Preparation of biotin derivatives by catalytic oxidative carbonylation of diamines
Yue
Zhang
,
Kara
Forinash
,
Courtney R.
Phillips
and
Lisa
McElwee-White
*
Department of Chemistry and Center for Catalysis, University of Florida, Gainesville, FL 32611-7200. E-mail: lmwhite@chem.ufl.edu
First published on 12th April 2005
Abstract
Biotin methyl ester and related heterocyclic ureas were synthesized by W(CO)6-catalyzed oxidative carbonylation of the corresponding diamines. This procedure provides an alternative to the use of phosgene and phosgene derivatives for formation of the urea moiety.
Introduction
Biotin (1b), also known as Vitamin H, is a cofactor for biochemical carboxylations. It is manufactured on a large scale as a feed additive for poultry and swine. In addition to its commercial value, biotin has been of great interest as a synthetic target. More than 40 total and formal syntheses have appeared since 1943, when the first racemic synthesis was reported.1 Many of these approaches involve conversion of a diaminotetrahydrothiophene moiety into the bicyclic urea core. Two general means of achieving this transformation are used repeatedly: 1) reaction of a tetrahydrothiophene diamine with phosgene2–4 and 2) hydrolysis of a bis(carbamate) derivative of the diamine so that generation of the first free amine triggers ring closure by nucleophilic attack on the remaining carbamate.5,6The strategies for biotin preparation reflect the most common approach to the formation of ureas, nucleophilic reaction of amines with phosgene or phosgene derivatives.7 Concerns about phosgene8 and the value of multiple methodologies for a given chemical transformation have led us to explore catalytic methods for the conversion of amines to ureas. We previously reported the catalytic oxidative carbonylation of primary and secondary amines to ureas using W(CO)6 as catalyst, I2 as the oxidant and CO as the carbonyl source.9–14 Tolerance of functional groups is broad and five- to eight-membered cyclic ureas are accessible from the corresponding diamines.11,13,14 The first application of this methodology to a functionalized target was formation of the core structure of the HIV protease inhibitors DMP 323 and DMP 450.15 We now report preparation of biotin methyl ester and related bicyclic systems by catalytic carbonylation of the corresponding diamines.
Results and discussion
Initial attempts to synthesize biotin by oxidative carbonylation of the ammonium salt 1a under the standard conditions were unsuccessful (eqn. (1)). Varying the solvent, temperature, reaction time and the amount of catalyst did not result in the formation of biotin. Since 1a was recovered as a solid at the end of the reaction, we concluded that the low solubility of 1a in CH2Cl2 was probably responsible for the failure to obtain the urea.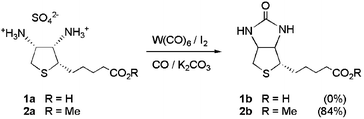 | (1) |
Based on previously reported functional group compatibility studies,13 the carboxylic acid moiety of 1a had been expected to present a problem in the direct synthesis of biotin. Thus, we carried out model studies using 6-aminocaproic acid as the substrate. The results were similar to those for compound 1a. Only trace amounts of the corresponding urea were detected in 13C NMR spectra of the reaction mixtures and approximately 90% of the starting material could be recovered. In contrast, when the methyl ester of 6-aminocaproic acid (3a) was carbonylated, 70% yield of the corresponding urea was obtained in 4 h at room temperature (eqn. (2)).
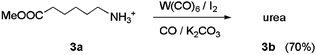 | (2) |
To obtain a biotin precursor that was soluble in organic solvents, carboxylic acid 1a was converted to its methyl ester 2a using SOCl2 in methanol.16 Carbonylation of 2a to biotin methyl ester (2b) at room temperature was complete in around 8 h. When the temperature was raised to 50 °C, the reaction time was decreased to 1 h and the yield of 2b was improved to 84%. Longer reaction times resulted in decomposition of the product.
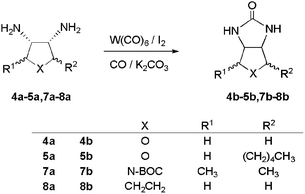
Related diamines such as the tetrahydrofuran derivatives 4a and 5a were also studied based on the promising results obtained from biotin methyl ester (Table 1, eqn. (3)). Unfortunately, carbonylation of 4a afforded “chainless oxybiotin” 4b in trace amounts. Attempts to optimize the reaction conditions by varying the amount of catalyst, or reaction time and temperature did not produce higher yields of 4b. The low solubility of diamine 4a in methylene chloride was clearly problematic and the solubility of urea 4b in water prevented use of the usual workup, which involves extraction. An alternative workup using ion exchange resins did not provide an improvement. Note that reaction of 4a with the phosgene derivative 1,1′-carbonyldiimidazole (CDI) also afforded 4b in low yield.
 | (3) |
| Amine | Urea | W(CO)6/I2 yielda | CDI yield |
|---|---|---|---|
| a Unless otherwise noted, all reactions were carried out in CH2Cl2 (40 mL) at room temperature under 80 atm of CO. Diamine (1 mmol), W(CO)6 (4 mol%), K2CO3 (3 mmol), I2 (1 mmol). b Yield based on diamine consumed (47%). c Yield based on diamine consumed (70%). d Conditions as above except pyridine (1.0 mL) was used as the base and the reaction was carried out at 50 °C. e Reaction not attempted. | |||
| 4a | 4b | Trace | 20% |
| 5a | 5b | 47% | 67% |
| 7a | 7b | 46% | 37% |
| 8a | 8b | 57%b | 56%c |
| 9a | 9b | 4%d | —e |
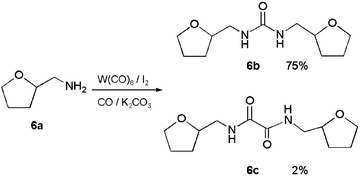
Since an alternative explanation of the low yields from 4a involved reactivity of tetrahydrofuran rings under the reaction conditions, the model compound tetrahydrofurfurylamine (6a) was subjected to catalytic carbonylation to test the stability of the ring (eqn. (4)). The yields of urea 6b were dependent upon the amount of K2CO3 in the reaction mixtures and reached a maximum value of 75% when 1.0 equiv. of K2CO3 was used. Oxamide 6c was obtained in 2% yield as a byproduct of this reaction.
 | (4) |
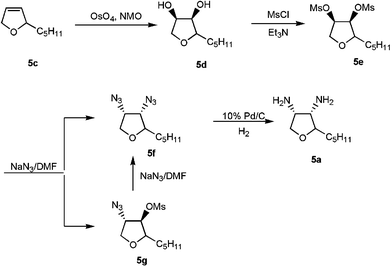
In order to test a tetrahydrofuran derivative with higher solubility in organic solvents, diamine 5a was prepared from the corresponding diol via the dimesylate and diazide (Scheme 1) and subjected to the carbonylation reaction under 85 atm of CO at room temperature for 30 h. The pure urea 5b was obtained in 47% yield as a white solid. Due to the sensitivity of diamine 5a, attempts to recover remaining starting material from the reaction mixtures failed.
 | ||
| Scheme 1 Synthetic route to 5a. | ||
Nitrogen-containing heterocycles were investigated by carbonylation of N-tert-butoxycarbonyl-2,5-dimethyl-3,4-diaminopyrrolidine 7a at room temperature for 8 h. Urea 7b was isolated in 46% yield after chromatography on silica gel. Longer reaction times and higher temperatures caused the yield of urea to decrease due to the formation of decomposition products. In comparison, conversion of 7a to 7b using the stoichiometric carbonylation reagent CDI afforded the urea in only 37% yield.
The carbocyclic substrates trans-1,2-diaminocyclohexane (8a) and cis-1,2-diaminocyclopentane (9a) were also subjected to the carbonylation conditions. The conversion of 8a to the corresponding urea 8b proceeded in moderate yield. Note that 8a had previously been reported to be a problem substrate for conversion to the urea with phosgene.17 Although the yield of 8b from catalytic carbonylation is modest, it is comparable to the yield from 8a and CDI. Carbonylation of cyclopentane derivative 9a was unsuccessful due to its instability and the insolubility of its diammonium salt in methylene chloride. Very low yields of 9b were obtained after chromatography and characterization by 1H NMR and HRMS.
In summary, catalytic oxidative carbonylation has been applied to the preparation of biotin methyl ester and related bicyclic systems as an alternative to phosgene or phosgene derivatives. Yields of the ureas are moderate to good and depend on the solubility of the diamine and urea in methylene chloride. Further applications of this methodology to the synthesis of complex targets are still under investigation.
Experimental
General
Solvents were purged with nitrogen and dried by passing the degassed solvent through a column packed with activated alumina.18 All other chemicals were purchased in reagent grade and used with no further purification unless stated otherwise. W(CO)6 was purified by chromatography on silica gel using hexane as eluent.5-(3,4-Diammoniumtetrahydrothiophen-2-yl)pentanoic acid, sulfate salt (1a)
The hydrolysis product of biotin was prepared in 78% yield as previously described.191H NMR (D2O): δ 4.23 (m, 2H), 3.86 (m, 1H), 3.36 (m, 1H), 3.01 (m, 1H), 2.39 (t, 2H), 1.86 (m, 1H), 1.62–1.35 (m, 5H); 13C NMR (D2O): δ 179.8, 57.2, 54.9, 47.6, 34.5, 30.4, 28.8, 28.1, 24.8; Anal. Calcd for C9H20N2O6S2: C 34.17, H 6.37, N 8.85; found: C 34.02, H 6.31, N 8.51.5-(3,4-Diammoniumtetrahydrothiophen-2-yl)pentanoic acid methyl ester, sulfate salt (2a)
MeOH (2 mL) was cooled in an ice–salt bath. Thionyl chloride (0.04 mL, 0.6 mmol) was added dropwise with stirring, followed by the diaminocarboxylic acid sulfate 1a (54 mg, 0.17 mmol). The resulting mixture was allowed to warm to room temperature and stirred for 3 days. The clear solution was then concentrated under vacuum to afford an off-white solid in quantitative yield. The product was identified by comparison to literature data.20Biotin methyl ester (2b)
The crude diamine 2a (82.5 mg, 0.25 mmol) was dissolved in 3 mL of H2O, neutralized to pH = 5 by adding 1 N KOH solution and diluted to a total of 5 mL. The solution was transferred to a glass-lined 300 mL Parr high pressure vessel, followed by addition of W(CO)6 (4 mg, 0.01 mmol), K2CO3 (138 mg, 1.00 mmol), I2 (63.5 mg, 0.250 mmol) and 15 mL of CH2Cl2. The carbonylation reaction was run at 50 °C for 70 min under 85 atm of CO. After the pressure was released, the aqueous solution was separated from the CH2Cl2 layer and extracted with CH2Cl2. The organic layers were combined, dried over MgSO4 and the solvent removed. The crude pale brown solid was rinsed with hexane to afford 54 mg (84% yield) of pure 2b. The product was identified by comparison to literature data.212-Pentyl-cis-3,4-dihydroxytetrahydrofuran (5d)
2-Pentyl-2,5-dihydrofuran (5c)22 (0.589 g, 4.20 mmol), 4-methylmorpholine N-oxide (2.95 g, 25.2 mmol) and OsO4 (4 wt% aq. solution, 1.59 mL, 0.252 mmol) were added to 16 mL of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 CH2Cl2∶THF and stirred at room temperature for 14 h. Then an aqueous solution of Na2S2O5
(20 mL) was added. The mixture was stirred for 1 h and the two layers were then separated. The aqueous layer was extracted with CH2Cl2 three times. The combined organic layers were washed with brine and dried over MgSO4. After removal of solvent, a dark brown residue was obtained. Chromatography on silica gel (1∶1 hexane∶EtOAc) afforded the pure 2-pentyl-cis-3,4-dihydroxytetrahydrofuran (5d) as a mixture of isomers in 59% yield. 1H NMR (CDCl3): δ 4.21 (m, 1H), 4.10 (m, 1H), 3.78–3.62 (m, 3H), 3.54 (d, 1H), 3.41 (d, 1H), 1.67–1.30 (m, 8H), 0.89 (t, 3H); 13C NMR (CDCl3): δ 82.8, 76.1, 72.7, 71.2, 33.5, 32.1, 25.7, 22.8, 14.2; Anal. Calcd for C9H18O3: C 62.04, H 10.41; found: C 62.10, H 10.61.
∶1 CH2Cl2∶THF and stirred at room temperature for 14 h. Then an aqueous solution of Na2S2O5
(20 mL) was added. The mixture was stirred for 1 h and the two layers were then separated. The aqueous layer was extracted with CH2Cl2 three times. The combined organic layers were washed with brine and dried over MgSO4. After removal of solvent, a dark brown residue was obtained. Chromatography on silica gel (1∶1 hexane∶EtOAc) afforded the pure 2-pentyl-cis-3,4-dihydroxytetrahydrofuran (5d) as a mixture of isomers in 59% yield. 1H NMR (CDCl3): δ 4.21 (m, 1H), 4.10 (m, 1H), 3.78–3.62 (m, 3H), 3.54 (d, 1H), 3.41 (d, 1H), 1.67–1.30 (m, 8H), 0.89 (t, 3H); 13C NMR (CDCl3): δ 82.8, 76.1, 72.7, 71.2, 33.5, 32.1, 25.7, 22.8, 14.2; Anal. Calcd for C9H18O3: C 62.04, H 10.41; found: C 62.10, H 10.61.
2-Pentyl-cis-3,4-di-O-methylsulfonyltetrahydrofuran (5e)
2-Pentyl-cis-3,4-dihydroxytetrahydrofuran (5d) (0.174 g, 1.00 mmol), Et3N (0.313 g, 3.09 mmol) and methylsulfonyl chloride (0.252 g, 2.20 mmol) were added to 10 mL of dry CH2Cl2 and stirred in an ice–acetone cold bath (−10 °C) for 1 h. Then the reaction mixture was diluted with CH2Cl2 and washed with 0.12 N HCl, saturated NaHCO3, and H2O. The solution was dried over MgSO4 and the crude product was obtained by evaporation of the solvent. Flash chromatography on silica gel (2∶1 hexane∶EtOAc) afforded a white solid as a mixture of isomers in 97% yield. 1H NMR (CDCl3): δ 5.16 (m, 1H), 4.68 (m, 1H), 4.25 (m, 1H), 4.00 (m, 2H), 3.13 (d, 6H), 1.75–1.30 (m, 8H), 0.89 (t, 3H); 13C NMR (CDCl3): δ 79.9, 78.9, 76.4, 70.6, 38.8, 32.6, 31.8, 25.2, 22.7, 14.2; Anal. Calcd for C11H22O7S2: C 39.98, H 6.71; found: C 40.25, H 6.74.
2-Pentyl-cis-3,4-diazidotetrahydrofuran (5f)
To 25 mL of dry DMF, 2-pentyl-cis-3,4-di-O-methylsulfonyltetrahydrofuran (5e) (0.783 g, 2.37 mmol) and NaN3 (2.16 g, 33.2 mmol) were added and heated at 150 °C for 24 h. After the mixture was cooled to room temperature, water was added to dissolve the pale brown solid. This solution was then extracted three times with 1∶1 Et2O∶hexane. The combined organic layers were dried over MgSO4 and the solvent was removed under vacuum. The pure diazide (5f) was obtained as a single diastereomer by chromatography on silica gel (8∶1 hexane∶EtOAc) in 46% yield as a colorless oil. Further elution with 4∶1 hexane∶EtOAc afforded the monosubstituted azide product 5g
(0.138 g, 21%). Compound 5g was dissolved in DMF (15 mL), and NaN3
(0.259 g, 4.0 mmol) was added to the solution. The mixture was stirred at 145 °C for 21 h. After the workup as described above and chromatography on silica gel, an additional amount of pure 5f
(0.031 g) was obtained. The total yield of 5f was 52%. 1H NMR (CDCl3): δ 4.24 (m, 1H), 4.01 (m, 2H), 3.88 (m, 1H), 3.80 (m, 1H), 1.73–1.30 (m, 8H), 0.90 (t, 3H); 13C NMR (CDCl3): δ 81.5, 68.7, 65.6, 63.2, 38.8, 31.9, 30.2, 25.9, 22.7, 14.2; Anal. Calcd for C9H16N6O: C 48.20, H 7.19, N 37.47; found: C 48.40, H 7.39, N 37.73.
2-Pentyl-cis-3,4-diaminotetrahydrofuran (5a)
2-Pentyl-cis-3,4-diazidotetrahydrofuran (5f) (0.244 g, 1.09 mmol) and 40 mg of 10% Pd/C were added to 6 mL of MeOH. The reaction mixture was degassed and refilled with H2 at 1 atm and then stirred at room temperature for 18 h until no starting material was evident by TLC. The mixture was filtered through Celite and solvent was removed to yield the crude product. Pure 5a was obtained as a single diastereomer by chromatography on silica gel (10% MeOH in CH2Cl2 with 1% Et3N) as a colorless oil in 82% yield. 1H NMR (CDCl3): δ 3.97 (t, 1H), 3.84 (m, 1H), 3.61 (m, 1H), 3.40 (t, 1H), 3.11 (m, 1H), 1.59–1.31 (m, 8H), 0.89 (t, 3H); 13C NMR (CDCl3): δ 82.4, 72.2, 55.9, 55.3, 32.2, 30.2, 26.1, 22.8, 14.2; Anal. Calcd for C9H20N2O: C 62.75, H 11.70, N 16.26; found: C 62.39, H 12.05, N 15.59.4-Pentyltetrahydrofuro[3,4-d]imidazol-2-one (5b)
To a glass lined 300 mL Parr high pressure vessel were added 2-pentyl-cis-3,4-diaminotetrahydrofuran (5a) (220 mg, 1.28 mmol), W(CO)6 (18 mg, 0.051 mmol), K2CO3 (530 mg, 3.84 mmol), I2 (325 mg, 1.28 mmol) and 45 mL of CH2Cl2. The reaction was run under 85 atm of CO at room temperature for 30 h. After the pressure was released, the solid was collected by filtration and washed with chloroform. The combined organic solutions were evaporated to dryness to afford a brown solid. The pure product (118 mg) was obtained as a single diastereomer by chromatography on silica gel (20∶1 EtOAc∶MeOH) as a white solid in 47% yield. mp: 105–107 °C; IR (CH2Cl2): νCO 1716 cm−1; 1H NMR (CDCl3): δ 5.55 (s, br, 1H), 5.33 (s, br, 1H), 4.37 (m, 1H), 4.20 (m, 1H), 3.90 (m, 2H), 3.54 (m, 1H), 3.45 (m, 1H), 1.72–1.30 (m, 8H), 0.89 (t, 3H); 13C NMR (CDCl3): δ 163.3, 83.1, 74.6, 59.3, 57.9, 32.0, 29.0, 26.2, 22.8, 14.2; Anal. Calcd for C10H18N2O2: C 60.58, H 9.15, N 14.13; found: C 60.85, H 9.30, N 13.74.
4-Pentyltetrahydrofuro[3,4-d]imidazol-2-one (5b) by reaction with CDI
2-Pentyl-cis-3,4-diaminotetrahydrofuran (5a) (0.183 g, 1.06 mmol) and 0.170 g (1.05 mmol) of 1,1′-carbonyldiimidazole (CDI) were added to 40 mL of dry THF and stirred at room temperature for 21 h. After the solvent was evaporated, the white residue was dissolved in CHCl3 and washed with 0.12 N HCl, followed by brine. The solution was then dried over MgSO4. The crude product was purified by chromatography on silica gel (20∶1 EtOAc∶MeOH). The pure urea was obtained in 67% yield as a white solid and identified by comparison to an authentic sample prepared as above.
N-tert-Butoxycarbonyl-2,5-dimethylpyrroline (7d)
To 5 mL MeOH were added 2,5-dimethylpyrroline (0.118 mL, 0.996 mmol), di-tert-butyl dicarbonate (0.24 g, 1.1 mmol) and NaHCO3 (250 mg). This mixture was subjected to ultrasonic irradiation for 4 h.23 Excess NaHCO3 was removed by filtration and the MeOH was evaporated under vacuum. The residue was redissolved in Et2O and a second filtration was carried out. Evaporation of Et2O afforded the pure product in 96% yield. The crude product was used for the next step without further purification. 1H NMR (CDCl3): δ 5.59 (m, 2H), 4.58–4.48 (m, 2H), 1.48 (s, 9H), 1.33 (d, 3H), 1.27 (d, 3H); 13C NMR (CDCl3): δ 153.9, 130.6, 130.3, 79.2, 60.3, 60.0, 28.8, 21.1, 19.7.N-tert-Butoxycarbonyl-2,5-dimethyl-3,4-dihydroxypyrrolidine (7e)
The oxidation product of N-tert-butoxycarbonyl-2,5-dimethylpyrroline (7d) was prepared as described in the literature.24 The pure product was obtained by chromatography (1∶1 hexane∶EtOAc) in 86% yield as a colorless oil. 1H NMR (CDCl3): δ 4.32 (m, 1H), 3.90 (m, 3H), 3.05 (m, 3H), 1.47 (s, 9H), 1.29 (m, 3H), 1.17 (d, 3H); 13C NMR (CDCl3): δ 154.6, 79.8, 70.9, 59.0, 55.3, 28.7, 18.5, 14.5; Anal. Calcd for C11H21NO4: C 57.12, H 9.15, N 6.06; found: C 57.45, H 9.40, N 6.01.
N-tert-Butoxycarbonyl-2,5-dimethyl-3,4-di-O-methylsulfonylpyrrolidine (7f)
To a solution of N-tert-butoxycarbonyl-2,5-dimethyl-3,4-dihydroxypyrrolidine (7e) (0.466 g, 2.02 mmol) in 10 mL anhydrous pyridine cooled in an ice–salt bath, was added dropwise methylsulfonyl chloride (0.694 g, 6.06 mmol). After addition, the solution was slowly warmed to room temperature and allowed to stir for 4 h. After the solvent was removed, the brown residue was dissolved in CH2Cl2–H2O and extracted with CH2Cl2. The combined organic solutions were then washed with 0.1 N HCl until the pH of the aqueous layer was acidic. After the organic solution was washed by brine and dried over MgSO4, the solvent was removed under vacuum to afford 7f as a yellow oil in quantitative yield. The compound was not purified before the next step. 1H NMR (CDCl3): δ 5.15 (m, 1H), 4.91 (m, 1H), 4.19 (m, 2H), 3.14 (m, 3H), 1.48 (s, 9H), 1.33 (m, 6H); 13C NMR (CDCl3): δ 154.6, 79.8, 70.9, 59.0, 55.3, 28.7, 18.5, 14.5.N-tert-Butoxycarbonyl-2,5-dimethyl-3,4-diazidopyrrolidine (7g)
To 20 mL of dry DMF were added N-tert-butoxycarbonyl-2,5-dimethyl-3,4-di-O-methylsulfonylpyrrolidine (7f) (1.161 g, 3.0 mmol) and NaN3 (2.73 g, 42.0 mmol). The mixture was heated to 120 °C for 21 h. After cooling to room temperature, deionized H2O and mixed solvent (3∶2 Et2O∶hexane) were added and the layers were separated. The organic phase was dried over MgSO4 and concentrated under vacuum to afford the crude product. The pure compound was obtained in 31% yield by chromatography on silica gel (5∶1 hexane∶EtOAc). 1H NMR (CDCl3): δ 4.29 (m, 1H), 4.08 (m, 1H), 3.87 (m, 2H), 1.47 (s, 9H), 1.35–1.27 (m, 6H); 13C NMR (CDCl3): δ 153.5, 80.3, 67.8, 62.8, 57.6, 54.9, 28.6, 19.6, 15.6; Anal. Calcd for C11H19N7O2: C 46.96, H 6.81, N 34.85; found: C 47.37, H 6.94, N 34.84.
N-tert-Butoxycarbonyl-2,5-dimethyl-3,4-diaminopyrrolidine (7a)
To a 7 mL MeOH solution of N-tert-butoxycarbonyl-2,5-dimethyl-3,4-diazidopyrrolidine (7g) (0.355 g, 1.26 mmol) was added 200 mg of 10% Pd/C. The slurry was degassed and refilled three times with H2 at 1 atm. The reaction mixture was stirred at room temperature until no starting material was evident by TLC. The catalyst was filtered through Celite and the solvent was removed to afford a pale yellow oil. Further purification by chromatography on silica gel (5% MeOH in CH2Cl2 with 1% TEA) afforded pure diamine 7a in 76% yield. IR (Nujol): 3373, 3318, 1694 cm−1; 1H NMR (DMSO-d6, 90 °C): δ 3.71 (dq, J = 8 Hz, J = 7 Hz, 1H), 3.55 (dq, J = 2 Hz, J = 7 Hz, 1H), 3.42 (dd, J = 8 Hz, J = 5 Hz, 1H), 2.95 (dd, J = 5 Hz, J = 2 Hz, 1H), 2.85 (s, br, 2H), 1.41 (s, 9H), 1.22 (d, J = 6 Hz, 3H), 1.14 (d, J = 6 Hz, 3H); 13C NMR (DMSO-d6, 90 °C): δ 153.0, 77.6, 59.2 (2 × C), 54.7, 52.6, 27.8, 18.4, 14.5; HRMS (LSIMS): Calcd for C11H24N3O2 (M+H)+ 230.1868, found 230.1880.4,6-Dimethyl-2-oxo-hexahydropyrrolo[3,4-d]imidazole-5-carboxylic acid tert-butyl ester (7b)
To a 300 mL glass lined Parr high pressure vessel were added W(CO)6 (16 mg, 0.045 mmol), 40 mL of CH2Cl2, N-tert-butoxycarbonyl-2,5-dimethyl-3,4-diaminopyrrolidine (7a) (0.26 g, 1.1 mmol), K2CO3 (0.47 g, 3.4 mmol), and I2 (0.288 g, 1.13 mmol). The vessel was then charged with 85 atm of CO and left to stir at room temperature for 8 h. The pressure was released and the pale yellow solution was filtered. The CH2Cl2 solution was washed with 0.1 N HCl and aqueous Na2S2O3 solution. After drying over MgSO4 and filtration, the resulting solution was then concentrated to afford a pale brown solid. The white solid urea was obtained in 46% yield after chromatography on silica gel (15∶1 EtOAc∶MeOH). mp: 232–234 °C; IR (CH2Cl2): νCO 1720, 1690 cm−1; 1H NMR (CDCl3): δ 5.42 (s, br, 1H), 5.23 (s, br, 1H), 4.28 (m, 1H), 3.86–4.00 (m, 3H), 1.47 (s, 9H), 1.35 (d, 3H), 1.19 (d, 3H); 13C NMR (CDCl3): δ 163.2, 154.8, 80.1, 62.8, 56.1, 28.7, 18.5, 15.9; Anal. Calcd for C12H21N3O3: C 56.45, H 8.29, N 16.46; found: C 56.40, H 8.56, N 16.34.
Octahydrobenzoimidazol-2-one (8b)
To a glass lined 300 mL Parr high pressure vessel were added W(CO)6 (28 mg, 0.08 mmol), 80 mL of CH2Cl2, trans-cyclohexane-1,2-diamine (8a) (0.23 g, 2.0 mmol), K2CO3 (0.828 g, 5.99 mmol), and I2 (0.508 g, 2.00 mmol). The vessel was then charged with 75 atm of CO and left to stir at 120 °C for 52 h. The pressure was released after cooling to room temperature and the brown solution was filtered. The CH2Cl2 solution was washed with 0.1 N HCl, dried over MgSO4 and filtered. The resulting solution was then concentrated to afford a pale brown solid. The white solid urea was obtained after chromatography on silica gel (EtOAc) in 58% yield based on conversion. Unreacted starting material was recovered as the diammonium salt in 53% yield. The urea was identified by comparison to literature data.17Hexahydrocyclopentaimidazol-2-one (9b)
To a glass lined 300 mL Parr high pressure vessel were added W(CO)6 (28 mg, 0.08 mmol), 80 mL of CH2Cl2, cis-cyclopentane-1,2-diammonium chloride (the hydrochloride salt of 9a) (0.454 g of the trihydrate, 2.00 mmol), pyridine (1.0 mL), and I2 (0.508 g, 2.00 mmol). The vessel was then charged with 87 atm of CO and left to stir at 50 °C for 14 h. The pressure was released after cooling to room temperature and the brown solution was filtered. The CH2Cl2 solution was washed with aqueous Na2S2O3 solution and the solvent was removed to afford a pale brown solid. The white solid urea was obtained after chromatography on silica gel (5% MeOH in EtOAc) in 4% yield. 1H NMR (CDCl3): δ 1.70 (m, 6H), 4.24 (m, 2H), 4.67 (s, 2H). HRMS (EI): Calcd for C6H10N2O (M+) 126.0793, found 126.0792.Acknowledgements
Funding was provided by the donors of the Petroleum Research Fund. K.F. and C.R.P. were supported by the US-France NSF-REU program at the University of Florida.References
- P. J. DeClercq, Chem. Rev., 1997, 97, 1755 CrossRef CAS.
- S. A. Harris, D. E. Wolf, R. Mozingo, G. E. Arth, R. C. Anderson, N. R. Easton and K. Folkers, J. Am. Chem. Soc., 1945, 67, 2096 CrossRef CAS.
- M. Marx, F. Marti, J. Reisdorff, R. Sandmeier and S. Clark, J. Am. Chem. Soc., 1977, 99, 6754 CrossRef CAS.
- P. N. Confalone, G. Pizzolato, E. G. Baggiolini, D. Lollar and M. R. Uskokovic, J. Am. Chem. Soc., 1977, 99, 7020 CrossRef CAS.
- P. N. Confalone, G. Pizzolato and M. R. Uskokovic, Helv. Chim. Acta, 1976, 59, 1005 CrossRef CAS.
- P. N. Confalone, G. Pizzolato and M. R. Uskokovic, J. Org. Chem., 1977, 42, 135 CrossRef CAS.
- A. F. Hegarty and L. J. Drennan, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn, and C. W. Rees, Pergamon, Oxford, 1995, pp. 499–526 Search PubMed.
- F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140 RSC.
- J. E. McCusker, K. A. Abboud and L. McElwee-White, Organometallics, 1997, 16, 3863 CrossRef CAS.
- J. E. McCusker, J. Logan and L. McElwee-White, Organometallics, 1998, 17, 4037 CrossRef CAS.
- J. E. McCusker, C. A. Grasso, A. D. Main and L. McElwee-White, Org. Lett., 1999, 1, 961 CrossRef CAS.
- J. E. McCusker, F. Qian and L. McElwee-White, J. Mol. Catal. A: Chem., 2000, 159, 11 CrossRef CAS.
- J. E. McCusker, A. D. Main, K. S. Johnson, C. A. Grasso and L. McElwee-White, J. Org. Chem., 2000, 65, 5216 CrossRef CAS.
- F. Qian, J. E. McCusker, Y. Zhang, A. D. Main, M. Chlebowski, M. Kokka and L. McElwee-White, J. Org. Chem., 2002, 67, 4086 CrossRef CAS.
- K.-G. Hylton, A. D. Main and L. McElwee-White, J. Org. Chem., 2003, 68, 1615 CrossRef CAS.
- M. Otsuka, T. Masuda, A. Haupt, M. Ohno, T. Shiraki, Y. Sugiura and K. Maeda, J. Am. Chem. Soc., 1990, 112, 838 CrossRef CAS.
- S. G. Davies and A. A. Mortlock, Tetrahedron, 1993, 49, 4419 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- K. Hofmann, D. B. Melville and V. du Vigneaud, J. Biol. Chem., 1941, 141, 207.
- T. M. Filippova, S. D. Mikhno, I. G. Suchkova, N. S. Kulachkina and V. M. Berezovskii, Khim. Geterotsikl. Soedin., 1983, 908 Search PubMed.
- H. Flaster and H. Kohn, Heterocycl. Chem., 1981, 18, 1425 CrossRef CAS.
- B. Schmidt and H. Wildemann, Eur. J. Org. Chem., 2000, 3145 CrossRef CAS.
- J. Einhorn, C. Einhorn and L. Luche Jean, Synlett, 1991, 37 CrossRef CAS.
- B. M. Trost and L. S. Kallander, J. Org. Chem., 1999, 64, 5427 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |