Ultra-deep desulfurization of transportation fuels via charge-transfer complexes under ambient conditions
Marc
Sévignon
a,
Mathieu
Macaud
a,
Alain
Favre-Réguillon
*ab,
Jürgen
Schulz
a,
Muriel
Rocault
a,
René
Faure
c,
Michel
Vrinat
d and
Marc
Lemaire
*a
aLaboratoire de Catalyse et Synthèse Organique, Université Claude Bernard Lyon1, UMR 5181, CPE, 43 bld du 11 Novembre 1918, 69622 Villeurbanne Cedex, France. E-mail: marc.lemaire@univ-lyon1.fr; Fax: +(33) 472431408; Tel: +(33) 472431407
bLaboratoire de Chimie Organique, UMR 7084, Conservatoire National des Arts et Métiers, 2 rue Conté, 75003, Paris. E-mail: favrereg@cnam.fr
cLaboratoire des Sciences Analytiques, UMR 5180, Bât. Raulin, 43 bld du 11 Novembre 1918, 69622 Villeurbanne Cedex, France
dInstitut de Recherches sur la Catalyse, UPR 5401, 2 avenue Albert Einstein, 69626 Villeurbanne Cedex, France
First published on 26th April 2005
Abstract
Very stringent regulations on the maximum sulfur content of gas oil have led to intense research into all aspects of desulfurization. Deep desulfurization of diesel fuels is particularly challenging due to the difficulty of removing refractory sulfur compounds, particularly 4,6-dialkyldibenzothiophenes, using conventional hydrodesulfurization processes (HDS). A novel approach to the potential desulfurization of fuels such as diesel is proposed. It relies on the ability of 4,6-dialkyldibenzothiophene to form charge-transfer complexes (CTC) with π-acceptor molecules. We present the synthesis of a new π-acceptor molecule, 4,5-dicyano-2,7-dinitrofluorenone, solution redox behaviour and the crystal structure of the charge-transfer complex with the refractory 4,6-dimethyldibenzothiophene. This π-acceptor compound was then immobilized on a hydrophobic support (poly(styrene-co-divinylbenzene)). The selectivity of the CTC process was confirmed by the analysis of the sulfur compounds trapped from straight run Arabian Light (SR) containing 13600 ppm S. The functionalized polymer can be used in multiple cycles for the removal of refractory S-containing compounds from hydrotreated SR. It can also be regenerated with toluene. The high selectivity of this material permits diesel fuel to be desulfurized to a level that meets future regulatory requirements, i.e. less than 10 ppm S, at ambient temperature and without hydrogen consumption.
Introduction
In recent years, deep desulfurization of diesel fuel has attracted much attention due to the gradual reduction of the statutory sulfur content in most western countries. In 2009, the maximum S-content within the European Union will be reduced to 10 ppm compared to today's value of 350 ppm.1,2 Heightened interest in ultra-clean fuels is also driven by the need for new emissions control technologies for IC engines (especially those for diesel fuels), and the use of on-board or on-site reforming of hydrocarbon fuels for new fuel cell vehicles.3There are three major types of transportation fuels: gasoline, jet fuels and diesel, which differ in composition and properties. The common types of sulfur compounds in the diesel fuel range are alkylated benzothiophenes (BTs), dibenzothiophene, and its alkylated derivatives (DBTs) (Fig. 1).
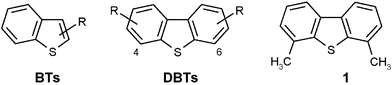 | ||
| Fig. 1 Types of sulfur compounds present in the diesel fuel range. | ||
The state of the art in desulfurization technology is hydrodesulfurization (HDS). In this process, the sulfur is removed from sulfur containing compounds by reaction with hydrogen, forming H2S on a sulfur type active phase catalyst. HDS is effective for a range of sulfur containing compounds that exhibit varying reactivities towards desulfurization. Reactivity is dependent upon the local environment of the sulfur atom in the molecule, and the overall shape of the molecule. The process is widely employed throughout the world and has been used for over 60 years. HDS is highly efficient at removing thiols, sulfides and disulfides, but is less effective for aromatic thiophenes and thiophene derivatives, especially those containing functional groups that hinder the sulfur atoms. Due to the current 350–500 ppm specifications mandated in several countries, the diesel fuel range available today has low total sulfur content but a disproportionately high concentration of refractory sulfur species.
Improvement of today's technology to enable production of ultralow sulfur content fuel requires that the difficulty of desulfurizing refractory sulfur compounds be overcome. Investigations have demonstrated that the sulfur compounds remaining in diesel fuels with a sulfur level lower than 500 ppm are the dibenzothiophenes with alkyl substituents at the 4- and/or 6- position, (Fig. 1). Both steric hindrance and electronic factors are responsible for the observed low reactivity. To meet the new regulations, the refining industry has demonstrated its capability to adapt hydrotreatment to the stringent regulations by improving catalytic activity and increasing the process severity, especially by increasing the hydrogen pressure or designing new reactor configurations.4 However, the selective elimination of this family of compounds from fuel is difficult due to their relatively low concentration in the feed and their low reactivity towards catalytic hydrodesulfurization.
The elimination of refractory sulfur compounds by non-catalytic processes is becoming a new challenge, because it could be accomplished at ambient temperature and pressure and without leading to an increase in the hydrogen consumption. New process concepts3 include design approaches for ultra-deep desulfurization focusing on adsorption,5–7 selective extraction using ionic liquids,8–10 oxidation-extraction,11,12 precipitation of S-alkylsulfonium salts13 and biodesulfurization.14
The planarity and electron-rich structure of 4,6-dimethyldibenzothiophene 1 (Fig. 1), lead us to expect that it would be capable of forming charge-transfer complexes (CTCs) with suitable π-acceptors. Charge-transfer complexes (CTCs) arising from the interaction of aromatic π-donor and π-acceptor molecules have been studied extensively15 and recently charge-transfer complex interactions have been evidenced by chemical force microscopy.16 CTCs have been used in supramolecular chemistry,17 for chiral recognition,18 and for selective organic transformation.19
An adsorption process is being explored in our laboratory for ultra-deep desulfurization of distillate fuels based on selective removal of electron rich refractory sulfur compounds like 4,6-dimethyl dibenzothiophene 1 (Fig. 1) by charge-transfer complex formation.20 However, diesel fuel contains a large variety of aromatic compounds, with and without heteroatoms, that compete with 1 in the formation of CTCs. Theoretical calculations21 and experimental studies on synthetic and real feed22–25 have shown that symmetrical π-acceptors based on polynitrosubstituted 9-fluorenones (e.g. 2,4,5,7-tetranitro-9-fluorenone 2, Fig. 2) efficiently and selectively complex dibenzothiophenes in preference to benzothiophenes and aromatic compounds.
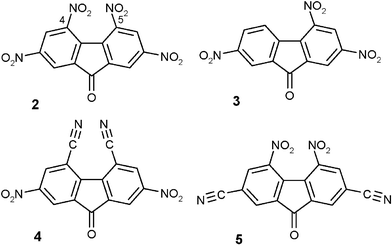 | ||
| Fig. 2 Structure of π-acceptor molecules. | ||
Polynitrosubstituted 9-fluorenones (Fig. 2) have been widely employed as acceptors for charge-transfer complex formation,16 as photosensitizers and in electron transport materials.26 The structure of acceptor 2 has been determined by single X-ray analysis and reveals significant steric strain between the nitro substituents in the 4- and 5- positions.27 Thus their acoplanarity with the fluorene nucleus probably leads to a decrease in the electron-acceptor efficiency of these groups on account of the reduction in their contribution to the conjugation effect,27 and could interfere with the formation of a CTC.21
With the objective of improving the selectivity and the efficiency of the complexation of 4,6-dialkyldibenzothiophene, we carried out the synthesis of planar symmetrical π-acceptors bearing less bulky electron-withdrawing groups in the 4- and 5- position. It was expected that the substitution of nitro groups in the 4- and 5- positions by linear cyano (compound 4, Fig. 2) groups would decrease the steric strain and thus the dihedral angle between the two aromatic rings. Then, we determined their redox properties and studied the crystal structures of the new fluorenone derivatives and the CTC complex formed by π-acceptor 4 and refractory compound 1. Compound 4 was then immobilized on an insoluble resin for the ultra-deep desulfurization of distillate fuels by solid–liquid extraction.
Results and discussion
Synthesis of the π-acceptor molecule 4
Acceptor 2 is known to react with various proton-containing nucleophiles with substitution of the nitro group.26–28 However, substitution occurs mainly in the 2- and 4- positions28 and introduction of the cyano group involves the use of HCN. We prefer to study a two step procedure starting from 2,7-dinitro-9-fluorenone 6 (Fig. 3), readily accessible from fluorenone. Methods for bromination of strongly deactivated aromatics derivatives are scarce.29–31 Compound 7 had previously been prepared using a mixture of mercury(II) nitrate and bromine.29 We found that 6 could be brominated in a concentrated H2SO4–CF3CO2H mixture with NBS31 to yield 4,5-dibromo-2,7-dinitrofluorenone 7 which was then converted into 4,5-dicyano-2,7-dinitrofluorenone 4 by reaction with CuCN in DMF solution.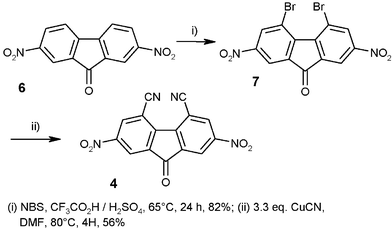 | ||
| Fig. 3 Synthesis of the acceptor 4. | ||
Electrochemistry
The redox properties of the π-acceptor 4 were compared to π-acceptor 2, 3 and 532 (Fig. 2). We conducted cyclic voltammetry (CV) measurements in acetonitrile at room temperature with Bu4N+BF4− as the supporting electrolyte (Fig. 4). The redox potentials of compounds 2, 3, 4 and 5 are summarized in Table 1.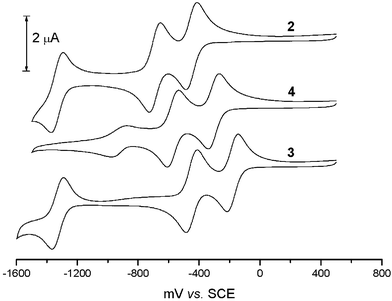 | ||
| Fig. 4 Cyclic voltammogram of compounds 2, 3 and 4 under the conditions stated in Table 1. | ||
| Compd | E 1red 1/2/V | E 1red 1/2/V | E 1red 1/2/V | EA (eV)a |
|---|---|---|---|---|
| Solvent acetonitrile; electrolyte 0.2 M Bu4N+BF4−; scan rate 100 mV s−1. All potentials given in the table were recalculated to Ag/AgCl scale.a Calculated using equation in ref. 32. | ||||
| 2 | −0.47 | −0.71 | −2.69 | 3.01 |
| 3 | −0.20 | −0.47 | −1.35 | 2.74 |
| 5 32 | −0.27 | −0.64 | −1.33 | 2.27 |
| 4 | −0.32 | −0.59 | −0.94 | 2.86 |
By analogy with compounds 2, 3 and 5, π-acceptor 4 shows clear amphoteric multiredox behaviour (Fig. 4, Table 1), consisting of 3 reversible single electron reduction waves.
Replacement of the nitro groups by cyano groups at the 4- and 5- positions slightly decreases the electron acceptor properties: the first reduction of compound 4 is shifted to a more positive value by 0.15 V (Table 1), but electron affinity (EA) calculations indicate that 4 is a rather strong acceptor, comparable to the strongest known acceptor of the fluorene series, i.e. 2,4,5,7-tetranitro-9-dicyanomethylene-fluorene32 or 3. The comparison of the reduction potentials for compounds 532 and 4 (Fig. 2) shows that the location of the cyano substituents (2,7 versus 4,5) does not exert a notable effect upon the electrochemical behavior of these compounds although a more planar structure, which is more favorable for CTC formation, was expected for compound 4.
Single crystal X-ray analysis
It was noted that the substituents in positions 4 and 5 had a strong influence on the crystal structure of compounds 2, 4 and 7, and in particular, on the torsion angles listed in Table 2.


The X-ray structure analysis of compound 2 has already been determined.33,34 The asymmetric unit consists of two molecules which differ slightly in their planarities and torsional angles. The two nitro groups in the 4- and 5- positions cannot remain in the fluorenyl plane and tend to move apart from one another. This repulsion causes rotation of these groups, inclination of C–N bonds and deformation of the fluorenone moiety. There are significant deviations from planarity of the fluorenone moiety of 2 in contrast to the planar molecule of 2,9-dinitrofluorenone 6.35 The dihedral angle values between the outer rings [C(4)–C(4a)–C(4b)–C(5)] (Fig. 5) of 2 are 13.5° and 7.7°33 (Table 2) although it is equal to 1.5° for 6.35 This deviation from planarity of the fluorenone moiety of 2 is expected to minimize the CT interactions between planar electron rich compounds 136 and 2.21 The same trend is amplified in compound 7, bearing bulky bromides in the 4- and 5- positions (Table 2), which has a dihedral angle value of 25.6°. In compound 4, introduction of the linear cyano groups in the 4- and 5- positions, strongly reduced the steric strain and the fluorenone moiety is almost planar. The dihedral angle between the outer rings [C(4)–C(4a)–C(4b)–C(5)] of compound 4 is 5.8° (Table 2).
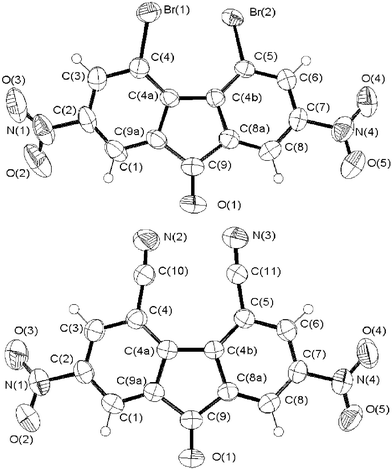 | ||
| Fig. 5 Structure of the π-acceptor 7 (top) and 4 (bottom). | ||
CT complexation in the solid state
Solution studies of the charge-transfer complexes formed between compound 1 and fluorenone derivatives are severely limited by their very low solubility.25 Single crystals of the charge-transfer complex were grown from a chloroform solution of equimolar amounts of compound 4 and 1. Fig. 6 shows the packing diagrams.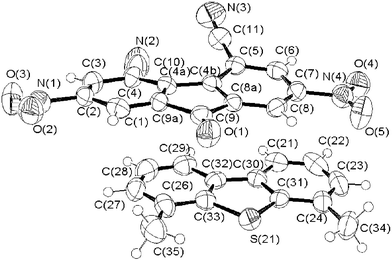 | ||
| Fig. 6 Structure of the charge-transfer complex between 4,6-dimethyldibenzothiophene 1 and π-acceptor 4. | ||
The donor 1 and π-acceptor molecule 4 lie parallel with interplanar separation of 3.4 Å, which corresponds to the value expected of a charge-transfer interaction.21 Surprisingly the interactions between 1 with π-acceptor 4 involve a distortion of the structure of 4. An increase in the dihedral angles values between the outer rings [C(4)–C(4a)–C(4b)–C(5)] from 5.8° to 14.1° was observed for 4 (Table 2) although almost no modification was noticed for compound 1. Fig. 7 shows the crystal packing of the CTC formed between compound 1 and π-acceptor 4. The CTC crystal is composed of alternate layers of donors and acceptors. These layers are not coplanar for the same entity.
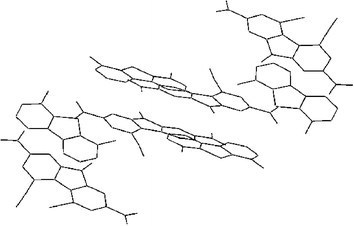 | ||
| Fig. 7 Crystal packing of the CTC between compound 1 and π-acceptor 4. H atoms are omitted. | ||
Immobilization of π-acceptor molecule on organic support
In order to check the feasibility of our concept for a deep desulfurization process, π-acceptor 4 was immobilized on a solid support according to Fig. 8. Formation of a hydrazone linker allows immobilisation on a solid support without substituting an electron-withdrawing group (nitro or cyano) at the fluorene moiety with an electron-donating group (alkoxy or alkyl). Furthermore, this didn't modify the symmetry of the π-acceptor and is expected to be stable under the experimental conditions.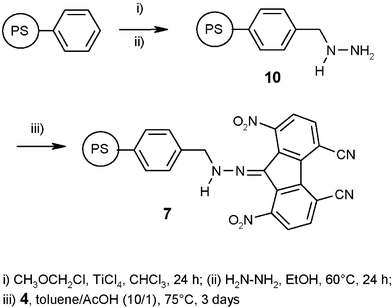 | ||
| Fig. 8 Immobilization of π-acceptor 4 on poly(styrene) resin 7. | ||
Poly(styrene) resins were chloromethylated (1.37 mmol Cl g−1) and then treated with an excess of hydrazine (35% wt) in EtOH at room temperature to give methyl hydrazine polymer-bound resin 10. π-Acceptor 4 was then added to resin 10 as a suspension in a toluene–acetic acid mixture at 75 °C. The excess of reagent was removed by soxhlet to yield resin 7. The loading of resin 7 was determined by elemental analysis to be 0.4 mmol of π-acceptor 4 per g.
Desulfurization of diesel fuel range with immobilized π-acceptor molecules
To examine the selectivity of resin 7 towards dibenzothiophene derivatives, we studied the complexation procedure with straight run oil (SR) (Table 3) with the following sulfur content: 13600 ppm, 8600 ppm (63%) including benzothiophene derivatives (BTs) and 5000 ppm (37%) including dibenzothiophene derivatives (DBTs) (Fig. 9). The SR was treated with resin 7 in a batch reactor with continuous stirring. The sulfur content of the residual feed was determined and was found to be 10600 ppm S, which corresponded to an elimination of about 23% of the total sulfur. Resin 7 was regenerated by washing with toluene. The selectivity of the process toward DBTs could be confirmed by analysis of the toluene fractions (Fig. 9). A benzothiophene/4,6-dimethyldibenzothiophene selectivity higher than 40 could be calculated from the sulfur chromatogram. About 70 wt% of the compounds trapped by resin 7 are dibenzothiophene derivatives and 30 wt% are polyaromatics without heteroatoms. This result highlights the selectivity of the CTC process towards refractory DBTs.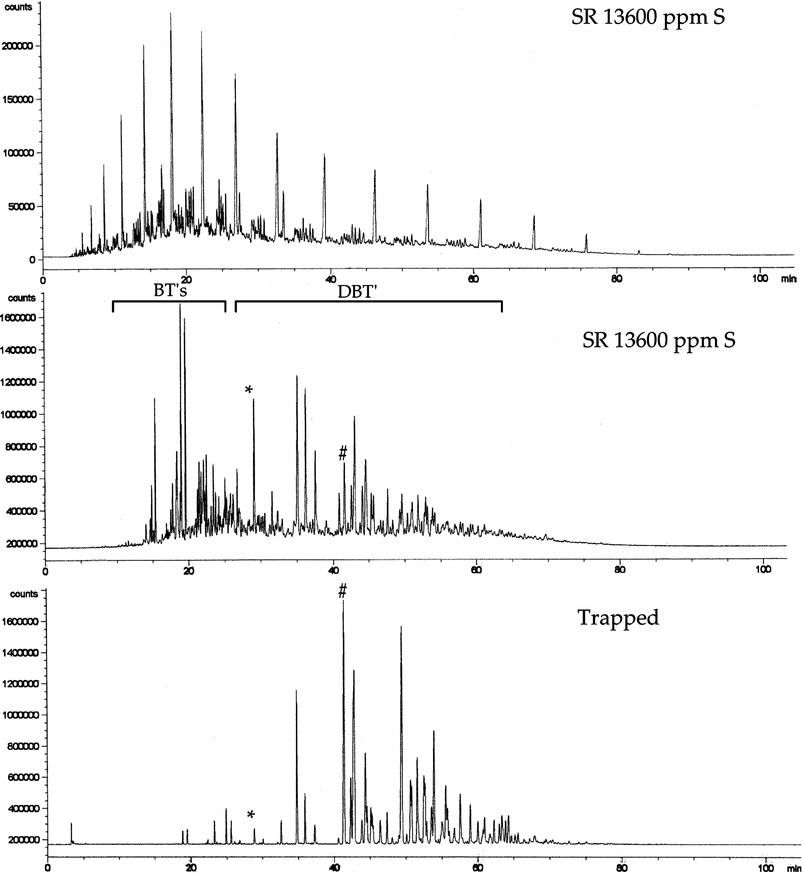 | ||
| Fig. 9 Carbon and sulfur chromatograms of SR oil and sulfur chromatogram of trapped compounds. * and # correspond to benzothiophene and 4,6-dimethyldibenzothiophene respectively. General conditions: ambient temp., contact time 24 h, ratio fuel/polymer = 2. | ||
| SR 13600 | SR 390 | SR 60 | |
|---|---|---|---|
| Density (288 K)/g l−1 | 853 | 838 | 835 |
| Sulfur (ppm) | 13600 | 390 | 60 |
| Total nitrogen (ppm) | 104 | 29 | 6 |
| Aromatics (wt%) | 32.1 | 26.2 | 23.4 |
In order to reach very low sulfur levels we used a multistage extraction process with hydrotreated SR. The desulfurized oil from the first extraction step was re-treated with regenerated resin 7. Resin 7 could be regenerated by back extraction with 5 resin volumes of toluene and could be reused up to 10 times without loss of activity.
This process was repeated up to four times with two hydrotreated SRs obtained after classical HDS and deep HDS (390 and 60 ppm S respectively). The results are summarized in Table 4.
As shown in Table 4, the results of experiments with hydrotreated SR are promising. In three steps, a fuel containing less than 50 ppm S was obtained starting from diesel with 390 ppm S. Resin 7 is effective even with low S content SR (60 ppm S) obtained after severe HDS. The sulfur compounds remaining in the feed are refractory DBTs and in four steps the future specification was obtained (<10 ppm S).
Conclusions
The results presented show a new approach to the ultra-deep desulfurization of liquid fuel, with high selectivity towards the refractory substituted dibenzothiophenes that are very difficult to remove by common hydrodesulfurization techniques. The new method is based on the selective adsorption of dibenzothiophenes by formation of charge-transfer complexes with immobilized π-acceptor molecules. The application of very mild process conditions (low pressure, ambient temperature, no hydrogen consumption) is an additional advantage of this new approach in comparison to traditional HDS.Experimental
Quantitative analysis of the total sulfur concentration was determined by energy X-ray fluorescence spectroscopy using a Horiba SLFA 1800 sulfur in oil analyzer, using 20 mm i.d. PTFE cells with 7 µm Mylar film windows. To eliminate the effect of the matrix on the gas oil, the apparatus was calibrated using a series of different gas oils containing between 100 and 13600 ppm of sulfur. Low sulfur and nitrogen concentrations were analyzed by UV fluorescence and by chemiluminescence, respectively, on an Antek 9000 Series nitrogen/sulfur analyzer equipped with a robotic liquid autosampler.Qualitative analysis of the sulfur-containing molecules was performed using a gas chromatograph associated with a sulfur specific detector (Sievers Model 355 B SCD). For qualification of the components, the BT and the DBT were commercially available and various alkyldibenzothiophenes have been synthesized as previously reported.37
The relevant properties of straight run oil are summarized in Table 3.
2,4,5,7-Tetranitro-9-fluorenone (2)
A solution containing 20 g (72 mmol) of 2,7-dinitrofluorenone (6) and 200 ml of concentrated sulfuric acid was added dropwise to a mixture of 120 ml concentrated nitric acid and 120 ml concentrated sulfuric acid at 60 °C. The temperature was gradually increased from 60 °C to 130 °C. The mixture was stirred for 5 hours at 130 °C, cooled, then poured into cold water. The yellow solid was washed with water and dried. The solid was purified by recrystallisation from acetic acid yielding yellow needles (79%). Rf: 0.53 (CH2Cl2). 1H NMR (200 MHz, δ DMSO d6): 8.97 (d, J = 2.26 Hz); 8.77 (d, J = 2.26 Hz), 13C NMR (50 MHz, δ DMSO d6): 187.61; 150.56; 146.31; 140.00; 137.11; 126.69; 123.37, FTIR (cm−1): 1732; 1540; 1340, Anal. Calcd. for C 43.35; H 1.12; N 15.55; O 39.98. Found: C 43.27; H 1.17; N 15.54; O 39.69%.2,4,7-Trinitro-9-fluorenone (3)
A solution containing 20.01 g (77 mmol) of 2,7-dinitrofluorenone (6) in 75 ml of concentrated sulfuric acid was added dropwise to a mixture of 11 ml concentrated nitric acid and 11 ml concentrated sulfuric acid at 60 °C. During the addition, the heating was gradually increased from 60 °C to 130 °C. At the end of the addition, the mixture was cooled and poured into cold water. After filtration, the yellow solid was washed with water and dried. The solid was purified by recrystallisation from acetic acid yielding yellow needles (57%). Rf: 0.58 (CH2Cl2). 1H NMR (200 MHz, δ DMSO d6): 8.99 (d, J = 1.88 Hz); 8.63 (dd, J = 8.67 and 2.26 Hz); 8.61 (d, J = 1.88 Hz); 8.43 (d, J = 2.26 Hz); 8.19 (d, J = 8.67 Hz), 13C NMR (50 MHz, δ DMSO d6): 187.18; 150.48; 149.70; 145.60; 144.13; 139.62; 138.65; 136.96; 131.58; 128.78; 126.77; 122.98; 119.79, FTIR (cm−1): 1732; 1542; 1343, Anal. Calcd. for C 49.59; H 1.68; N 13.27; O 35.59. Found: C 49.54; H 1.60; N 13.33; O 35.53%.4,5-Dibromo-2,7-dinitrofluorenone (7)
77 g (433 mmol) of N-bromosuccinimide was added in small portions over 4 hours, with agitation, to a solution containing 15 g (54 mmol) of 2,7-dinitrofluorenone (6), 225 ml of trifluoroacetic acid and 90 ml of concentrated sulfuric acid at 65 °C. The mixture was stirred for 24 hours at 65 °C (until the bromine vapour had disappeared). After hydrolysis and filtration, the yellow–orange precipitate formed was washed with water and purified by recrystallisation from acetic acid yielding yellow needles (82%). 1H NMR (200 MHz, δ CDCl3) 8.74 (d, J = 2.07 Hz); 8.59 (d, J = 2.07 Hz), 13C NMR (50 MHz, δ DMSO d6): 185.43; 149.09; 146.97; 139.19; 136.07; 117.43, FTIR (cm−1): 1730; 1540; 1343; 580, Anal. Calcd. for: C 36.48; H 0.94; N 6.55; Br 37.34; O 18.69. Found: C 36.47; H 0.93; N 6.59; Br 37.39; O 18.58%.4,5-Dicyano-2,7-dinitrofluorenone (4)
9.56 g (22.3 mmol) of 4,5-dibromo-2,7-dinitrofluorenone (7) was dissolved in 100 ml of anhydrous DMF at 80 °C. 6.56 g (73.3 mmol) of copper cyanide was added in portions, and the mixture stirred at 80 °C for 4 hours. A solution containing 35 g (215 mmol) of iron(III) chloride, 150 ml of water and 17 ml of concentrated hydrochloric acid was then added and the mixture was stirred at 70 °C for 3 hours. The mixture was cooled to ambient temperature, filtered and the precipitate was washed with water. Sodium chloride was added, and the aqueous phase was extracted with ethylacetate. The combined organic phases were dried over Na2SO4 and concentrated. 500 mg of brown solid was recovered. The precipitate was washed with toluene in a soxhlet. After evaporation of solvent, a further 4.2 g of brown solid was recovered. The brown solids were combined and purified by column chromatography with dichloromethane to give a brown solid (56%). 1H NMR (200 MHz, δ DMSO d6): 9.10 (d, J = 2.25 Hz); 8.67 (d, J = 2,18 Hz), 13C NMR (50 MHz, δ DMSO d6): 185.83; 150.33; 147.32; 138.38; 136.79; 123.32; 116.50; 109.85, FTIR (cm−1): 3063; 2244; 1731; 1530; 1350, Anal. Calcd. for: C 56.26; H 1.26; N 17.50; O 24.98. Found: C 56.13; H 1.25; N 17.50; O 24.94%.Crystal structure determination
Single crystals suitable for X-ray diffraction were grown from chloroform. The crystallisation a 1 ∶ 1 mixture of 1 and 4 from chloroform resulted in a 1 ∶ 1 molecular complex. Needles were stable in air for several weeks at room temperature. X-Ray diffraction experiments (see Table 5) were carried out on a Kappa CCD Nonius diffractometer using graphite-monochromated MoKα radiation. The intensity data were measured at 295 K. The structures were solved using direct method and refined by full-matrix least-squares on F2 using SHELXTL.38| Parameter | 7 | 4 | CTC 4/1 |
|---|---|---|---|
| Formula | C13H4Br2N2O5 | C15H4N4O5 | C29H16N4O5S |
| M | 428.00 | 320.22 | 532.52 |
| System | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n | P21/n |
| a/Å | 10.3660(2) | 14.1690(8) | 7.7310(4) |
| b/Å | 16.0720(5) | 6.4510(4) | 15.1430(9) |
| c/Å | 8.3120(2) | 15.2380(11) | 21.1520(14) |
| β/° | 103.372(2) | 110.614(2) | 98.189(2) |
| V/Å3 | 1347.26(6) | 1303.64(14) | 2451.0(3) |
| Z | 4 | 4 | 4 |
| ρ/g cm−3 | 2.110 | 1.632 | 1.443 |
| µ/mm−1 | 6.043 | 0.128 | 0.182 |
| θ range/° | 2.38–31.00 | 2.44–27.69 | 1.66–24.39 |
| Number reflections measured | 14094 | 5902 | 13094 |
| Number of independent reflections | 4181 | 2562 | 3983 |
| R int | 0.078 | 0.049 | 0.135 |
| Final R value (I > 2 σ(I)) | 0.063 | 0.054 | 0.068 |
| Final R (all data) | 0.115 | 0.090 | 0.147 |
CCDC reference numbers 253625 (for compound 4), 253627 (for compound 7) and 253626 (for CTC 4/1). See http://www.rsc.org/suppdata/gc/b5/b502672e/ for crystallographic data in CIF or other electronic format.
Synthesis of Merrifield resin
A 250 mL reactor was charged with 60 g of poly(styrene-co-divinylbenzene) resin (Aldrich 42,698-9) (300–800 µm) and a solution of 185 mL of chloroform and 12.5 g (155 mmol) of chloromethyl-methyl ether39 at 20 °C. A solution prepared by cautiously adding 8 mL (68.5 mmol) of tin(IV) chloride to 12.5 g (155 mmol) of chloromethyl-methyl ether and 25 mL of chloroform was added dropwise with gentle stirring to the cooled mixture over a 30 min period. After 24 hours at ambient temperature, the resin was collected by filtration and washed successively with dioxan–water (1 ∶ 1), dioxan–water–HCl (5 ∶ 5 ∶ 1), water, dioxan, THF and finally pentane. Anal. Found: C 82.25; H 6.74; Cl 6.69%. The accessible chlorine on the resin was determine by Volhard titration40 and was found to be 1.37 mmol Cl g−1, FTIR (cm−1): 3017, 2925, 1600, 1510, 1450, 1280, 1120, 610.Synthesis of resin 10
74 ml (820 mmol) of hydrazine as a 35% weight in water was slowly added to 40 g of chloromethyl resin (1.37 mmol Cl g−1) in 60 ml of ethanol at ambient temperature. At the end of the addition, the mixture was mechanically stirred for 24 hours at 60 °C. The resultant resin was then filtered, washed with water then with ethanol. Anal. Found: C 80.97; H 6.79; N 1.41; Cl 1.86, Accessible chlorine: 0.15 mmol Cl g−1 FTIR (cm−1): 3018, 2927, 1608, 1452.Synthesis of resin 7
5.01 g (14 mmol) of 4,5-dicyano-2,7-dinitrofluorenone (4) was dissolved in 650 ml of toluene and 10 ml of acetic acid, then 10 g of resin 10 was added. The mixture was mechanically stirred for 3 days at 75 °C. The resin was then washed for 48 hours with toluene in a soxhlet, then with pentane and was dried at 60 °C yielding brown resins. Anal. Found: C 81.39; H 6.30; N 1.93; Cl 1.86; O 7.75, accessible chlorine: <0.05 mmol Cl g−1The loading capacity was calculated based on nitrogen elemental analysis and was found to be 0.4 mmol π-acceptor 4 per g. FTIR (cm−1): 3020, 2925, 1700, 1606, 1540, 1347.
Acknowledgements
This research was supported by Ademe, FSH, IFP and TotalFinaElf. The authors acknowledge IFP for the donation of hydrotreated diesel fuel.References
- Directive 2003/17/EC, 3 March 2003, Official Journal of the European Union.
- EPA420-R-00-026, December 2000, United States Environmental Protection Agency.
- C. Song and X. Ma, Appl. Catal. B, 2003, 41, 207–238 CrossRef CAS.
- M. Breysse, G. Djega-Mariadassou, S. Pessayre, C. Geantet, M. Vrinat, G. Pérot and M. Lemaire, Catal. Today, 2003, 84, 129–138 CrossRef CAS.
- A. J. Hernandez-Maldonado, S. D. Stamatis, R. T. Yang, A. Z. He and W. Cannella, Ind. Eng. Chem. Res., 2004, 43, 769–776 CrossRef CAS.
- R. T. Yang, A. J. Hernandez-Maldonado and F. H. Yang, Science, 2003, 301, 79–81 CrossRef CAS.
- B. Castro, M. J. Whitcombe, E. N. Vulfson, R. Vazquez-Duhalt and E. Barzana, Anal. Chim. Acta, 2001, 435, 83–90 CrossRef CAS.
- J. Eβer, P. Wasserscheid and A. Jess, Green Chem., 2004, 6, 316–322 RSC.
- S. Zhang and Z. C. Zhang, Green Chem., 2002, 4, 376–379 RSC.
- A. Boesmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494–2495 RSC.
- W.-H. Lo, H.-Y. Yang and G.-T. Wei, Green Chem., 2003, 5, 639–642 RSC.
- Y. Shiraishi, H. Hara, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 1589–1595 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, Y. Taki, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2001, 40, 1225–1233 CrossRef CAS.
- D. J. Monticello, Curr. Opin. Biotechnol., 2000, 11, 540–546 CrossRef CAS.
- R. S. Mulliken and W. B. Person, Molecular Complexes, Wiley, New York, 1969 Search PubMed.
- R. Gil, J.-C. Fiaud, J.-C. Poulin and E. Schulz, Chem. Commun., 2003, 2234–2235 RSC.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651–669 RSC.
- H. Okubo, D. Nakano, S. Anzai and M. Yamaguchi, J. Org. Chem., 2001, 66, 557–563 CrossRef CAS.
- M. Lemaire, A. Guy, D. Imbert and J. P. Guette, J. Chem. Soc., Chem. Commun., 1986, 741–742 RSC.
- T. Koltai, M. Macaud, A. Milenkovic, E. Schultz, M. Lemaire and M. Vrinat, Catal. Lett., 2002, 83, 143–148 CrossRef CAS.
- A. Milenkovic, D. Loffreda, E. Schulz, H. Chermette, M. Lemaire and P. Sautet, Phys. Chem. Chem. Phys., 2004, 6, 1169–1180 RSC.
- M. Lemaire, E. Schulz, M. Sevignon, M. Macaud, A. Favre-Reguillon, M. Thomas and R. Loutaty, PCT Int. Appl., 2002024836, (Total Raffinage Distribution S. A., Fr.; Centre National de la Recherche Scientifique; Institut Francais du Petrole), 2002.
- A. Milenkovic, M. Macaud, E. Schulz, T. Koltai, D. Loffreda, M. Vrinat and M. Lemaire, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 459–463 CrossRef CAS.
- A. Milenkovic, E. Schulz, V. Meille, D. Loffreda, M. Forissier, M. Vrinat, P. Sautet and M. Lemaire, Energy Fuels, 1999, 13, 881–887 CrossRef CAS.
- V. Meille, E. Schulz, M. Vrinat and M. Lemaire, Chem. Commun., 1998, 305–306 RSC.
- D. F. Perepichka, M. R. Bryce, I. F. Perepichka, S. B. Lyubchik, C. A. Christensen, N. Godbert, A. S. Batsanov, E. Levillain, E. J. L. McInnes and J. P. Zhao, J. Am. Chem. Soc., 2002, 124, 14227–14238 CrossRef CAS.
- L. G. Kuz'mina, I. F. Perepichka, D. F. Perepichka, J. A. K. Howard and M. R. Bryce, Cryst. Rep., 2002, 47, 251–261 Search PubMed.
- I. F. Perepichka, A. F. Popov, T. V. Orekhova, M. R. Bryce, A. M. Andrievskii, A. S. Batsanov, J. A. K. Howard and N. I. Sokolov, J. Org. Chem., 2000, 65, 3053–3063 CrossRef CAS.
- D. Hellwinkel and G. Haas, Justus Liebigs Ann. Chem., 1978, 1913–1915 CrossRef CAS.
- A. M. Andrievskii, M. V. Gorelik, S. V. Avidon and E. S. Al'tman, Russ. J. Org. Chem., 1993, 29, 1519–1524.
- J. Duan, L. H. Zhang and W. R. Dolbier, Jr., Synlett, 1999, 1245–1246 CrossRef CAS.
- I. F. Perepichka, L. G. Kuz'mina, D. F. Perepichka, M. R. Bryce, L. M. Goldenberg, A. F. Popov and J. A. K. Howard, J. Org. Chem., 1998, 63, 6484–6493 CrossRef CAS.
- R. G. Baughman, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 933–936 CrossRef.
- L. A. Chetkina, O. V. Semidetko, V. K. Belskii, A. N. Sobolev and A. M. Andrievskii, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 931–933 CrossRef.
- R. G. Baughman, Cryst. Struct. Commun., 1982, 11, 479–484 CAS.
- V. Meille, E. Schulz, M. Lemaire, R. Faure and M. Vrinat, Tetrahedron, 1996, 52, 3953–3960 CrossRef CAS.
- M. Macaud, A. Milenkovic, E. Schulz, M. Lemaire and M. Vrinat, J. Catal., 2000, 193, 255–263 CrossRef CAS.
- G. M. Sheldrick, SHELXTL: Structure Analysis Program, Bruker AXS Software, Madison, WI, 1998.
- J. Stadlwieser, Synthesis, 1985, 490 CrossRef CAS.
- G.-S. Lu, S. Mojsov, J. P. Tam and R. B. Merrifield, J. Org. Chem., 1981, 46, 3433–3436 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |