Liquid–liquid behaviour of ionic liquid–1-butanol–water and high pressure CO2-induced phase changes
Vesna
Najdanovic-Visak
,
Luís P. N.
Rebelo
and
Manuel
Nunes da Ponte
*
Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Apartado 127, 2780–901 Oeiras, Portugal. E-mail: mnponte@itqb.unl.pt; Fax: +351– 21 441 12 77; Tel: +351–21 4469 444
First published on 11th April 2005
Abstract
The liquid–liquid equilibrium of the [C4mim][NTf2]–1-butanol system presents upper critical solution temperature (UCST) behaviour. We report the influence of added water as well as the effect of hydrostatic pressure on the cloud points. Similarly to previously studied systems that involved RTILs in our laboratories, water is shown to be a very good co-solvent with alcohols. If a small amount of water is present in the [C4mim][NTf2]–1-butanol system it decreases the UCST as much as 1.5 K per mol% of water added to butanol. The hydrostatic pressure effect on cloud points is rather modest (≈−3.5 × 10−3 K bar−1). As for liquid–vapour equilibria, a new apparatus which employs a high-pressure variable-volume cell, was conceived and built. The apparatus was tested for the well-known system 1-butanol–CO2. Demixing pressures of the ternary and quaternary mixtures, (1-butanol–water–CO2) and ([C4mim][NTf2]–1-butanol–water–CO2), were determined for a few compositions and temperatures. The demixing pressure is strongly controlled by the water concentration.
Introduction
Over the last decade, environmental chemists have focused their attention on coupling traditional and alternative green solvents in order to design benign media for a cleaner chemistry and sustainable technology. The successful use of room temperature ionic liquids (RTILs) as solvents has been demonstrated for a wide range of organic reactions including acid catalyzed and transition metal catalyzed transformations.1Previous works2–4 demonstrated that the addition of water to mixtures of RTILs–alcohol markedly increases mutual solubility (co-solvent effect). The ratio of water to alcohol content can be profitably used as a tool for fine-tuning desired situations of total miscibility, partial miscibility, or almost complete phase separation. In contrast, pressurized carbon dioxide (CO2) acts as an anti-solvent5 for the ternary RTIL–ethanol–water mixture. Thus, several switches in the number of phases are possible, allowing for reactions to be carried out in a single phase, leading to increased rates as well as catalyst immobilization, and, at a later stage, easy product separation produced by biphasic conditions.
Although RTILs are generally considered as environmentally acceptable media, the subsequent product recovery using additional traditional volatile and flammable molecular organic solvents would reduce the green nature of processes involving ionic liquids. Thus, supercritical carbon dioxide (scCO2) can alternatively be used to extract reaction products. Recent advances in the implementation of the concept of combining these two completely different solvents have been reported.6
First, Blanchard et al.7 reported that mixtures of scCO2 with an ionic liquid show gas–liquid equilibrium behaviour whereby carbon dioxide can dissolve significantly into the IL-rich liquid phase, but, in practice, no ionic liquid dissolves in the gas phase. The latter corresponds to a quite different situation as compared to the solubility of common organic solvents in scCO2. The same group has also shown8 that organic compounds can be extracted from RTILs using scCO2. Based on these principles, researchers have successfully extracted organic compounds from RTILs.9–11 The type of phase behaviour of RTIL–organics–supercritical carbon dioxide mixtures is, in fact, the same as for systems already observed fifty years ago by Francis,12 who gave an extensive account of the phase behaviour of binary and ternary mixtures containing carbon dioxide. A number of systems that initially are completely miscible and are separated into two liquid phases by addition of CO2 were presented. Moreover, the same effect has been observed by Niehaus et al.13
Knowledge of the phase behaviour of binary and ternary mixtures containing RTILs is absolutely essential for the design of any process involving either chemical reactions or separations in RTIL media. Therefore, it is not surprising that in recent years scientific efforts have been made aiming at understanding the underlying factors that dictate the phase behaviour of RTIL–CO2 mixtures.7,14 In order to understand the nature of the interactions of RTIL–CO2 systems, Kazarian et al.15 reported a molecular-level insight of the state of CO2 dissolved in ionic liquids using in situ ATR-IR spectroscopy. They found evidence for a weak Lewis acid–base interaction between CO2 and the anions of RTILs, concluding that it is the anion that dominates the interactions with CO2.
Despite all these studies, there is a serious lack of solubility data for RTIL–CO2 systems. Therefore, information on ternary mixtures, which is paramount for the design of any technological process, is even scarcer. Few communications on this matter have so far been published. For instance, the T, p, x phase behaviour of the [C4mim][PF6]–methanol–CO2 mixture was reported9,16 as well as phase behaviour of the [C4mim][BF4]–water–CO2 mixture.17 It was also shown that CO2 can extract water from various RTILs without dissolution of the latter in the CO2-rich phase.18,19 Recently, the solubilities of [C4mim][PF6] and [C4mim][BF4] in scCO2 with and without organics were presented.20,21 Our group5 demonstrated that it is possible to separate [C4mim][PF6] from water–ethanol mixtures using CO2. As carbon dioxide is added to [C4mim][PF6]–ethanol–water, a third phase starts to form between the liquid and the gas phase.
Therefore, only [C4mim][PF6] or [C4mim][BF4] in combination with organics and/or water and supercritical CO2, have so far been studied. Since it was proven that these RTILs are potentially harmful2,22 the scientific community has, more recently, refocused its attention to ionic liquids that do not undergo hydrolysis.23 For this reason, in this work, we focus our interest on [C4mim][NTf2].
In this work, the liquid–liquid phase diagram of [C4mim][NTf2]–1-butanol as well as the effect of water added to the same system on the cloud points have been determined using either an already existing24 He–Ne light scattering apparatus or visual determinations. The influence of pressure on the cloud point has been determined for two compositions.
For vapour–liquid equilibria, a new apparatus, which employs a high-pressure stainless steel variable-volume cell for the determination of cloud points, has been designed, built, and tested. Aiming at testing the method and apparatus, the 1-butanol–CO2 system has been chosen as it is already well known and has been studied by several authors.25 Subsequently, the phase behaviour of the quaternary [C4mim][NTf2]–1-butanol–water–CO2 system was studied at different temperatures, pressures, and overall compositions.
Experimental
Materials
[C4mim][NTf2] was prepared according to reported procedures26 and purified at the Department of Chemistry, Universidade Nova de Lisboa. Its purity (estimated at 99.8%) was checked using NMR spectroscopy. Immediately prior to experiments, samples of [C4mim][NTf2] were dried under vacuum (0.1 Pa) at moderate temperature (60 °C) for several days. 1-Butanol (purity better than 99.0%) was purchased from Merck and was further dried with 3 Å molecular sieves. Water was distilled and deionized using a Milli-Q water filtration system from Millipore. All liquid solutions were gravimetrically prepared with an estimated uncertainty of 0.05% for a typical non-diluted weight percentage. Carbon dioxide (CO2) of minimum purity 99.99% was obtained from Air Liquide and was used without further purification.Experimental apparatus
Since this work deals with both liquid–liquid and vapour–liquid equilibria, different apparatus have been employed. In the case of liquid–liquid equilibrium measurements, both a naked eye technique and a laser light scattering technique have been used. In the case of vapour–liquid equilibrium measurements, a new apparatus which employs a high-pressure variable-volume cell has been designed, built and tested (see below).The influence of pressure on the cloud points was obtained by a He–Ne laser light scattering technique using an apparatus which operates up to 50 bar. It consists of a thick-walled Pyrex glass tube cell (internal volume ≈ 1.0 cm3, optical length ≈ 2.6 mm) connected to a pressurization line with a mercury plug acting as a buffer. The apparatus, as well as the methodology used for the determination of phase transitions have recently been described in detail.24 The cloud-point temperature accuracy is typically ±0.01 K in the range 240 < T/K < 400 while for pressure, accuracy is ±0.1 bar up to 50 bar.
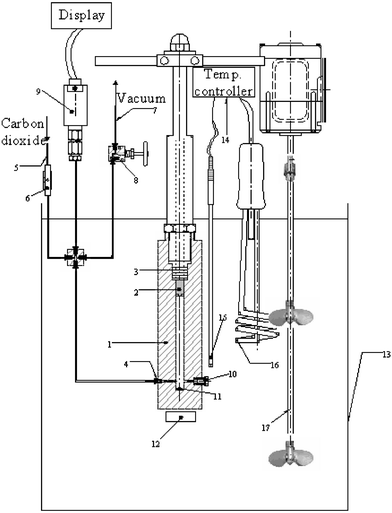 | ||
| Fig. 1 New apparatus for solubility measurements: (1)–cell, (2)–piston, (3)–Teflon O-ring, (4) and (10)–cell inlets, (5)–CO2 inlet, (6)–valve, (7)–vacuum line, (8)–valve, (9)–pressure transducer, (11)–magnetic stirrer, (12)–magnetic plate, (13)–bath, (14)–temp.controller, (15)–RTD probe, (16)–heater, and (17)–stirrer. | ||
The cloud-point stage corresponds to a sudden decrease of the transmitted light. It is defined as the pressure, at a given temperature, at which incipient phase separation occurs. At that moment, the solution loses its transparency and becomes turbid. The optical path was designed in a similar manner to that proposed by Rebelo et al.27 An inlet optical cable guides the light from the laser (He–Ne laser light 632.8 nm) to the cell through sapphire windows. The dimensions of both sapphire windows are 14 mm, and 8 mm, for the diameter and thickness, respectively. One terminal of the outlet cable receives the transmitted light through the cell, the other one is connected to a photodiode. The latter converts light intensity into voltage, enabling one to monitor the intensity of the transmitted light.
Each cloud point is determined in accordance with the following procedure: first, vacuum is applied for at least 2 hours and then the piston of the cell is fixed and a known amount of liquid sample is loaded into the cell. The cell is then filled with CO2 up to the desired initial pressure. The mass of CO2 that entered the cell can be estimated by knowing the available volume for CO2 (the volume of the cell minus that of the loaded liquid content) as well as the initial pressure and temperature. In this way we neglect that some gas may have dissolved before recording pressure. Taking into consideration that the time required to charge the gas is relatively short, the associated error is probably very small. The volume of the cell at several marked positions of the piston was previously calibrated based on the ideal-gas expansion of nitrogen at room temperature. The density of CO2 was calculated using its equation of state.28 The uncertainty in density resulting from the uncertainty in the measurement of pressure (±0.7 bar) varied with the pressure and temperature conditions. The error bar in the calculated composition of the mixture had a maximum value of ±0.04 mole fraction of CO2.
In contrast to the phase behaviour of the CO2–1-butanol mixture, systems of high pressure carbon dioxide and room temperature ionic liquids show two distinct phases, even up to very high pressures.7 Therefore, in the case of CO2–1-butanol and/or water, the objective is to determine the transition from one to two-phase regions, while in the case of the RTIL–1-butanol–water–CO2 systems the aim is to detect the formation of the third phase, which precipitates from the upper CO2-rich phase. Consequently, it is clear why the experimental procedure for detecting cloud points has to be different, mainly with respect to the loading of samples. This is presented in Fig. 2.
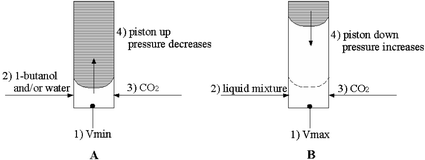 | ||
| Fig. 2 Experimental procedure for (A) CO2–1-butanol and/or water; (B) RTIL–1-butanol–water–CO2. The numbers 1), 2)… represent the chronological order of the experimental operations. | ||
The experimental procedure for CO2–1-butanol and/or water systems uses the following steps (Fig. 2A): the piston is fixed in such a position as to provide the minimum volume of the cell; 1-butanol and/or water as well as CO2 are loaded into the cell. At this stage, the system is in the one-phase region and slow depressurization can take place, as described in the next paragraph. On the contrary, the method used in the case of mixtures which besides other components contain RTILs, consists of the following steps (Fig. 2B): the piston is fixed in a position that ensures the maximum volume of the cell; the liquid sample is placed into the cell and CO2 is introduced up to the desired pressure, all together forming three phases; after closing the cell, the piston is moved down (pressure increases) in order to bring the system to the two-phase region.
The cell is immersed into the controlled-temperature bath and heated up to a chosen temperature. The solution is stirred in order to homogenize the mixture and achieve dissolution. At this point, stirring is stopped and pressure is slowly decreased/increased by moving the piston. This is done until a cloud point is obtained, meaning that the two-phase region is observed. Changes in the pressure and intensity of transmitted light are recorded.
Results and discussion
Liquid–liquid equilibrium of [C4mim][NTf2]–1-butanol–water mixtures
Fig. 3 illustrates the atmospheric pressure phase diagrams of the [C4mim][NTf2]–1-butanol system in terms of IL mass fraction. This system presents upper critical solution temperature (UCST) behaviour, similar to other systems which involve RTILs and alcohols as solvents.2,3,29,30 Our results have been compared with the data reported by Crosthwaite et al.30 (also presented in Fig. 3). To rationalize the data with the aim of permitting interpolation or extrapolation of the phase diagrams and approximate determination of the critical coordinates, Tc; wc, a scaling-type equation,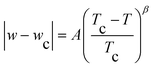 | (1) |
![Atmospheric pressure liquid–liquid equilibrium diagram of ([C4mim][NTf2]–1-butanol), where wt represents mass fraction. This work: ●
(visually detected); *
(laser light scattering technique). ○ Crosthwaite et al.30 The insert shows the same diagram using a different variable—mole fraction of [C4mim][NTf2]. The solid lines represent the fit to experimental data using the scaling-type eqn. (1) while the dashed lines represent extrapolation.](/image/article/2005/GC/b500854a/b500854a-f3.gif) | ||
| Fig. 3 Atmospheric pressure liquid–liquid equilibrium diagram of ([C4mim][NTf2]–1-butanol), where wt represents mass fraction. This work: ● (visually detected); * (laser light scattering technique). ○ Crosthwaite et al.30 The insert shows the same diagram using a different variable—mole fraction of [C4mim][NTf2]. The solid lines represent the fit to experimental data using the scaling-type eqn. (1) while the dashed lines represent extrapolation. | ||
The coordinates of all experimental cloud points of this system, [C4mim][NTf2]–1-butanol and/or water, are reported in Table 1. The influence of pressure on the cloud points (demixing temperatures) has been obtained for two concentrations of the binary [C4mim][NTf2]–1-butanol mixture and one concentration of the ternary [C4mim][NTf2]–1-butanol–water. All these mixtures show a small pressure effect.
| w IL | x H2O | p/bar | T/K | dT/dp × 103/K bar−1 |
|---|---|---|---|---|
| 0.1312 | 0.5319 | 1 | 302 | — |
| 0.1333 | 0.5101 | 1 | 276 | — |
| 0.1527 | 0.2220 | 1 | 254 | — |
| 0.1554 | 0.1646 | 1 | 261 | — |
| 0.1571 | 0.1241 | 1 | 272 | — |
| 0.1596 | 0.0585 | 1 | 283 | — |
| 0.1616 | 0 | 1 | 294 | — |
| 0.2028 | 0 | 1 | 296 | — |
| 2.2 | 297.12 | |||
| 10.7 | 297.09 | |||
| 0.2220 | 0 | 19.9 | 297.06 | −3.37 |
| 30.1 | 297.02 | |||
| 41.4 | 296.99 | |||
| 0.2361 | 0 | 1 | 297 | — |
| 0.2813 | 0 | 1 | 298 | — |
| 0.2937 | 0.8016 | 1 | 355 | — |
| 2.9 | 282.32 | |||
| 12.9 | 282.28 | |||
| 0.2960 | 0.0990 | 21.6 | 282.27 | −3.45 |
| 31.1 | 282.22 | |||
| 40.2 | 282.19 | |||
| 0.2986 | 0.7938 | 1 | 346 | — |
| 0.3017 | 0.7888 | 1 | 340 | — |
| 0.3045 | 0.7840 | 1 | 334 | — |
| 0.3066 | 0.7805 | 1 | 330 | — |
| 0.3084 | 0 | 1 | 299 | — |
| 0.3128 | 0.7693 | 1 | 310 | — |
| 0.3144 | 0.7664 | 1 | 304 | — |
| 0.3336 | 0 | 1 | 299 | — |
| 0.3353 | 0.0452 | 1 | 292 | — |
| 0.3371 | 0.0768 | 1 | 287 | — |
| 0.3390 | 0.1078 | 1 | 282 | — |
| 0.3414 | 0.1431 | 1 | 276 | — |
| 0.3420 | 0.1458 | 1 | 275 | — |
| 0.3623 | 0.6496 | 1 | 259 | — |
| 0.3888 | 0 | 1 | 299 | — |
| 0.4177 | 0.3793 | 1 | 259 | — |
| 0.4197 | 0.3644 | 1 | 260 | — |
| 0.4215 | 0.0000 | 1 | 299 | — |
| 0.4236 | 0.3327 | 1 | 262 | — |
| 0.4287 | 0.2875 | 1 | 264 | — |
| 0.4337 | 0.2377 | 1 | 269 | — |
| 0.4386 | 0.1831 | 1 | 276 | — |
| 0.4407 | 0 | 1 | 299 | — |
| 0.4432 | 0 | 1 | 299 | — |
| 0.4456 | 0.0933 | 1 | 287 | — |
| 0.4473 | 0.0684 | 1 | 290 | — |
| 0.4494 | 0.0378 | 1 | 294 | — |
| 0.4509 | 0.0135 | 1 | 298 | — |
| 0.4517 | 0 | 1 | 299 | — |
| 0.4928 | 0 | 1 | 299 | — |
| 2.0 | 299.09 | |||
| 14.8 | 299.03 | |||
| 0.5107 | 0 | 25.7 | 299.00 | −3.73 |
| 41.3 | 298.95 | |||
| 42.6 | 298.95 | |||
| 0.5259 | 0 | 1 | 299 | — |
| 0.6004 | 0 | 1 | 299 | — |
| 0.7607 | 0 | 1 | 291 | — |
In many ways, the mixture [C4mim][NTf2]–1-butanol and/or water demonstrates behaviour similar to other studied mixtures which involve RTILs, alcohols and/or water. Besides the UCST behaviour and asymmetry of the phase diagram when plotted on a mole fraction basis, there is a significant water–1-butanol co-solvent effect. Similarly to the cases of [C4mim][PF6]–ethanol2,3 and [C4mim][NTf2]–i-butanol3
(Tc
= 303.5 K; wc
= 0.43; β
= 0.257; A
= 1.010) mixtures, there is a sharp drop in the demixing temperature as water is added to the [C4mim][NTf2]–alcohol mixture (Fig. 4). This occurs up to a point (typically 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio of water–alcohol) where further addition of water induces an increase in the demixing temperature. For some concentrations of ternary mixtures a transition from one liquid phase to solid has been observed (stars in Fig. 4).
:1 mole ratio of water–alcohol) where further addition of water induces an increase in the demixing temperature. For some concentrations of ternary mixtures a transition from one liquid phase to solid has been observed (stars in Fig. 4).
![Effect of addition of water on the demixing temperature of [C4mim][NTf2]–1-butanol mixtures: near-critical (■ initial wIL
= 0.4407, ● initial wIL
= 0.4517) and off-critical (▲ initial wIL
= 0.1616); * represents transitions from homogeneous liquid mixture to the solid. The lines are drawn as guides to the data.](/image/article/2005/GC/b500854a/b500854a-f4.gif) | ||
| Fig. 4 Effect of addition of water on the demixing temperature of [C4mim][NTf2]–1-butanol mixtures: near-critical (■ initial wIL = 0.4407, ● initial wIL = 0.4517) and off-critical (▲ initial wIL = 0.1616); * represents transitions from homogeneous liquid mixture to the solid. The lines are drawn as guides to the data. | ||
Although not numerous, the set of cloud-points data taken from Table 1 enables one to construct a semi-quantitative ternary phase diagram (Fig. 5) at 288 K and atmospheric pressure. All experimental data have been interpolated to the temperature of 288 K. Mutual solubilities of (1-butanol–water) at 288 K were taken from Stephenson and Stuart31 while the solubilities of [C4mim][NTf2]–water were used as given by Crosthwaite et al.30 While the system ([C4mim][NTf2]–water) is basically immiscible,30 both ([C4mim][NTf2]–1-butanol) and (1-butanol–water) mixtures show partial miscibility. When all three components are mixed together the ternary system becomes more and more miscible at least up to a certain composition. As mentioned above, the same pattern of behaviour was found in several mixtures of (RTILs–alcohol–water) whereby all combinations tested so far involve RTILs based on the 1-butyl-3-methylimidazolium cation. Ionic liquids do not expand significantly upon addition of carbon dioxide,14f a fact that suggests the existence of free-volumes or void space. This feature may allow water and alcohol to be dissolved together in the IL whilst maintaining the bulk alcohol–water hydrogen-bonding structure.4
![Ternary phase diagram of the ([C4mim][NTf2]–1-butanol–water) system at 288 K and atmospheric pressure (compositions in mole fraction).](/image/article/2005/GC/b500854a/b500854a-f5.gif) | ||
| Fig. 5 Ternary phase diagram of the ([C4mim][NTf2]–1-butanol–water) system at 288 K and atmospheric pressure (compositions in mole fraction). | ||
Vapour–liquid equilibrium of 1-butanol–CO2 mixtures—testing the new high-pressure cell
In order to check the new apparatus for determining cloud points, vapour–liquid equilibrium of an already studied system 1-butanol–CO2 has been determined for three different compositions. Table 2 reports the demixing pressures at several temperatures.| T/K | p/bar |
|---|---|
| x CO2 = 0.442 | |
| 313.15 | 63.9 |
| 323.15 | 71.7 |
| 333.15 | 80.0 |
| x CO2 = 0.598 | |
| 298.15 | 57.2 |
| 313.15 | 75.8 |
| 323.15 | 96.5 |
| 333.15 | 102.7 |
| x CO2 = 0.769 | |
| 313.15 | 77.9 |
| 323.15 | 94.5 |
| 333.15 | 107.6 |
Fig. 6 represents comparisons made with literature data25 on the same system. These literature data are interpolated with respect to composition. In view of the fact that all authors have used quite different methods for the detection of cloud points (static-analytic, flow-analytic, and high-pressure densitometer methods) there is reasonable agreement between our experimental data and those found in the literature.
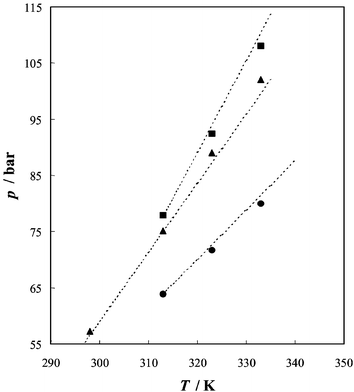 | ||
| Fig. 6 Experimental p–T cloud points for the system (1-butanol–CO2), for three different concentrations: ● xCO2 = 0.442; ▲ xCO2 = 0.599; and ■ xCO2 = 0.769. xCO2 is the overall mole fraction of carbon dioxide. The lines represent average values of results given in ref. 25. Above the line the system is in the one-phase region, and below it in the two-phase domain. | ||
Vapour–liquid equilibrium of the quaternary ([C4mim][NTf2]–1-butanol–water–CO2) mixture
For these VLE experiments, the new high-pressure cell described in the Experimental section was used. Phase transitions were observed either visually (through the sapphire windows) or using the light scattering method. Cloud-point data for all mixtures are presented in Table 3.| No | Estimated composition | Cloud-point data | |||
|---|---|---|---|---|---|
| x IL | x 1-butanol | x CO2 calc. | T/K | p/bar | |
| 1 | 0.046 | 0.257 | 0.628 | 302.25 | 68.5 |
| 313.15 | 96.5 | ||||
| 323.15 | 137.9 | ||||
| 333.15 | 158.6 | ||||
| 2 | 0.025 | 0.295 | 0.587 | 295.6 | 213.7 |
| 3 | 0.016 | 0.087 | 0.868 | 296.70 | 218.4 |
| 298.65 | 417.1 | ||||
| 298.75 | 448.2 | ||||
| 299.13 | 489.5 | ||||
| 299.35 | 517.1 | ||||
| 300.67 | 558.5 | ||||
| 301.10 | 596.4 | ||||
| 301.44 | 701.5 | ||||
| 301.60 | 723.9 | ||||
| 313.15 | 965.3 | ||||
| 4 | 0.016 | 0.078 | 0.859 | 296.15 | 806.7 |
| 5 | 0 | 0.363 | 0.526 | 297.15 | 59.6 |
| 6 | 0 | 0.343 | 0.509 | 297.15 | 641.2 |
| 7 | 0 | 0.352 | 0.517 | 297.15 | 339.6 |
In order to compare the behaviour of mixtures with and without the ionic liquid, the study of the ternary mixture (1-butanol–water–CO2) was included in this work. This was done to overcome the lack of vapour–liquid equilibrium data on this ternary system at about room temperature.
Only in the case of the quaternary mixture No 2 (see Table 3) it was possible to obtain a visual observation of transitions from the two-phase to the three-phase regions. In the other cases, the limited visual field (vacant diameters of sapphire windows are approximately 6 mm, while the length of the cell varies from 42 to 193 mm) prevented similar observations. Fig. 7 shows the development of phases as pressure changes for mixture No 2 at room temperature (295.6 K). In Fig. 7 (I) the system is in the two-phase region; the lower phase is the RTIL-rich phase and the upper one is the CO2-rich phase. By moving the piston backwards, pressure was slowly decreased up to the moment when the first sign of turbidity was observed (Fig. 7 (II)). This cloud-point pressure was recorded. From this point on, if we allow the system to stabilize (for about 10 minutes), it will turn into a transparent three-phase system with clear menisci, as in Fig. 7 (IIa). A further decrease in pressure provoked the reappearance of turbidity (Fig. 7 (III)) and, again, waiting for several minutes resulted in three clear phases as shown in Fig. 7 (IIIa). Note that the volume of the intermediate phase has increased (Fig. 7 (IV) and (V)). For all other mixtures, cloud points were observed using the light scattering technique as described in the Experimental section.
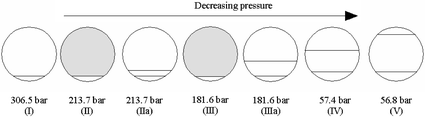 | ||
| Fig. 7 Phase changes as pressure decreases at room temperature for mixture No 2 in Table 3 (see text). | ||
In order to compare the behaviour of the quaternary ([C4mim][NTf2]–1-butanol–water–CO2) and ternary (1-butanol–water–CO2) systems Fig. 8 has been drawn (pressure of phase changes, p, as a function of the overall mole fraction of water with respect to butanol–water, xwater in 1-butanol). In the case of mixtures 1 and 3, an extrapolation of pressure of the phase change was performed to T = 297.15 K (the temperature at which the experiments for the ternary mixture were carried out). The dashed line in Fig. 8 reflects the demixing pressure of the ternary (1-butanol–water–CO2) system, whereby the system is in the one-phase region above the line and in the two-phase region below it. The full line in Fig. 8 represents the demixing pressure of the quaternary ([C4mim][NTf2]–1-butanol–water–CO2) system, whereby above the line the system is in the two-phase region and below it in the three-phase region. An interesting observation follows from Fig. 8: the pressure of the transition is strongly dependent on the xwater in 1-butanol. Both the ternary and quaternary mixtures show similar trends—along with an increase of water fraction, the demixing pressure increases and the systems become increasingly immiscible. Note that there is a difference between the slopes for the quaternary and ternary mixtures (see Fig. 8).
![Demixing pressure p of [C4mim][NTf2]–1-butanol–water–CO2
(●) and 1-butanol–water–CO2
(○) as a function of the overall mole fraction of water with respect to 1-butanol at 297.15 K. All points correspond to data reported in Table 3. The lines (full line for the quaternary mixture and dashed line for the ternary one) are drawn as guides to the data.](/image/article/2005/GC/b500854a/b500854a-f8.gif) | ||
| Fig. 8 Demixing pressure p of [C4mim][NTf2]–1-butanol–water–CO2 (●) and 1-butanol–water–CO2 (○) as a function of the overall mole fraction of water with respect to 1-butanol at 297.15 K. All points correspond to data reported in Table 3. The lines (full line for the quaternary mixture and dashed line for the ternary one) are drawn as guides to the data. | ||
It remains to be seen whether the two lines from Fig. 8 would coincide or not if the mole fraction of water with respect to butanol–water, xwater in 1-butanol in the upper phase (CO2-rich phase) could be used instead of overall values. In order to undoubtedly clarify and substantiate the role of the ionic liquid on the phase behaviour, another analytical method that allows for the sampling of phases must be used. Investigations to elucidate this point are currently under way in our laboratories.
Conclusion
Similar to other systems which involve RTILs and alcohols as solvents, the [C4mim][NTf2]–1-butanol system presents upper critical solution temperature (UCST) behaviour. For the same system, the influence of hydrostatic pressure on cloud points is rather modest. Although the [C4mim][NTf2]–1-butanol system shows partial mutual solubility and the [C4mim][NTf2]–water system is almost totally immiscible, the mutual solubility of the ternary [C4mim][NTf2]–1-butanol–water system is enhanced. Thus, water shows a strong co-solvent effect with alcohols.A new apparatus that employs a high-pressure variable-volume cell, based on the synthetic method, was built, tested and used in order to determine the vapour–liquid equilibrium of ternary (1-butanol–water–CO2) and quaternary ([C4mim][NTf2]–1-butanol–water–CO2) systems. The results for both mixtures suggest that the demixing pressure is strongly controlled by the water concentration.
Acknowledgements
This work was financially supported by Fundação para a Ciência e Tecnologia, (FC&T), Portugal, under contract POCTI/EQU/35437/00. V. N.-V. is grateful to FC&T for a PhD grant. We thank Prof. Carlos A. M. Afonso, Department of Chemistry, Universidade Nova de Lisboa, Portugal, for the synthesis of [C4mim][NTf2].References
- (a) H. Olivier-Bourbigou and L. J. Magna, J. Mol. Catal. A: Chem., 2002, 182, 419 CrossRef; (b) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS; (c) J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214, 11 CrossRef CAS; (d) J. D. Holbrey and K. R. Seddon, Clean Prod. Processes, 1999, 1, 223 CrossRef; (e) M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391 CrossRef CAS; (f) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3773 CrossRef; (g) J. Dupont, C. S. Consorti and J. J. Spencer, Braz. Chem. Soc., 2000, 11, 337 Search PubMed; (h) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS; (i) D. B. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157 CrossRef CAS.
- V. Najdanovic-Visak, J. M. S. S. Esperança, L. P. N. Rebelo, M. Nunes da Ponte, H. J. R. Guedes, K. R. Seddon and J. Szydlowski, Phys. Chem. Chem. Phys., 2002, 4, 1701 RSC.
- V. Najdanovic-Visak, J. M. S. S. Esperança, L. P. N. Rebelo, M. Nunes da Ponte, H. J. R. Guedes, K. R. Seddon, H. C. de Sousa and J. Szydlowski, J. Phys. Chem. B, 2003, 107, 12797 CrossRef CAS.
- R. P. Swatloski, A. E. Visser, W. M. Reichhert, G. A. Broker, L. M. Farina, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2001, 2070 RSC.
- V. Najdanovic-Visak, A. Serbanovic, J. M. S. S. Esperanca, H. J. R. Guedes, L. P. N. Rebelo and M. N. da Ponte, ChemPhysChem, 2003, 4, 520 CrossRef CAS.
- S. V. Dzyuba and R. A. Bartsch, Angew. Chem., Int. Ed., 2003, 42, 148 CrossRef CAS.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28 CrossRef.
- L. A. Blanchard and J. F. Brennecke, Ind. Eng. Chem. Res., 2001, 40, 287 CrossRef CAS.
- A. M. Scurto, S. N. V. K. Aki and J. F. Brennecke, J. Am. Chem. Soc., 2002, 124, 10276 CrossRef CAS.
- (a) F. Liu, M. B. Abrams, R. T. Baker and W. Tumas, Chem. Commun., 2001, 433 RSC; (b) R. A. Brown, P. Pollet, E. McKoon, C. A. Eckert, C. L. Liotta and P. G. J. Jessop, J. Am. Chem. Soc., 2001, 123, 1254 CrossRef CAS; (c) M. F. Sellin, P. B. Webb and D. J. Cole-Hamilton, Chem. Commun., 2001, 781 RSC.
- L. C. Branco, A. Serbanovic, M. Nunes da Ponte and C. A. M. Afonso, Chem. Commun., 2005, 107 RSC.
- A. W. Francis, J. Phys. Chem., 1954, 58, 1099 CrossRef CAS.
- (a) D. E. Niehaus and R. M. Wightman, Anal. Chem., 1991, 63, 1728 CrossRef CAS; (b) D. Niehaus, M. Philips, A. Michael and R. M. Wightman, J. Phys. Chem., 1989, 93, 6232 CrossRef CAS.
- (a) L. A. Blanchard, Z. Gu and J. F. Brennecke, J. Phys. Chem. B, 2001, 105, 2437 CrossRef CAS; (b) A. P. S. Kamps, D. Tuma, J. Z. Xia and G. J. Maurer, J. Chem. Eng. Data, 2003, 48, 746 CrossRef CAS; (c) A. Shariati and C. J. J. Peters, J. Supercrit. Fluids, 2004, 29, 43 CrossRef CAS; (d) A. Shariati and C. J. J. Peters, J. Supercrit. Fluids, 2004, 30, 139 CrossRef CAS; (e) J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315 CrossRef CAS; (f) S. N. V. K. Aki, B. R. Mellein, E. M. Saurer and J. F. Brennecke, J. Phys. Chem. B, 2004, 108, 20355 CrossRef CAS; (g) M. Costantini, V. A. Toussaint, A. Shariati, C. J. Peters and I. Kikic, J. Chem. Eng. Data, 2005, 50, 52 CrossRef CAS; (h) M. C. Kroon, A. Shariati, M. Costantini, J. Van Spronsen, G.-J. Witkamp, R. A. Sheldon and C. J. Peters, J. Chem. Eng. Data, 2005, 50, 173 CrossRef CAS.
- S. G. Kazarian, B. J. Briscoe and T. Welton, Chem. Commun., 2000, 2047 RSC.
- Z. M. Liu, W. Z. Wu, B. X. Han, Z. X. Dong, G. Y. Zhao, J. Q. Wang, T. Jiang and G. Y. Yang, Chem.–Eur. J., 2003, 9, 3897 CrossRef CAS.
- Z. Zhang, W. Wu, Z. Liu, B. Han, H. Gao and T. Jiang, Phys. Chem. Chem. Phys., 2004, 6, 2352 RSC.
- A. M. Scurto, S. N. V. K. Aki and J. F. Brennecke, Chem. Commun., 2003, 572 RSC.
- Z. Zhang, W. Wu, H. Gao, B. Han, B. Wang and Y. Huang, Phys. Chem. Chem. Phys., 2004, 6, 5051 RSC.
- W. Z. Wu, J. M. Zhang, B. X. Han, J. W. Chen, Z. M. Liu, T. Jiang, J. He and W. J. Li, Chem. Commun., 2003, 1412 RSC.
- W. Z. Wu, W. J. Li, B. X. Han, T. Jiang, D. Shen, Z. F. Zhang, D. H. Sun and B. Wang, J. Chem. Eng. Data, 2004, 49, 1597 CrossRef CAS.
- (a) A. E. Visser, R. P. Swatloski, W. M. Reichert, S. T. Griffin and R. D. Rogers, Ind. Eng. Chem. Res., 2000, 39, 3596 CrossRef CAS; (b) R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361 RSC.
- J. F. Brennecke, Chem. Eng. News, 2001, 79, 86.
- H. C. Sousa and L. P. N. Rebelo, J. Chem. Thermodyn., 2000, 32, 355 CrossRef CAS.
- (a) D. W. Jennings, R. J. Lee and A. S. Teja, J. Chem. Eng. Data, 1991, 36, 303 CrossRef CAS; (b) C. M. J. Chang, Fluid Phase Equilib., 1992, 74, 235 CrossRef CAS; (c) K. Ishihara, A. Tsukajima, H. Tanaka, M. Kato, T. Sako, M. Sato and T. Hakuta, J. Chem. Eng. Data, 1996, 41, 324 CrossRef CAS; (d) T. Hiaki, H. Miyagi, T. Tsuji and M. Hongo, J. Supercrit. Fluids, 1998, 13, 23 CrossRef CAS; (e) G. Silva-Oliver and L. A. Galicia-Luna, Fluid Phase Equilib., 2001, 182, 145 CrossRef CAS; (f) H. I. Chen, H. Y. Chang and P. H. Chen, J. Chem. Eng. Data, 2002, 47, 776 CrossRef CAS.
- (a) C. M. Gordon, J. D. Holbrey, R. Kennedy and K. R. Seddon, J. Mater. Chem., 1998, 8, 2627 RSC; (b) P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS; (c) T. Kitazume, F. Zulfiqar and G. Tanaka, Green Chem., 2000, 2, 133 RSC; (d) A. E. Visser, R. P. Swatloski and R. D. Rogers, Green Chem., 2000, 2, 1 RSC; (e) J. Dupont, C. S. Consorti, P. A. Z. Suarez, R. F. de Souza, S. L. Fulmer, D. P. Richardson, T. E. Smith and S. Wolff, Org. Synth., 2002, 79, 236.
- L. P. N. Rebelo, Z. P. Visak, H. C. de Sousa, J. Szydlowski, R. Gomes de Azevedo, A. M. Ramos, V. Najdanovic-Visak, M. Nunes da Ponte and J. Klein, Macromolecules, 2002, 35, 1887 CrossRef CAS.
- R. Span and W. Wagner, J. Phys. Chem. Ref. Data, 1996, 25, 1509 CAS.
- (a) M. Wagner, O. Stanga and W. Schröer, Phys. Chem. Chem. Phys., 2003, 5, 3943 RSC; (b) C.-T. Wu, K. N. Marsh, A. V. Deev and J. A. Boxall, J. Chem. Eng. Data, 2003, 48, 486 CrossRef CAS; (c) K. N. Marsh, A. Deev, A. C. T. Wu, E. Tran and A. Klamt, Korean J. Chem. Eng., 2002, 19, 357 Search PubMed; (d) A. Heintz, J. K. Lehmann and C. Wertz, J. Chem. Eng. Data, 2003, 48, 472 CrossRef CAS.
- J. M. Crosthwaite, S. N. V. K. Aki, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2004, 108, 5113 CrossRef CAS.
- R. Stephenson and S. Stuart, J. Chem. Eng. Data, 1986, 31, 56 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |