Tailbiter: a new amide foldamer†
Christopher A.
Hunter
*,
Andrea
Spitaleri
and
Salvador
Tomas
*
Centre for Chemical Biology, Krebs Institute for Biomolecular Science, Department of Chemistry, University of Sheffield, Sheffield, UK S3 7HF. E-mail: C.Hunter@shef.ac.uk; S.Tomas@shef.ac.uk; Fax: +44 (0)114 2229346; Tel: +44 (0)114 2229476
First published on 1st July 2005
Abstract
Hydrogen bond directed folding of a synthetic polyamide was studied in chloroform solution, and the three-dimensional structure of the foldamer determined using 1H NMR chemical shifts.
Biological macromolecules are composed of one or more linear oligomers that fold into a functional form. The information that governs the final structure is encoded in the sequence of monomers and the precise functionality that they display, but accurate prediction of such structures remains a major challenge in structural biology.1 Recently, chemists have begun to develop small molecule systems that fold in a similar way, and these may help to answer more complex questions,2 or find applications in artificial molecular assemblies.3–7 We have been studying the properties of a family of oligoamides composed of alternating repeats of isophthalic acid and bisaniline building blocks. These systems have a rich supramolecular chemistry in non-polar solvents, forming macrocyclic receptors, catenanes, knots and double-stranded zipper complexes via a combination of amide–amide hydrogen bonds and aromatic interactions.8–11 Here we report the serendipitous discovery of a new member of the family that folds into a well-defined, compact, three-dimensional structure, governed by a combination of hydrogen bonding and aromatic interactions.
The study of such systems requires methods for determining the solution structure of the folded state, which is usually in equilibrium with other, less populated unfolded or misfolded states. Moreover, small molecule foldamers are prone to crystallise in a conformation dictated by crystal packing contacts rather than in the conformation found in solution.12 Just as with large structures, like proteins, NMR spectroscopy provides the most detailed information about the solution structures of foldamers.13 Although the two-dimensional spectra of small synthetic systems are often easier to interpret, the constraints imposed by NOE contacts can be ambiguous, and are often not enough to get a clear picture of the three-dimensional structure. We have previously shown that complexation-induced changes in 1H NMR chemical shift (Δδ) can be used to determine the high resolution, three-dimensional structure of intermolecular complexes in solution.14,15 Here we demonstrate the utility of this approach for the solution structure determination of a synthetic foldamer, where folding-induced changes in 1H NMR chemical shift are used as the Δδ parameters.16
Compound 1 was synthesised as part of a programme to quantify the intermolecular interactions between the terminal functional groups of double-stranded zipper complexes using chemical double mutant cycles (Scheme 1).17 All other symmetric compounds of this type that we have previously prepared had relatively simple 1H NMR spectra in chloroform. In contrast, the 1H NMR spectrum of 1b in deuterochloroform solution is surprisingly complicated (Fig. 1, (a)). The two-fold symmetry of the chemical structure is not reflected in the spectrum, indicating that the molecule adopts a stable conformation that makes the two ends non-equivalent on the 1H NMR timescale. Compound 1a is much less soluble than 1b and was not studied in detail, but the 1H NMR spectra of the two compounds are very similar, indicating that the large solubilising groups (R) are not implicated.
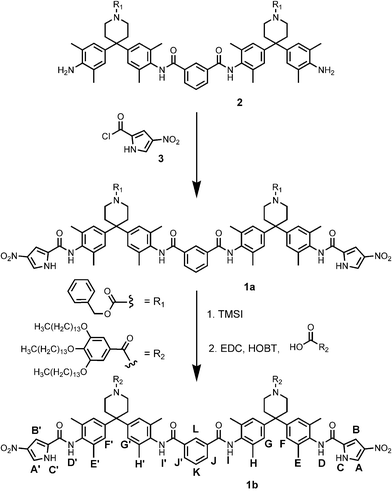 | ||
| Scheme 1 Synthesis of 1a and 1b. The proton labelling scheme is also shown. The syntheses of 2 and 3 were reported previously.17 | ||
 | ||
| Fig. 1 Aromatic region of the 1H NMR spectrum of 1b (a) in pure CDCl3 and (b) in CDCl3 ∶ DMSO-d6, 5 ∶ 1 v/v. See Scheme 1 for the proton labelling scheme. | ||
The overall profile of the 1H NMR spectrum of 1b remains unchanged over a wide concentration range (10 µM to 10 mM), which shows that the unusual spectrum is not due to the formation of intermolecular complexes but a consequence of unimolecular folding. Most signals show negligible changes in chemical shift as a function of concentration, but the signals due to the amide protons D and D′ move 0.5 ppm downfield, indicative of hydrogen bonding. These changes are ascribed to aggregation at high concentrations, and the data could be fitted to an aggregation isotherm with a self-association constant of 31 ± 7 M−1. These observations suggest that there is some aggregation due to hydrogen bonding between amide groups exposed on the surface of the folded structure, but that this process does not perturb the folding of 1b.
The 1H NMR signals of 1b are sharp and well-resolved at a concentration of 1 mM in deuterochloroform, and were assigned using COSY and ROESY experiments. Some signals show very large changes in chemical shift compared with those observed for related compounds containing the same functional groups, 4 and 5. Table 2 summarises the folding induced change in chemical shift, Δδ, calculated by assuming that the chemical shifts measured for 4 and 5 at low concentration, where there is no aggregation, correspond to the chemical shifts of the related protons in the unfolded state of 1b at room temperature in deuterochloroform (see Fig. 2). The signals due to the amide and pyrrole protons show large downfield shifts that are indicative of hydrogen bonding and the large upfield shifts observed for the pyrrole protons are indicative of interactions with the face of an aromatic ring. The importance of hydrogen bonding in this system was confirmed by addition of DMSO, which dramatically simplified the 1H NMR spectrum (Fig. 1, (b)). In the presence of DMSO, the two ends of 1b are equivalent, and the chemical shifts of all of the signals are comparable to those observed for the related protons in reference compounds 4 and 5. Thus DMSO is able to reverse the folding process by competing for the hydrogen bonding interactions that drive the folding of 1b.
| Proton | k i/×10−4 s−1 | Proton | k/×10−4 s−1 | P (= ki/k) |
|---|---|---|---|---|
| a The concentration of oligoamide was 1 mM in all cases. The error in P is ± 20% | ||||
| c | 36 | C | 1.0 | 36 |
| c | 36 | C′ | 0.8 | 45 |
| d | 8.8 | D | 4.0 | 2.2 |
| d | 8.8 | D′ | 5.1 | 1.7 |
| i | 2.1 | I | 0.5 | 4.2 |
| i | 2.1 | I′ | 0.5 | 4.2 |
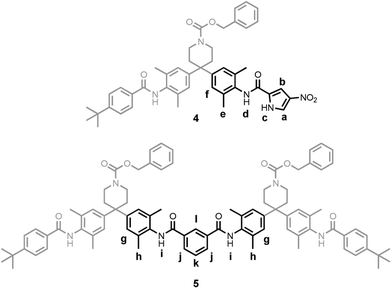 | ||
| Fig. 2 Reference compounds 4 and 5. The lower case labels indicate the protons used as unfolded reference points for the corresponding upper case labels in 1b. | ||
H–D exchange experiments further underline the importance of hydrogen bonding. Deuteromethanol (60 mM) was added to a 1 mM solution of 1b in deuterochloroform, and the 1H-NMR spectrum recorded over a period of several hours. The decrease in the intensity of the NH signals relative to A′ was fitted to pseudo first-order kinetics for the initial stages of the exchange process to obtain a rate constant (k). Compounds 4 and 5 were used to obtain the corresponding intrinsic rate constants for the exchange of the related protons under the same conditions (ki). These rate constants were used to determine the folding-induced protection factors (P) for the exchangeable protons of 1b (P = ki/k), the results of which are summarised in Table 1. Protons C and C′ are strongly protected, suggesting that they are buried in the folded structure and involved in strong hydrogen bonding interactions. Protons I and I′ show moderate protection, due to the formation of weaker hydrogen bonds or limited solvent exposure. D and D′ are clearly much more exposed to the solvent bulk and exchange more rapidly, consistent with the relatively small Δδ values in Table 2 and their involvement in the aggregation behaviour described above. These observations suggest that the exchange process does not involve a concerted, cooperative unfolding that exposes all of the buried groups to solvent, rather that local interactions can open independently, giving rise to different protection factors in different regions of the structure.
| Proton | Δδexp | Δδcalc | Error |
|---|---|---|---|
| A | −1.29 | −1.25 | −0.04 |
| A′ | 0.06 | 0.06 | 0.00 |
| B | −2.52 | −2.58 | 0.06 |
| B′ | 0.05 | 0.09 | −0.04 |
| C | 2.92 | 2.85 | 0.06 |
| C′ | 2.51 | 2.50 | 0.01 |
| I | 0.63 | 0.42 | 0.21 |
| I′ | 0.94 | 0.92 | 0.02 |
While these data provide good evidence for the presence of a hydrogen bonded, folded conformation in solution, more information is required to elucidate the precise structure. For this, we turn to the Δδ values in Table 2. Our genetic algorithm based software was used to convert these data into an optimised three-dimensional structure for which theoretically calculated Δδ values are matched to those observed experimentally. The result is shown in Fig. 3, and Table 2 compares the calculated Δδ values for this structure with the experimental values (RMS deviation = 0.03 ppm, excluding the solubilising groups that were not included in the calculation). One nitropyrrole moiety is folded inside a perfectly complementary binding pocket created by the rest of the molecule and is held in place by a combination of hydrogen bonds and aromatic interactions. The structure in Fig. 3 clearly explains all of the observations listed above. One set of pyrrole protons (A and B) sit over the faces of aromatic rings, while the others (A′ and B′) are on the outside of the structure and are relatively unperturbed by folding. The pyrrole protons C and C′ are involved in strong hydrogen bonds with the amide carbonyl groups, amides D and D′ are exposed to solvent on the outside of the structure, and amides I and I′ are involved in weak hydrogen bonds to the nitro oxygens. The differences in NH protection with respect to H–D exchange can be accounted for from the strengths of the individual interactions involved: a pyrrole–amide interaction is worth 6 kJ mol−1 in free energy in chloroform—corresponding to P = 10, and an amide–nitro interaction is worth 2 kJ mol−1 giving P = 2.‡ Thus the exchange mechanism involves the disruption of individual hydrogen bond interactions rather than unfolding of the whole structure. Unlike proteins, in small molecule foldamers of this type, no protons are completely buried and removed from contact with the solvent.
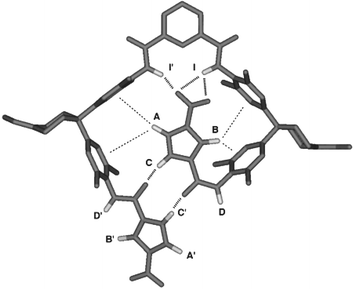 | ||
| Fig. 3 The three-dimensional structure of foldamer 1b, calculated using the folding-induced changes in chemical shift. | ||
Notes and references
- T. E. Creighton, Proteins: Structures and Molecular Properties, Freeman, New York, 1997 Search PubMed.
- M. S. Searle and B. Ciani, Curr. Opin. Struct. Biol., 2004, 14, 458 CrossRef CAS.
- V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2798 CrossRef CAS.
- L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528 CrossRef CAS.
- M. S. Cubberley and B. L. Iverson, Curr. Opin. Chem. Biol., 2001, 5, 650 CrossRef CAS.
- H. Yin, G. Lee, K. A. Sedey, J. M. Rodriguez, H.-G. Wang, S. M. Sebti and A. D. Hamilton, J. Am. Chem. Soc., 2005, 127, 5463 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS.
- C. A. Hunter, J. Chem. Soc., Chem. Commun., 1991, 749 RSC.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS.
- O. Safarowsky, M. Nieger, R. Frohlich and F. Vogtle, Angew. Chem., Int. Ed., 2000, 39, 1616 CrossRef CAS.
- A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856 CrossRef CAS.
- I. L. Karle, S. Prasad and P. Balaram, J. Pept. Res., 2004, 63, 175 CrossRef CAS.
- K. Wuthrich, Angew. Chem., Int. Ed., 2003, 42, 3340 CrossRef.
- C. A. Hunter and M. J. Parker, Chem.–Eur. J., 1999, 5, 1891 CrossRef CAS.
- A. Spitaleri, C. A. Hunter, J. F. McCabe, M. J. Paker and S. L. Cockroft, CrystEngComm, 2004, 6, 489 Search PubMed.
- M. Iwadate, T. Asakura and M. P. Williamson, Eur. J. Biochem., 1998, 257, 479 CrossRef CAS.
- C. A. Hunter and S. Tomas, Chem. Biol., 2003, 10, 1023 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Synthesis and characterisation of 1a and 1b, complete listing of experimental and calculated 1H NMR Δδ data for the folding of 1b. See http://www.rsc.org/suppdata/cc/b5/b506093a/ |
| ‡ The strength of a hydrogen bonding interaction can be estimated using ΔΔGH-bond = −(α − αS)(β − βS), where α and β are the H-bond parameters for the two groups involved (pyrrole α = 3.0, amide α = 2.9, β = 8.3, or nitro β = 3.7), and αS and βS are the corresponding parameters for the solvent (2.2 and 0.8 respectively for chloroform).18 We used this free energy to estimate P = e(−ΔΔGH-bond/RT). The solute H-bond parameters are based on the parent functional groups, and because we neglected the effect of methanol, the calculated values of P are qualitative estimates. Nevertheless, they are comparable to the experimental values. |
| This journal is © The Royal Society of Chemistry 2005 |