-off and cylinder gas), and find that levels of Kr contamination can vary largely both between batches and also within vessels as the Ar levels become depleted. However, we consistently find that the Kr signals are more stable on a short timescale (tens of minutes) using compressed Ar cylinder-gas, rather than as boil-off from liquid Ar dewars, and we now use the former source exclusively with our instruments. As long as Kr levels remain relatively constant on the scale of a given analysis, the ‘on peak zero’ methodology is highly effective at removing gas-related interferences, independent of any bias estimates, and is used in all the analyses reported here. In practice the ‘on peak zero’ is implemented using the Nu Plasma analysis software, with all gas flows and instrument parameters remaining constant and the laser firing, but with the laser shutter closed, thus preventing ablation of the sample. After measurement of this baseline, the laser shutter is opened and, once the signal has stablised, integration is initiated from within the Nu Plasma time-resolved software suite.Two other interferences are also relevant to the measurement of Sr-isotope ratios in modern carbonates. The first is that of 87Rb on 87Sr; fortunately, however, in most carbonates the Rb/Sr ratio is extremely low and so correction for this interference is minor. In most of the samples considered in this study, 87Rb constitutes less than 100 ppm of the signal on mass 87 and an effective correction can be made by monitoring 85Rb and peak-stripping the appropriate ion current from the signal on mass 87, assuming a natural 87Rb/85Rb ratio of 0.3856. At this level the effect of mass bias on the Rb correction is minimal and thus we assume Rb mass bias is identical to that of Sr. When analysing materials with considerably higher Rb contents (producing thousands of ppm interference on mass 87), however, slight differences in mass bias behaviour between Sr and Rb could become significant. In this case we would advocate experimentation with materials of known 87Sr/86Sr but variable Rb/Sr ratio to help define the relationship between Sr and Rb mass bias in detail.
We also observe significant interferences from Ca argide or dimer species in the laser ablation of carbonate materials. Although some previous authors have noted the potential for such interferences,3,10 we do not believe that anyone has previously attempted to apply an on-line correction for these effects. As in the case of Kr, although the interferences are small, the effects can be significant. In particular, although such interferences are unlikely to be sufficient in themselves to influence the accuracy of 87Sr/86Sr ratios beyond the levels of within-run uncertainties, they can easily produce moderate shifts in the 84Sr/86Sr ratio. This is important inasmuch as this ratio, considered invariant in nature, is often used in both solution-mode MC-ICP-MS and TIMS analysis as an internal monitor of data quality, in particular of the efficacy of the mass bias correction. The ability to monitor this ratio in laser ablation studies is equally important, providing the analyst with the only assessment of confidence in the bias corrections applied to raw 87Sr/86Sr ratios during ablation of real samples. Typically, during laser ablation of carbonates, we observe total Ca argide/dimer signals (all masses) of around 100 mV, resulting in a shift in 84Sr/86Sr from accepted values (ca. 0.056511) to nearer 0.0575 (see Fig. 1), although larger shifts may be observed with different laser/ICP-MS combinations. Fortunately, with the Nu Plasma we have sufficient collectors to enable monitoring of the 42Ca40Ar/42Ca40Ca peak at mass 82 and, from this, we can strip appropriate contributions from the Sr masses 84, 86 and 88. As shown in Fig. 1, this correction both reduces scatter and returns all 84Sr/86Sr ratios back to expected values, thus allowing the continued use of this ratio in assessment of data quality. We also note that, although it is not clear what proportion of these interferences can be attributed to argides as opposed to dimers, the relative isotopic abundances of these molecules are very similar for the masses of interest, and so the same correction will accommodate both, at the levels of interference likely to be encountered by the analyst.
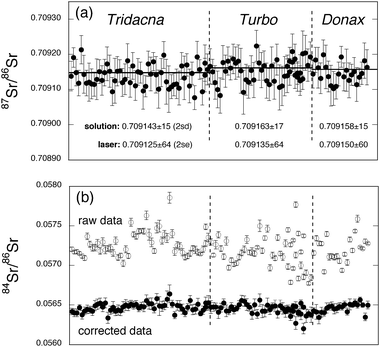 | ||
| Fig. 1 In situ Sr-isotope analyses for three different modern sea-water carbonates, recorded during five separate analytical sessions. Each datum represents a single 60 s acquisition and is shown with associated within-run errors (2 s.e.). The horizontal lines in panel (a) represent the corresponding solution-based analysis conducted on Sr separated from the sample by cation exchange chromatography. These 87Sr/86Sr values can be compared numerically with the mean of the laser ablation data in the lower part of the figure. Panel (b) demonstrates the effect of the interference corrections on the 84Sr/86Sr ratio, with corrected data showing less variability and clustering around the accepted value (∼0.0565). | ||
Finally, although rarely an issue for modern carbonates, which usually show very low rare earth element (REE) abundances, we note the potential for doubly charged REE interferences in the Sr mass region. It is possible that these may be encountered in the analysis of ancient carbonates, which often contain far higher REE contents owing to the incorporation of other mineral phases either upon burial, or during diagenesis.12 While the protocol for the correction for such interferences is beyond the scope of this paper we also note that it is a relatively simple matter to monitor 89Y simultaneously (see Table 2) as an REE proxy to alert the analyst to the potential presence of doubly charged REE.
Validation of the analytical protocol
Before embarking upon the possibility of high-resolution line scans, it is important to determine the effectiveness of our analytical protocol using simple spot analyses of materials of known Sr isotopic composition. Although we have in the past characterised the widely-used NIST 610-612-614 glass reference materials for Sr isotopic composition,13 these are inappropriate for this purpose since (a) the matrix is radically different from carbonates and (b) Rb/Sr ratios in the NIST glasses are unrealistically high (orders of magnitude higher than most carbonates). Unfortunately, at this time, no carbonate reference materials are available which are certified homogeneous at the scale of ∼100 μm, and of known Sr isotope composition. Given the general dearth of suitable reference materials, however, one solution is to measure a variety of modern marine carbonates that can be assumed (and tested) to be in isotopic equilibrium with modern sea-water (itself a well-mixed reservoir). While some previous authors have used corals for this purpose (e.g., ref. 14), we have found that the skeletal microstructure of most corals makes for less-than-ideal ablation and this, coupled with the fine-scale growth zonation in Sr content, render corals unsuitable. As an alternative we have utilised mollusc shells which are generally much denser and often display less fine-scale variation in Sr content. Fig. 1 shows the results of spot analyses conducted on 3 different modern marine carbonates (two bivalve shells from the genera Tridacna and Donax, and a gastropod operculum of the genus Turbo). In order to enable comparison with the laser analyses, solution-MC-ICP-MS analyses of Sr, separated from these samples by ion exchange chemistry, were conducted; these demonstrate that the samples are in approximate Sr-isotopic equilibrium with present day sea-water (∼0.709![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 170 on our instrument)—see the horizontal lines in Fig. 1. Each data point in Fig. 1 represents a single laser analysis undertaken with a 105 μm diameter spot and 5 Hz repetition rate. Laser output energy was adjusted to provide a total Sr signal of between 5 and 10 V, measured on Faraday cups, and each analysis represents a 60 s integration after a 60 s ‘on peak’ baseline measurement with the laser shutter closed. All of these analyses are static spot analyses, drilling at a rate of some 0.1–0.2 μm per pulse, resulting in ablation pits between 30–60 μm in depth. As a result pit aspect ratios are well below the level of 2 ∶ 1 (depth/width) suggested by Woodhead et al.5 as a practical limit for depth-profiling applications with this system. In addition, although we note the concerns of some authors regarding variations in elemental fractionation with depth, and its limits on particular applications (e.g., ref. 15), since no elemental ratios are determined in this analysis routine, and all isotope ratios are internally normalised, elemental fractionation with depth has no effect on the quality of our Sr-isotope ratio determinations. Data processing for static spot analyses is undertaken via the Nu Plasma on-line software. Fig. 1 is a compilation of data from five different analytical sessions, covering a period of several months, and demonstrates that corrections for Kr, Rb and Ca argide/dimer interferences result in 87Sr/86Sr ratios closely approaching solution-ICP-MS ratios determined on separated Sr, and modern sea-water values.
170 on our instrument)—see the horizontal lines in Fig. 1. Each data point in Fig. 1 represents a single laser analysis undertaken with a 105 μm diameter spot and 5 Hz repetition rate. Laser output energy was adjusted to provide a total Sr signal of between 5 and 10 V, measured on Faraday cups, and each analysis represents a 60 s integration after a 60 s ‘on peak’ baseline measurement with the laser shutter closed. All of these analyses are static spot analyses, drilling at a rate of some 0.1–0.2 μm per pulse, resulting in ablation pits between 30–60 μm in depth. As a result pit aspect ratios are well below the level of 2 ∶ 1 (depth/width) suggested by Woodhead et al.5 as a practical limit for depth-profiling applications with this system. In addition, although we note the concerns of some authors regarding variations in elemental fractionation with depth, and its limits on particular applications (e.g., ref. 15), since no elemental ratios are determined in this analysis routine, and all isotope ratios are internally normalised, elemental fractionation with depth has no effect on the quality of our Sr-isotope ratio determinations. Data processing for static spot analyses is undertaken via the Nu Plasma on-line software. Fig. 1 is a compilation of data from five different analytical sessions, covering a period of several months, and demonstrates that corrections for Kr, Rb and Ca argide/dimer interferences result in 87Sr/86Sr ratios closely approaching solution-ICP-MS ratios determined on separated Sr, and modern sea-water values.
Based upon a total of 123 analyses, accuracy† is estimated to be approximately 50 ppm, with the best results (46 ppm) obtained for the Donax and the worst (57 ppm) for the Turbo operculum. It is a useful exercise to compare these results with the quality of data obtained by routine thermal ionisation mass spectrometry (TIMS) procedures, the usual benchmark for Sr-isotope analyses. Taking two instruments with which we have considerable experience (Thermo Finnigan MAT 261 and 262), a total of 96 and 189 Sr-isotope analyses obtained over a several year period on the NIST SRM 987 standard Sr provided accuracy of ∼28 ppm and ∼53 ppm, respectively, the latter being somewhat compromised by regular shifts from positive to negative ion mode on this machine, which appeared to have a detrimental effect on stability. Although the most recent generation of TIMS instruments claim substantial improvement in accuracy, long-term data are not yet available with which to substantiate this claim. Clearly, however, our in situ determinations appear to have only slightly degraded accuracy compared with routine TIMS analysis despite requiring additional interference corrections for Kr, Rb and Ca argides/dimers, which are not present or are insignificant in the TIMS method.
External precision, expressed as 2 s.e. is approximately ± 0.000090, i.e., ca. 125 ppm, with typical internal precision (2 s.e.) around half this value. Since a laser spot analysis of this type consumes a few nanograms of Sr at most, this compares very favourably with a typical TIMS analysis consuming several tens to hundreds of nanograms Sr, and typically taking 20–30 min. We note, however, that TIMS is still the preferred technology for high accuracy Sr-isotope analysis where spatial resolution is not an issue.
The issue of resolution: depth profiling versus line scans
Two possibilities exist for extracting spatially-resolved information by laser ablation analysis; gradually drilling into a sample (depth profiling) or line scans (raster analysis).16 Since ablation pits are generally many tens of micrometres in diameter and yet individual laser pulses may only ablate a few tenths of a micrometre at a time, it is clear that, theoretically, depth profiling is capable of producing far better spatial resolution. Woodhead et al.5 have provided an example of this methodology in resolving complex isotopic stratigraphies in the mineral zircon. In some cases, however, sample morphology is not conducive to depth profiling (e.g., relatively large, thin samples) and, in this case, line scans must be considered as an alternative analytical strategy.In such cases excimer lasers offer considerable advantages when compared, for example, with Nd-YAGs. Rather than focussing the laser beam, in excimer-based systems the laser ‘spot’ is usually an image of an aperture, illuminated by a rather broad laser beam (24 × 8 mm in our case). It is then a relatively simple matter to substitute apertures of different size and shape and thus produce ablation pits of variable geometry. Using the ‘knife edge’ aperture noted above we can ablate narrow ‘slits’ down to a few micrometres wide, and up to several hundred micrometres long. If the long axis of the slit is aligned parallel to the zonation/perpendicular to the growth axis of a sample, very high resolution ablation profiles can be produced.
When attempting analyses of this kind, raw data are exported and processed off-line using a program developed specifically for this purpose (Kemp et al., in preparation), which provides features unavailable in the Nu Plasma time-resolved software suite (in particular the ability to view corrected isotope ratios versus time). Individual 0.2 s data slices are corrected for interferences and mass bias exactly as documented above for static analyses, and then displayed in a time-resolved output. The user then has complete control over where to perform larger integrations for statistical purposes.
One current limitation of this methodology using our system is that the orientation of the ablated slit relative to the sample is fixed. As a result, growth structures in samples of interest have to be specifically aligned within the sample holder. It is possible that future refinements of the system will allow rotation of the laser aperture relative to the sample but such movement is likely to be limited by the dimensions of the laser beam itself. Alternatively a rotating sample stage could be constructed.
An example application of the methodology
Otoliths (earstones) are aragonitic, auditory structures located in the semi-circular canals of all bony fishes.17 New material is added daily and thus the concentric structure of otoliths provides a valuable record of the environment experienced over the lifetime of the fish.18 As a result, otolith microchemistry has become a powerful tool for the investigation of diadromy,19 larval dispersal,20 natal homing,21 juvenile nursery habitats22,23 and stock discrimination.24,25One of the most widely utilized tracers in otolith research has been the ratio Sr ∶ Ca, which has been extensively applied for investigating migration between marine and freshwater environments. However, the uptake of these and other elements by fish can be influenced not only by environmental factors such as temperature, salinity and water chemistry but also by biotic factors such as genetics, growth rate and the condition of the fish.26–29 Consequently, a number of authors have recently investigated the use of Sr isotope analysis as a means of tracking the chemistry of waters in which fish grow, as isotope ratios for most high atomic mass elements are not subject to significant biological fractionation.
Ingram and Weber30 analysed Sr-isotope ratios in whole otoliths from chinook salmon in an attempt to distinguish between wild and hatchery fish. Although bulk sampling was a success for their application, they note in conclusion that “subsampling could also supply information on migration if otolith chemistry is related to the otolith banding chronology”. Waight et al.10 made a laser ablation MC-ICP-MS profile across an otolith from an Arctic char but encountered analytical problems manifest as correlated changes in 87Sr/86Sr and the invariant ratio 84Sr/86Sr. This they attributed to uncorrected interferences, most probably of the type documented above. Most recently Milton and Chenery31 obtained Sr-isotope transects across otoliths from the tropical shad, which demonstrated the viability of the technique. Their analyses, however, used a 250 μm wide ablation path and 10 μm s−1 stage translation, requiring relatively large otoliths and limiting spatial resolution.
Although these studies provide encouraging evidence for the utility of Sr isotope analysis in otolith research, there are clearly unresolved methodological issues that can be addressed using a high-resolution time-resolved analysis routine. Because growth bands are deposited concentrically, recording important environmental information, otoliths tend to be ‘pancake shaped’ inasmuch as the growth layers are highly compressed in vertical section. As a result depth profiling is unsuitable (the laser would quickly drill through the whole structure) and slow line scans are the only viable alternative. In addition, in most fish species the otoliths of the larval and juvenile stages are extremely small (tens to a few hundreds of micrometres diameter), requiring very high resolution.
In Fig. 2 we show a typical analysis using the protocol described above applied to an otolith. We ablate a slit of length 147 μm, but only ca. 3 μm wide, using a repetition rate of 20 Hz. This results in Sr beams of approximately 2 and 6 V in freshwater and marine carbonate segments, respectively. The sample was translated beneath the laser beam at a rate of approximately 2 μm s−1, providing around 2000 individual data points, each representing a single 0.2 s integration. These data demonstrate a clear shift in 87Sr/86Sr ratio between core and edge of the otolith, which appears to mirror variation in Sr content (as indicated by signal size). Integration of the data for the core (larval) region of the otolith provides an 87Sr/86Sr ratio of 0.709161 ± 50 (2 s.e.), identical within error to the modern sea-water value (actual sea-water samples measured by solution ICP-MS on our instrument gave a mean of 0.709168 during the period of this work). Sr-isotope signatures near the edge of the otolith appear to be better resolved on the left hand side (Fig. 2), presumably because here the laser slit is oriented almost exactly parallel to the micrometre-scale growth banding. Integration of these data provides an 87Sr/86Sr ratio of 0.706534 ± 121 (2 s.e.), clearly distinct from the sea-water signal, and representing a period of otolith growth in a freshwater environment. A well-defined plateau is not achieved on the right hand edge, which we attribute to the fact that growth banding crosses the laser slit obliquely on this side. Nevertheless, the deviation from the sea-water values in the core is immediately apparent, and a similar baseline level is ultimately achieved at the extreme edge of the otolith. Note that 84Sr/86Sr ratios remain constant throughout the transect, and show no relation to either 87Sr/86Sr or Sr signal.
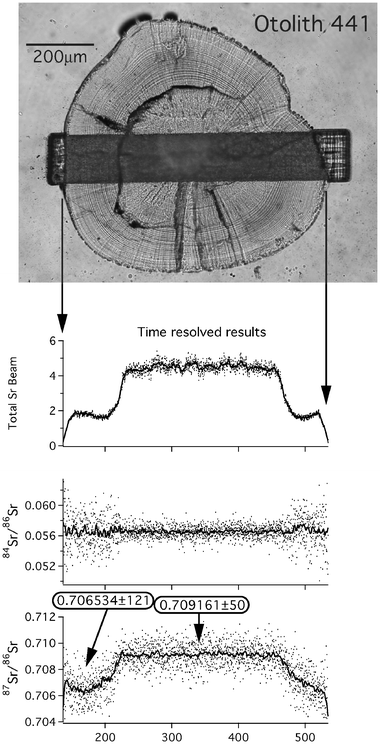 | ||
| Fig. 2 An example of high-resolution Sr-isotope analysis. A slit, approximately 147 × 3 μm in size, is slowly traversed across the sample. Each datum in the time-resolved plots represents a single 0.2 s integration, with a weighted running average also plotted for clarity. The strong Sr concentration contrasts between carbonate deposited in marine and freshwater are clearly reflected in the size of the Sr beam. 87Sr/86Sr ratios also vary systematically between core and edge, with compositions for the core and left hand edge shown in boxes. The edge composition is not well resolved on the right hand side since in this case the ablation slit runs oblique to the growth zonation of the otolith. | ||
We believe that this analytical strategy overcomes many of the spatial resolution issues that have hampered previous studies; the system provides the ability to resolve small changes in isotope ratio over very fine length-scales. As a result it might be possible, for example, to detect differences in Sr-isotope ratios among different freshwater end-members, i.e., identifying the natal source of fishes. Furthermore, the increased resolution allows greater precision in the determination of the timing of migration (i.e., the age of the fish) between marine and freshwater environments, compared with what might be achieved with a rastered spot analysis.
Conclusions
We have demonstrated that a high sensitivity laser ablation system, coupled with appropriate interference corrections, is capable of producing Sr-isotope ratio determinations in carbonate materials in situ with levels of precision and accuracy closely approaching those obtained from solution-based MC-ICP-MS or TIMS analyses using many times more material and considerably more time in both chemical separation and mass spectrometry.In addition many interesting research avenues are opened up with an in situ analytical capability. Using the aperture-imaging optics of an ArF excimer laser, operated in line-scan mode, and appropriate time-resolved software, it is possible to investigate Sr-isotopic variations on the scale of tens of micrometres.
Acknowledgements
The HelEx laser ablation system was constructed by Mike Shelley; both he and Steve Eggins are thanked for their support of the Melbourne University ICP-MS facility. Rob Hale provided the polished galaxiid otolith. Roger Kemp was invaluable in producing the data reduction template used in processing the time-resolved analyses. John Hellstrom provided much helpful advice in the early stages of this project. Constructive comments from two anonymous reviewers are greatly appreciated.References
- J. N. Christensen, A. N. Halliday, D.-C. Lee and C. M. Hall, Earth Planet. Sci. Lett., 1995, 136, 79 CrossRef CAS
.
- J. Davidson, F. Tepley, III, Z. Palacz and S. Meffan-Main, Earth Planet. Sci. Lett., 2001, 184, 427 CrossRef CAS
- M. Bizzarro, A. Simonetti, R. K. Stevenson and S. Kurszlaukis, Geochim. Cosmochim. Acta, 2003, 67, 289 CrossRef CAS
- S. M. Eggins, L. P. J. Kinsley and J. M. G. Shelley, Appl. Surf. Sci., 1998, 127–129, 278 CrossRef CAS
- J. Woodhead, J. Hergt, M. Shelley, S. Eggins and R. Kemp, Chem. Geol., 2004, 209, 121 CrossRef CAS
- S. Kuper and J. Brannon, Appl. Phys. Lett., 1992, 60, 1633 CrossRef CAS
- S. M. Eggins and M. G. Shelley, Geostand. Newsl., 2002, 26, 269 CAS
- D. Bleiner and D. Gunther, J. Anal. At. Spectrom., 2001, 16, 449 RSC
- S. Ehrlich, I. Gavrieli, L.-B. Dor and L. Halicz, J. Anal. At. Spectrom., 2001, 16, 1389 RSC
- T. J. Waight, J. Baker and D. Peate, Int. J. Mass Spectrom., 2002, 221, 229 CrossRef CAS
- M. F. Thirlwall, Chem. Geol., 1991, 94, 85 CrossRef CAS
- M. R. Palmer and H. Elderfield, Geochim. Cosmochim. Acta, 1986, 50, 409 CrossRef CAS
- J. D. Woodhead and J. M. Hergt, Geostand. Newsl., 2001, 25, 261 CrossRef CAS
- P. M. Outridge, S. R. Chenery, J. A. Babaluk and J. D. Resit, Environ. Geol., 2002, 42, 891 Search PubMed
-
P. R. D. Mason and A. J. G. Mank, Mineralogical Association of Canada Short Course Series, 2001, vol. 29, p. 93 Search PubMed
- M. Sanborn and K. Telmer, J. Anal. At. Spectrom., 2003, 18, 1231 RSC
- S. E. Campana, M. Sanborn, K. Telmer and J. D. Neilson, Can. J. Fish. Aquat. Sci., 1985, 42, 1014
-
S. E. Campana, Marine Ecology Progress Series, 1999, vol. 188, p. 263 Search PubMed
- D. H. Secor, Fish. Bull., 1992, 90, 798 Search PubMed
- S. E. Swearer, J. E. Caselle, D. W. Lea and R. R. Warner, Nature, 1999, 402, 799 CrossRef CAS
- S. R. Thorrold, C. Latkoczy, P. K. Swart and C. M. Jones, Science, 2001, 291, 297 CrossRef CAS
-
B. M. Gillanders and M. J. Kingsford, Marine Ecology Progress Series, 1996, vol. 141, p. 13 Search PubMed
-
G. E. Forrester and S. S. Swearer, Marine Ecology Progress Series, 2002, vol. 241, p. 201 Search PubMed
- J. S. Edmonds, M. J. Moran, N. Caputi and M. Morita, Can. J. Fish. Aquat. Sci., 1989, 46, 50 CAS
- S. E. Campana, G. A. Chouinard, J. M. Hanson, A. Fréchet and J. Brattey, Fish. Res., 2000, 46, 343 CrossRef
- R. J. Dodd, J. Palaeont., 1967, 41, 1313 Search PubMed
- J. M. Kalish, J. Exp. Mar. Biol. Ecol., 1989, 41, 749
- R. L. Radke and D. J. Schafer, Aust. J. Mar. Freshwater Res., 1992, 43, 935 Search PubMed
- D. H. Secor and J. R. Rooker, Fish. Res., 2000, 46, 359 CrossRef
- B. L. Ingram and P. K. Weber, Geology, 1999, 27, 851 CrossRef
- D. A. Milton and S. R. Chenery, Can. J. Fish. Aquat. Sci., 2003, 60, 1376 CrossRef
Footnote |
| † For a group of analyses, accuracy (expressed in ppm) is defined as A
=
[ΣA2i/(N
− 1)]1/2 where Ai
= 1000000 ×
(R′i
−
Rtr)/Rtr, is the measured ratio for the sample, corrected for interferences and mass bias, and Rtr is the true ratio (in this case the analysis of Sr separated from ca. 10 mg of the sample by cation exchange chemistry, and measured by solution MC-ICP-MS). Precision is quoted as 2 s.e.. |
| This journal is © The Royal Society of Chemistry 2005 |