Oxidation of 2,4,6-trichlorophenol by hydrogen peroxide. Comparison of different iron-based catalysts†
Gábor
Lente
*a and
James H.
Espenson
b
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, Debrecen, Hungary. E-mail: lenteg@delfin.unideb.hu; Fax: +36-52-489-667; Tel: +36-52-512-900 Ext.: 2373
bAmes Laboratory and Department of Chemistry, Iowa State University, Ames, IA, USA. E-mail: espenson@ameslab.gov; Fax: +1-515-294-5233; Tel: +1-515-294-5370
First published on 25th November 2004
Abstract
A systematic study of the stoichiometric and kinetic efficiencies of catalysts in the oxidation of 2,4,6-trichlorophenol by hydrogen peroxide was carried out. Six different iron-based activators were used: Fe3+(aq), Fe(TPPS)+, cis-[(cyclam)Fe]3+, trans-[(cyclam)Fe]3+, [Fe(TPA)]2+, and [Fe(6-Me2-TPA)2+ (ligand abbreviations: TPPS = meso-tetra(4-sulfonatophenyl) porphine, cyclam = 1,4,8,11-tetraazacyclotetradecane, TPA = tris(2-pyridylmethyl)amine, 6-Me2-TPA = bis[(6-methyl-2-pyridyl)methyl](2-pyridylmethyl)amine). The stoichiometric efficiencies of the catalyst were characterized by comparing the total organic carbon content, chloride ion concentration, and remaining oxidant concentration to the ideal stoichiometry where carbon dioxide, water and inorganic chloride are the only products. The kinetic efficiencies of the catalysts were compared using the rate of chloride ion formation and H2O2 consumption. It was shown that Fe(TPPS)+ is the best catalyst. Illumination by visible light significantly enhances the rate of oxidation.
Introduction
Polychlorinated phenols are used as wood preservatives, pesticides, fungicides, herbicides, insecticides or disinfectants, and they are also present in the waste of paper mills. These compounds are highly toxic, persistent, and regarded as priority pollutants for which efficient chemical treatment processes are needed.1–4 Oxidative degradation is probably the most advantageous reaction type for this purpose and several different methods have been reported including H2O2-dependent homogeneous catalysis,1–12 TiO2-based systems for photodegradation,13–16 ozonization,17 and bacterial methods.18–202,4,6-Trichlorophenol (TCP) is one of the most significant pollutants among chlorinated phenols and is often used to test the efficiency of oxidation methods.1,3 These methods usually use H2O2 or KHSO5 as a stoichiometric oxidant. Aqueous H2O2 is generally regarded as an environmentally friendly, “green” reagent21–27 partly because it gives water as the sole by-product of oxidation. However, H2O2 does not oxidize TCP or other chlorinated phenols in the absence of catalysts.28 The activators are usually transition metal complexes, most often iron(II) or iron(III) complexes involving N-donor ligands.1–12
Collins and co-workers recently reported the use of an oxidatively robust iron(III) complex of a TAML (tetra amido macrocyclic ligand) for the oxidation of TCP.3 Meunier and co-workers used iron tetrasulfophthalocyanine1 and iron(III) porphine derivatives.5,6 These complexes have a common feature: all of them have 4 N donor atoms in a planar arrangement surrounding the iron center. Iron-based non-heme activators for H2O2 are also being developed and studied for the epoxidation of olefins.29–33 We postulated that these might also be successful in the oxidation of TCP. Que and co-workers used iron complexes of tripodal polypyridyl ligands and they found characteristic differences between the catalytic activity of high-spin and low-spin iron(II) complexes which otherwise were very similar.30–32 Iron(III) cyclam complexes have also been successfully used to activate H2O2 toward epoxidation of olefins.29 Two separable isomers of the iron(III) cyclam complex are known: the cis complex is high-spin, the trans complex is low-spin.34
In this work, we present a systematic study of the efficiencies of iron catalysts in the aqueous oxidation of TCP by H2O2. Six different complexes were used (HS: high-spin, LS: low-spin): Fe(TPPS)+ (LS), cis-[(cyclam)Fe]3+ (HS), trans-[(cyclam)Fe]3+ (LS), [Fe(TPA)]2+ (LS), [Fe(6-Me2-TPA)]2+ (HS), and Fe(H2O)63+ (HS) for comparison. The structural formulas of the ligands are shown in Chart 1. We report the development of several methods to characterize and compare the stoichiometric and kinetic efficiencies of the different catalysts.
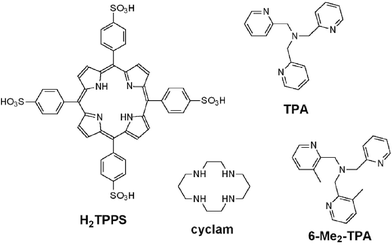 | ||
| Chart 1 Ligands used in this study. | ||
Results and discussion
General considerations
In an ideal case, the total oxidation of TCP by H2O2 should produce only water, carbon dioxide, and inorganic chloride:| C6H3Cl3O + 11H2O2 → 6CO2 + 11H2O + 3H+ + 3Cl− | (1) |
One method often used to characterize the final organic product distribution is GC-MS.3 This method usually requires considerable sample treatment, which, together with possible secondary reactions on the GC column, can potentially introduce considerable error.35 In addition, reliable quantification of GC-MS data requires time consuming calibration for each organic component measured. Used with caution, GC-MS can still be a highly valuable source of information on the organic products. Our GC-MS experiments did not prove any formation of highly toxic dioxins at any time, which is of particular concern in chlorophenol oxidations in general.3 This is probably the only really important piece of information from GC-MS studies for the purposes of this work, as the overall stoichiometric efficiency involves mostly inorganic substances (see eqn 1), which cannot be analyzed by this method. After describing our observations on the choice of oxidant and the possible use of buffers, this paper will summarize our efforts to develop methods to characterize the stoichiometric and kinetic efficiency of catalytic TCP oxidation with special emphasis on the environmentally relevant aspects of the process.
Choice of oxidant
The stoichiometric oxidant of a green oxidation process should be readily available and give minimal amounts of by-products. H2O2 is ideal in this respect because its reduced form is water. One possible problem with the use of H2O2 is that under certain conditions it decomposes and iron complexes, which are used as activators, often catalyze this decomposition.36,37 This may lead to disadvantageous consumption of additional peroxide reagent during the oxidation process.We also tested KHSO5 as an oxidizing agent for TCP in a few experiments. KHSO5 can often be used as an inexpensive alternative to H2O2 in organic transformations, and its use was preferred even in TCP oxidation in a few earlier studies.5 Although reactions with KHSO5 were usually faster than those with H2O2 and the same catalyst, a major problem was revealed by monitoring the concentration of Cl− formed in the reaction. After its initial formation, a decrease was detected in Cl− concentration during the oxidation process. To identify the reaction responsible for the decrease, experiments were carried out using Cl−, KHSO5 and Fe(TPPS)+ as a catalyst (without the addition of TCP). A substantial decrease was detected in Cl− concentration (see Electronic Supplementary Information†), and the formation of chlorine was confirmed. Thus, KHSO5 oxidizes the Cl− formed in the primary reaction to Cl2 under catalytic conditions. This procedure is disadvantageous because it consumes additional oxidizing agent and produces a toxic gas. The use of KHSO5 also has the inherent disadvantage of forming considerable amounts of K2SO4 as a by-product. It is concluded that H2O2 is a more environmental friendly oxidizing agent for the oxidation of chlorinated phenols.
Possible use of buffers
Buffers are often used in laboratory studies on the oxidation of chlorophenols.1,2,5–7 However, the use of buffers in real waste treatment methods would be rather disadvantageous. It is thus arguable that the laboratory studies should be done without additives such as buffers. In fact, the use of buffers in laboratory studies is a potential source of misleading conclusions for two common, but often overlooked reasons: possible oxidation of the buffer and complexation of the catalyst by the buffer. Because the buffer is typically present at a concentration at least an order of magnitude higher than any substrate, these processes can be very significant even if they are relatively slow or the interactions are not very strong. Indeed, the non-innocent role of phosphate buffers during TCP oxidation has been reported earlier.7In our experiments, we have shown that the presence of citric acid, a common buffer compound,5,6 inhibits the Fe(TPPS)+-catalyzed oxidation of TCP (see ESI†). In agreement with the considerations presented at the beginning of this section, we did not use buffers in this study. In a few examples, the acidity of the solutions was monitored with a pH electrode. These measurements showed that the original pH of the TCP solutions (about 5.5) decreased during the oxidation to 3.2–2.8 depending on the exact initial concentrations. This showed that acid is produced in the reaction as shown in eqn. 1. However, this pH change did not cause any dramatic change in the activity of the catalysts used in this study.
2H NMR experiments
One possibility to characterize the stoichiometric efficiency of TCP oxidation is to monitor the hydrogens of the original TCP throughout the process. 1H NMR is generally not suitable for this purpose because the catalysts are usually paramagnetic, and H2O2 is not readily available in a deuterated form. However, the use of isotopically-labelled TCP-d2 and 2H NMR offers clear advantages.38,39 Note that 2H is a quadrupolar nucleus and 2H NMR spectra are thus not sensitive to the presence of paramagnetic catalysts at the concentration levels used in this study.2H NMR experiments were carried out using a saturated aqueous solution of TCP-d2 (∼2.2 mM), in which the only detectable signals were those of TCP-d2 and HDO, the latter from the natural isotope distribution of hydrogen in solvent water. After the addition of H2O2 and the catalyst, the intensity of the signal of TCP-d2 decreased, the intensity of the HDO signal increased gradually. Only the HDO signal was detectable after ca. 12 h. This shows that all the hydrogens present in the initial TCP were oxidized to water or became exchangeable (OH or CO2H) protons. No separate signal corresponding to a 2H-containing intermediate was detected in these experiments.
A few 2H NMR experiments were carried out with the standard CD3CN added directly to the solution rather than used as an external standard. In these experiments, clear evidence was found for the oxidation of CD3CN as well: the intensity of its signal decreased whereas the intensity of the HDO signal increased. This observation further emphasizes that oxidation of added organic substances including buffers or standards may be a serious concern during TCP oxidation.
Comparison of the efficiencies of different catalyst
The overall stoichiometric and kinetic efficiency of the six different catalysts mentioned in the introduction were compared in systematic studies. It is known that the oxidation of TCP and other chlorophenols is influenced by light, including the room lights of a laboratory.28 Most of the experiments were carried out in the dark to avoid the interference of light. However, some measurements were also carried out under deliberate illumination to study the effect of light on the stoichiometric and kinetic efficiencies.Stoichiometric efficiencies
A series of experiments under standardized conditions (1.63 mM TCP, 136 mM H2O2, and 30 mg L−1 of catalyst) was carried out to compare the stoichiometric efficiencies of the catalyst studied in this work. The mass concentration of the different catalysts was kept constant in these measurements because this is more likely to be important for possible applications than the molar concentration. These conditions were chosen primarily because of solubility issues, the most serious limitation being the solubility of TCP (around 2 mM).Before performing the test runs, separate experiments, in which the concentration of a TCP stock solution was followed spectrophotometrically, were carried out to exclude any possible aqueous self- or light-induced decomposition of TCP. In these experiments, there was no measurable decrease in the TCP concentration over 2–3 months.
Samples were prepared by adding a solution of H2O2 to a solution of TCP, and the reaction was initiated by adding the catalyst. To make sure that all processes go to completion, the samples were stored at room temperature in the dark for 10 days before analysis in a sealed glass container that was opened three times during this period to allow gases to escape. After this time, the samples were analyzed for total organic carbon (TOC) content, Cl− concentration and H2O2 concentration. The catalyst Fe(TPPS)+ was used in two batches of experiments. In the first, the samples were stored in the dark similarly to other samples. In the second, the samples were illuminated with a 500 W halogen lamp for the first day and then stored in room light.
The TOC of the oxidized sample was considerably lower than that of the original sample in each case. This clearly indicates that carbon dioxide (or less likely carbon monoxide) was formed during the process. Results for the different catalysts are shown in Fig. 1, where the decrease of TOC is shown relative to the initial TCP sample. The higher the decrease, the more carbon dioxide is formed and the more efficient the oxidation is. The best catalyst is Fe(H2O)63+, with a TOC decrease of 58%; the others gave slightly lower decreases, 35–45%. The TOC decrease values given in Fig. 1 were calculated assuming that the TOC reduction occurs only because of TCP decomposition, and not because of the oxidation of the organic ligand present in the catalyst (which accounted for only a small fraction, less than 10% of the TOC of the original solution). The observational basis of this assumption was that we did not see significant loss of TOC in experiments where no TCP was used, i.e. the organic-containing iron-catalyst was reacted with hydrogen peroxide.
![Decrease in total organic carbon content during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. Initial concentrations: [TCP]
= 1.63 mM, [H2O2]
= 136 mM, [catalyst]
= 30 mg L−1; reaction time: 10 days; room temperature.](/image/article/2005/GC/b411269e/b411269e-f1.gif) | ||
| Fig. 1 Decrease in total organic carbon content during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. Initial concentrations: [TCP] = 1.63 mM, [H2O2] = 136 mM, [catalyst] = 30 mg L−1; reaction time: 10 days; room temperature. | ||
The chloride yield, defined as the amount of chloride ion in the final solution relative to the original total organic chlorine content, was also used to characterize the stoichiometric efficiencies. Fig. 2 shows that chloride yields are generally better than 90% and in two cases are quantitative (i.e., 100%) within experimental error. This shows that chlorine is removed from the organic compounds quite efficiently and little chlorinated organics or chlorinated aromatics remain in the solution after the oxidation. The only exception is Fe(6-Me2TPA)2+, where the chloride yield is only 75%. This value is significantly different from that determined for Fe(TPA)2+ (90%). The difference is not unexpected as these catalysts are known to have different reactivity patterns in epoxidation studies.31
![Chloride ion yield during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. Initial concentrations: [TCP]
= 1.63 mM, [H2O2]
= 136 mM, [catalyst]
= 30 mg L−1; reaction time: 10 days; room temperature.](/image/article/2005/GC/b411269e/b411269e-f2.gif) | ||
| Fig. 2 Chloride ion yield during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. Initial concentrations: [TCP] = 1.63 mM, [H2O2] = 136 mM, [catalyst] = 30 mg L−1; reaction time: 10 days; room temperature. | ||
The concentration of H2O2 remaining in the samples was also determined by iodometric titration.40Fig. 3 shows the amount of H2O2 consumed relative to the amount originally added. Under the standardized initial concentrations, the quantitative H2O2 consumption according to eqn. 1 is 13% and the determined values are usually close to or somewhat lower than this limit. The oxidant consumption was much higher in two cases: 25% for Fe3+(aq) and 64% for Fe(TPA)2+. Extra consumption of H2O2 is possible through its decomposition to water and dioxygen. It is concluded that a considerable amount of H2O2 is lost to catalytic decomposition when Fe3+(aq) or Fe(TPA)2+ is used as a catalyst. In the case of Fe(TPPS)+ and the two cyclam complexes, data for TOC, chloride ion, and oxidant consumption show that catalytic decomposition cannot be a major source of H2O2 loss.
![Hydrogen peroxide consumpsion during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. Initial concentrations: [TCP]
= 1.63 mM, [H2O2]
= 136 mM, [catalyst]
= 30 mg L−1; reaction time: 10 days; room temperature.](/image/article/2005/GC/b411269e/b411269e-f3.gif) | ||
| Fig. 3 Hydrogen peroxide consumpsion during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. Initial concentrations: [TCP] = 1.63 mM, [H2O2] = 136 mM, [catalyst] = 30 mg L−1; reaction time: 10 days; room temperature. | ||
In summary, the H2O2 consumption is the only really significant difference between the catalysts in terms of overall stoichiometric efficiency. The catalyst systems based on tripodal polypyridyl ligands—Fe(TPA)2+ and Fe(6-Me2TPA)2+—are probably less efficient than the others because Fe(TPA)2+ consumes a significant quantity of H2O2 in reactions not involved in the catalytic oxidation, whereas Fe(6-Me2TPA)2+ leaves considerable chlorinated organics in the system. It should also be noted that the stoichiometric efficiency of Fe3+(aq) is comparable to other catalysts. Light does not have significant effects on the stoichiometric efficiency.
Kinetic efficiencies
The kinetics of the oxidation process could be followed by two independent methods: measurement of the rates of Cl− formation and H2O2 consumption. All catalysts were studied in experiments using the same standardized conditions, but the initial concentrations were somewhat different from those used in the stoichiometric efficiency studies. The mass concentration of the different catalysts was kept constant.The rate of chloride ion formation was characterized in terms of the half-times (t1/2) necessary to reach a chloride ion concentration corresponding to half the total organic chlorine content of the original solution. It should be noted that this quantity is not completely analogous to half lives because the chloride ion formation did not follow an exponential (first-order) curve. Still, t1/2 values are suitable for the semi-quantitative comparison of different catalysts, as shown in Fig. 4. Three of the catalysts are similar, with t1/2ca. 16–18 h. Fe(6-Me2-TPA)2+ is slower than other systems, t1/2ca. 24 h, whereas Fe(H2O)63+ is somewhat faster, t1/2 = 12.6 h. Without doubt Fe(TPPS)+ is the fastest (t1/2 = 4.5 h), and illumination with visible light further lowers t1/2 to <1 h. These experiments were done using the different catalysts with the same mass concentrations. If molar concentrations are considered, the advantage of Fe(TPPS)+ is even larger because it has the highest molecular weight. It is also seen that light considerably enhances the rate of chloride ion formation and most likely the rate of overall oxidation.
![The time (t1/2) needed to reach 50% Cl− yield during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. [TCP]
= 1.61 mM; [H2O2]
= 95 mM; [catalyst]
= 50 mg L−1; μ
= 0.1 M (NaNO3); T
= 25.0 °C.](/image/article/2005/GC/b411269e/b411269e-f4.gif) | ||
| Fig. 4 The time (t1/2) needed to reach 50% Cl− yield during the oxidation of 2,4,6-trichlorophenol with H2O2 using different catalysts. [TCP] = 1.61 mM; [H2O2] = 95 mM; [catalyst] = 50 mg L−1; μ = 0.1 M (NaNO3); T = 25.0 °C. | ||
It was also possible to follow the time course of H2O2 concentration by taking samples from a reaction mixture and determining the concentration of H2O2. The results from an experiment series with Fe3+(aq) as a catalyst are shown in Fig. 5. Experiment a was carried out under usual TCP oxidation conditions. In experiment b, no TCP was added, and the rate of H2O2 consumption was clearly faster, an interesting fact that will be interpreted later. Experiment c was carried out without the addition of the catalyst. No H2O2 consumption was detected in this experiment showing once again that TCP is not oxidized by H2O2 without catalysts.
![Concentration of H2O2 as a function of time during the oxidation of 2,4,6-trichlorophenol with H2O2 catalyzed by Fe3+(aq). [TCP]
= 1.01 mM (a, c), 0 (b); [Fe3+(aq)]
= 160 µM (a, b), 0 (c); T
= 25.0 °C.](/image/article/2005/GC/b411269e/b411269e-f5.gif) | ||
| Fig. 5 Concentration of H2O2 as a function of time during the oxidation of 2,4,6-trichlorophenol with H2O2 catalyzed by Fe3+(aq). [TCP] = 1.01 mM (a, c), 0 (b); [Fe3+(aq)] = 160 µM (a, b), 0 (c); T = 25.0 °C. | ||
This series of experiments was repeated with every catalyst. The ratios of the initial rates measured in the presence (vTCP) and absence (vblank) of TCP were used as a semi-quantitative indicator. Fig. 6 shows these ratios for every catalyst. Ratios span a large range from 54 for Fe(TPPS)+, through 5–10 for the cyclam complexes, to 0.16–0.21 for Fe3+(aq), Fe(TPA)2+ and Fe(6-Me2-TPA)2+. The last three catalysts had the same value within experimental error.
![Initial rate ratios of H2O2 consumption in the presence (vTCP, [TCP]
= 1.0 mM) and absence (vblank) of TCP. T
= 25.0 °C. Note the logarithmic scale on the y axis.](/image/article/2005/GC/b411269e/b411269e-f6.gif) | ||
| Fig. 6 Initial rate ratios of H2O2 consumption in the presence (vTCP, [TCP] = 1.0 mM) and absence (vblank) of TCP. T = 25.0 °C. Note the logarithmic scale on the y axis. | ||
Fe(TPA)2+ induced a sudden burst of H2O2 decomposition with intense formation of bubbles. This initial burst was over in a few minutes and then a relatively slow decrease in H2O2 concentration was detected (see ESI†). No similar results were obtained with Fe(6-Me2-TPA)2+, which again shows that these two catalysts are dissimilar. The slowly decreasing region was used for evaluation with Fe(TPA)2+, because chloride ion kinetic measurements show without doubt that oxidation of the substrate occurs in this stage. The initial burst of H2O2 decomposition can likely be attributed to the catalyst in its original form accelerating the decomposition of H2O2 efficiently, but then being transformed to another form during the first few minutes.
It is well known that the decomposition of hydrogen peroxide is sensitive to experimental conditions and the possible presence of impurities. The consistency of the results and the general reliability of the procedures used was also checked in two ways. First, the stock concentration of the stock solutions of hydrogen peroxide, which were prepared from commercially available 30% hydrogen peroxide, was followed using iodometric titration. In a ca. 1 M stock solution, a gradual decrease in hydrogen peroxide concentration was observed. The overall decrease was 3.5% relative to the original concentration in 125 days, implying that decomposition because of the impurities present in the water used is probably not significant on the time scale of the initial rate studies (typically a few hours). Second, a test directly connected to the conditions of the initial rate measurements was also performed. Samples with hydrogen peroxide were prepared without a catalyst and in one case with phosphate-buffered Fe(TPPS)+, where the porphyrin forms a catalytically inactive dimer. No measurable loss of hydrogen peroxide was detected in either case for 5–6 hours.
Mechanistic considerations
Two fundamentally different mechanisms can be envisioned for TCP oxidation in these systems. The first is when the catalyst basically catalyzes the decomposition of H2O2 and the reactive intermediates produced (e.g. hydroxyl radicals in Fenton-type reactions) cause the oxidative damage to the ligand.36 In this case, the catalyst itself does not play a direct role in the oxidation of the substrate. The rate of H2O2 consumption should be similar in the presence and absence of an oxidizable substrate. However, the initial pH is about 2 units lower in the presence of TCP because of its acidity (pKa = 6.15). Decomposition of hydrogen peroxide by Fenton-type catalysts is known to be slower in more acidic solutions.37 Thus, the rate of H2O2 consumption can actually be lower in the presence of substrate. This general mechanism can finally result in a H2O2 consumption that is much larger than needed for the oxidation because the useful oxidation only happens after the first step of catalytic decomposition. This is clearly seen with Fe3+(aq) and Fe(TPA)2+ from the H2O2 consumption results (Fig. 3). In the case of Fe(6-Me2-TPA)2+ the oxidation of the substrate is significantly less complete than with other catalysts (see Fig. 2), so catalytic decomposition may be a major source of H2O2 loss despite the relatively low overall oxidant consumption. It should also be explained at this point why H2O2 is present at the end of the oxidation process. The most likely reason is that the H2O2-decomposing activity of the catalysts is diminished by the increasing acidity and/or through complex formation with dicarboxylic acids formed as the final products of the oxidation.A second possible mechanism is a catalytic cycle involving the catalyst, H2O2 and TCP. The substrate is oxidized in a direct reaction with an active high oxidation state iron complex. In this case, the catalytic cycle cannot operate without substrate and the rate of H2O2 consumption is clearly expected to be larger in the presence of substrate than in its absence. Fe(TPPS)+ and the cyclam complexes seem to follow this pattern. They are inefficient catalysts of H2O2 decomposition as shown by the high ratios in Fig. 6. The actual catalytic cycle and the species participating in it may be different for the different catalysts and further studies are needed to identify them.
Attempted use of methyltrioxorhenium(VII) as a catalyst
To obtain more general mechanistic information, an attempt to use methyltrioxorhenium (MTO, CH3ReO3), a well known and efficient activator for hydrogen peroxide,41 to catalyze the oxidation of TCP was also made. The mechanisms of MTO-catalyzed oxidations are based on the formation of peroxo complexes CH3ReO2(O–O), and CH3ReO(O–O)2, and it is understood that MTO activates H2O2 exclusively toward oxygen transfer.41 Our results showed that MTO does not catalyze the oxidation of TCP or other chlorophenols by H2O2 at all. This was confirmed in both water and CH3CN. In these experiments, only independently known and slow catalyst decomposition was observed and the chlorophenol was intact after a day. Thus it is concluded that oxygen transfer cannot be among the first steps of the oxidation of chlorophenols. This agrees with earlier mechanistic proposals, where phenoxyl radical formation through either hydrogen atom transfer or deprotonation of the phenol followed by electron transfer is the usual initial process.6 The results presented here also have an important implication for future studies: the mechanistic conclusions drawn using the same catalysts for the epoxidation of olefins,29–33 in which oxygen transfer is the most important step, are not necessarily valid for TCP oxidation.Conclusion
In this work, we defined quantitative and semi-quantitative measures of stoichiometric and kinetic efficiencies to characterize the catalytic oxidation of TCP. These definitions can be used for other similar oxidation systems as well. The overall stoichiometric efficiencies are similar for the six different catalysts used in this work, but kinetic observations clearly favor Fe(TPPS)+. The results suggest that heme-like iron catalysts involving planar coordination of four nitrogen donor atoms may be the most successful ones to activate H2O2 toward chlorophenol oxidation. Light has an accelerating effect on the reaction.Experimental
Materials
Commercial 2,4,6-trichlorophenol was purified by vacuum sublimation. Iron(III) meso-tetra(4-sulfonatophenyl) porphine chloride was purchased from Frontier Scientific (http://www.porphyrin.com). Fe(NH4)(SO4)2·12H2O was used as received. The complexes cis-[cyclamFeCl2]Cl,42trans-[(cyclamFeCl2)]BF4,42 [Fe(TPA)(CH3CN)2](ClO4)2,30 and [Fe(6-Me2-TPA)(CH3CN)2](ClO4)230 were prepared according to literature methods and characterized by 1H NMR and/or UV-vis spectroscopy. 2,4,6-Trichlorophenol-d2 was prepared as described earlier.35 H2O2 was determined by iodometric titration.40 Ion exchanged and ultrafiltered water from a Millipore MILLI-Q purification system was used to prepare solutions.Instruments
A Shimadzu UV-3101PC scanning spectrophotometer and a Shimadzu TOC-5000A total organic carbon analyzer were used in this study. 2H NMR spectra were recorded in natural isotope distribution H2O and referenced to the signal of standard CD3CN (λ = 1.55 ppm).38,39 GC-MS experiments were performed on a Finnigan Magnum ion trap mass spectrometer equipped with a 30 m DB-5 column using He carrier gas. A Weiss Research CL3005 combination chloride ion selective electrode and a Hanna Instruments pH302 pH-meter were used. The electrode was calibrated every day using standard NaCl solutions. The lamp used in this study was a commercially available Regent 500 watt halogen lamp (Regent Lighting Corporation, Burlington, NC, USA).Acknowledgements
This research was supported by the Center for Catalysis, Institute for Physical Research and Technology, Iowa State University. Some experiments were conducted with the use of the facilities of the Ames Laboratory of the US Department of Energy, which is operated by Iowa State University of Science and Technology under contract W-7405-Eng-82. We acknowledge helpful discussions with Prof. W. S. Jenks and Mr. Youn-chul Oh. GL also wishes to thank the Fulbright Program and Hungarian funding agency OTKA (grant No. T042755) for financial support.References
- A. Sorokin, J. L. Séris and B. Meunier, Science, 1995, 268, 1163 CrossRef CAS.
- B. Meunier, Science, 2002, 296, 270 CrossRef CAS.
- S. Sen Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. Horwitz, K. W. Schramm and T. J. Collins, Science, 2002, 296, 326 CrossRef.
- T. J. Collins, Acc. Chem Res., 2002, 35, 782 CrossRef CAS.
- G. Labat, J. L. Séris and B. Meunier, Angew. Chem., Int. Ed. Engl., 1990, 29, 1471 CrossRef.
- R. S. Shukla, A. Robert and B. Meunier, J. Mol. Catal. A: Chem., 1996, 113, 45 CrossRef CAS.
- A. Hadasch, A. Sorokin, A. Rabion and B. Meunier, New J. Chem., 1998, 22, 45 RSC.
- R. P. Ferrari, E. Laurenti and F. Totta, J. Biol. Inorg. Chem., 1999, 4, 232 CrossRef CAS.
- M. Sanchez, A. Hadasch, R. T. Fell and B. Meunier, J. Catal., 2001, 202, 177 CrossRef CAS.
- M. Fukushima, A. Sawada, M. Kawasaki, H. Ichikawa, K. Morimoto and K. Tatsumi, Environ. Sci. Technol., 2003, 37, 1031 CrossRef CAS.
- G. Lente and J. H. Espenson, Int. J. Chem. Kinet., 2004, 36, 449 CrossRef CAS.
- G. Lente and J. H. Espenson, New J. Chem., 2004, 28, 847 RSC.
- R. Bauer, G. Waldner, H. Fallmann, S. Hager, M. Klare, T. Krutzler, S. Malato and P. Maletzky, Catal. Today, 1999, 53, 131 CrossRef CAS.
- X. Li, J. W. Cubbage, T. A. Tetzlaff and W. S. Jenks, J. Org. Chem., 1999, 64, 8509 CrossRef CAS.
- X. Li, J. W. Cubbage and W. S. Jenks, J. Org. Chem., 1999, 64, 8525 CrossRef CAS.
- K. Mogyorósi, A. Farkas, I. Dékány, I. Ilisz and A. Dombi, Environ. Sci. Technol., 2002, 36, 3618 CrossRef CAS.
- F. J. Benitez, J. Beltran-Hereida, J. L. Acero and F. J. Rubio, Ind. Eng. Chem. Res., 1999, 38, 1341 CrossRef CAS.
- G. V. B. Reddy, M. D. S. Gelpke and M. H. Gold, J. Bacteriol., 1998, 180, 5259.
- O. Zaborina, H. J. Seitz, I. Sidorov, J. Eberspächer, E. Alexeeva, L. Golovleva and F. Lingens, J. Basic. Microbiol., 1999, 39, 61 CrossRef CAS.
- L. Padilla, V. Matus, P. Zenteno and B. Gonzalez, J. Basic Microbiol., 2000, 40, 243 CrossRef CAS.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC.
- D. Bogdal, M. Lukasiewicz and J. Pielichowski, Green Chem., 2004, 110 RSC.
- Y. Usui and K. Sato, Green Chem., 2003, 373 RSC.
- X. Hao, O. Yamazaki, A. Yoshida and J. Nishikido, Green Chem., 2003, 524 RSC.
- J. Peng, F. Shi, Y. Gu and Y. Deng, Green Chem., 2003, 224 RSC.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 1 RSC.
- A. Severeyns, D. E. De Vos and P. A. Jacobs, Green Chem., 2002, 380 RSC.
- G. Lente and J. H. Espenson, Chem. Commun., 2003, 1162 RSC.
- W. Nam, R. Ho and J. S. Valentine, J. Am. Chem. Soc., 1991, 113, 7052 CrossRef CAS.
- Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que, Jr., J. Am. Chem. Soc., 1997, 119, 4197 CrossRef CAS.
- K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que, Jr., J. Am. Chem. Soc., 2002, 124, 3026 CrossRef CAS.
- M. Fujita, M. Costas and L. Que, Jr., J. Am. Chem. Soc., 2003, 125, 9912 CrossRef CAS.
- J. U. Rohde, J. H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Jr., Science, 2003, 299, 1037 CrossRef CAS.
- P. K. Chan and C. K. Poon, J. Chem. Soc., Dalton Trans., 1976, 858 RSC.
- G. Lente and J. H. Espenson, J. Photochem. Photobiol. A: Chem., 2004, 163, 249 CrossRef CAS.
- E. Neyens, J. Hazard. Mater., 2003, B98, 33 CrossRef.
- M. L. Kremer, J. Phys. Chem. A, 2003, 107, 1734 CrossRef CAS.
- A. Durazzo and M. M. Abu-Omar, Chem. Commun., 2002, 66 RSC.
- G. S. Owens, A. Durazzo and M. M. Abu-Omar, Chem. Eur. J., 2002, 8, 3053 CrossRef CAS.
- S. B. Brown, P. Jones and A. Suggett, Anal. Chim. Acta, 1968, 43, 343 CrossRef CAS.
- J. H. Espenson, Chem. Commun., 1999, 479 RSC.
- L. C. G. Vasconcellos, P. Oliveira, E. E. Castellano, J. Ellena and I. S. Moreira, Polyhedron, 2001, 20, 493 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: additional experimental observations. See http://www.rsc.org/suppdata/gc/b4/b411269e/ |
| This journal is © The Royal Society of Chemistry 2005 |