Synthesis of huge macrocycles using two calix[4]arenes as templates†
Yudong
Cao
a,
Leyong
Wang
a,
Michael
Bolte
b,
Myroslav O.
Vysotsky
a and
Volker
Böhmer
*a
aAbteilung Lehramt Chemie, Fachbereich Chemie, Pharmazie und Geowissenschaften, Johannes Gutenberg – Universität, D-55099 Mainz, Germany. E-mail: vboehmer@mail.uni-mainz.de
bFachbereich Chemie und pharmazeutische Wissenschaften, Institut für Organische Chemie, J.-W. Goethe Universität, D-60439 Frankfurt/Main, Germany
First published on 1st June 2005
Abstract
Macrocycles with up to 100 atoms have been synthesised using two calix[4]arenes as templates: first, (3,5-dialkenyloxy)phenyl groups are attached to the wide rim of a calix[4]arene via urea links, then the alkenyl groups are connected via a metathesis reaction using a tetratosylurea calix[4]arene for their correct prearrangement and finally the urea functions are cleaved to detach the newly formed macrocycles.
The prearrangement of different reactive molecules or different functional groups within a molecule often is an important and decisive factor for the formation of a desired reaction product.1 Such preorganisation may be achieved by a suitable template (e.g. a single molecule) to which the reacting units are covalently attached or (more or less strongly) bound via reversible links. “Classical” biochemical examples for such matrix or template syntheses are the replication and transcription of DNA as well as the translation of RNA into a specific peptide sequence.2
Purely synthetic examples comprise the formation of large macrocycles, where the reacting units are bound to the template via metal–ligand coordination,3 ion–dipole interactions4 or through easily cleavable functional groups.5 Templates are especially welcome in the synthesis of topologically interesting molecules,6 huge “hosts”7 or even nanotubes.8 The formation of self-assembled molecular capsules may depend also on the presence of a suitable guest as template.9
We recently showed10 that the heterodimerisation of tetraarylurea calix[4]arenes 4 with tetratosylurea calix[4]arene115 (Y = C5H11) can be used to preorganise alkenyl residues attached to the four urea functions in such a way, that the metathesis reaction between their double bonds12 (followed by hydrogenation) leads to the formation of the respective multimacrocyclic calix[4]arene derivatives 6 as the only identifiable products (yields of 60–95% after purification). If eight alkenyl groups are attached, two per urea function, molecules result, in which a second macrocycle is attached via four (urea) links to the wide rim of the calix[4]arene (Scheme 1). This second macrocycle keeps the calix[4]arene in the cone-conformation even in the case of tetramethyl ethers, which usually prefer the partial cone conformation. This follows unambiguously from VT-NMR spectra and is demonstrated by the single crystal X-ray structure shown in Fig. 1.‡ The calix[4]arene skeleton of 6 assumes the pinched cone conformation with two opposite aromatic units nearly parallel to each other (dihedral angles 2.4°/5.8°) and the other two oriented outwards (dihedral angles 109.1°/103.7°).13
 | ||
| Scheme 1 Synthesis of huge macrocycles: (i) DPPA, TEA, toluene, 70 °C, 2 h; (ii) toluene, 90 °C, 2 h; (iii) dichloromethane–benzene (1∶1), r.t., 2 days; (iv) a) Grubbs' Catalyst (40%), r.t., 2–6 days; b) H2, PtO2, THF, r.t., 12 h; (v) chromatographic purification; (vi) acetic acid, reflux, 24 h. | ||
 | ||
| Fig. 1 Single crystal X-ray structure of the tetraloop compound 6 (m = 10, Y = CH3); one of two crystallographically independent molecules is shown from two perspectives. | ||
Multimacrocyclic compounds 6 have been used as building blocks for hitherto unknown multicatenanes.6d However, it should be possible also to cleave the urea functions to detach the newly formed macrocycle from the original calix[4]arene 3. The total reaction sequence depicted in Scheme 1 then describes the synthesis of single, but huge macrocycles, the sizes of which are determined by the attachment of four molecules of 2 to the calix[4]arene 3 and by the length of the alkenyl chains in 2.
After various unsuccessful attempts to hydrolyse the urea groups under alkaline (sodium hydroxide in CH3OH, reflux, 24 h) or acidic (hydrochloric or sulfuric acid in EtOH, reflux, 24 h) conditions, we found that this cleavage is possible by refluxing compounds 6 in acetic acid (Scheme 1).§ The macrocyclic tetraacetamides 7 were thus obtained in yields between 50 and 75% after simple chromatographic purification. The calixarene used as template could also be isolated in the form of its tetraacetamide 8 (which of course can be easily prepared directly from 3).
The structure of macrocyclic acetamides 7 was unambiguously confirmed through 1H-NMR and ESI-MS. Typically a singlet for NH at 9.78 ppm, a doublet for ArH at 6.77 ppm, a triplet for OCH2 at 3.87 ppm and a singlet for C(O)CH3 at 1.99 ppm are found in the expected ratio of 1∶2∶4∶3. The peak M + Na found in all cases as the base peak confirms the size of the macrocycle.
It should be pointed out that in the examples described above calix[4]arenes are used as a template in a twofold way. The strategy may be generalised as follows. In a first step (or sequence of reaction steps) a bifunctional molecule 2 (in red, Scheme 1) is covalently attached via a third function (here the urea group as link) to the wide rim of a calix[4]arene 3 which acts as a molecular skeleton (in black). A second calix[4]arene, the tetratosylurea 5 (in blue), forms a heterodimer with 4viareversible hydrogen bonds. Thus, the functional groups of 2 are arranged in an appropriate way, which ensures their correct intramolecular connection within 4. In polar, hydrogen bond breaking solvents the dimeric assembly easily dissociates. Subsequent cleavage of the urea functions leads to giant macrocycles 7 with up to now 52, 60, 76 and 100 atoms which otherwise would be difficult to prepare (if at all). Larger rings should be available analogously.
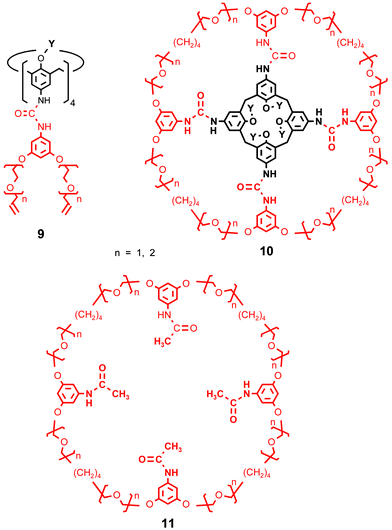
This general strategy can be modified in various ways. Tetraurea calix[4]arenes 9 have been successfully converted into the tetraloop compounds 10, from which huge macrocycles 11 containing oligoethylene oxide units were available in analogy with Scheme 1. This suggests that further structural elements can be used to construct macrocyclic molecules in the described way.
In addition, calix[4]arenes may be substituted in p-position by various urea residues.13 If then calix[4]arenes consisting of such different phenolic units A, B, etc.:

Finally, suitable bifunctional fragments might be attached to the urea residues in alternative ways (e.g. via easily cleavable ester links), and reactions different from metathesis may also be used for the ring closure (step iv), as long as these reactions are possible under conditions in which the tetraurea dimers exist.
This work was supported by the Deutsche Forschungsgemeinschaft (Bo 523/14-4, SFB 625) and the Fonds der Chemischen Industrie.
Notes and references
- Templated Organic Synthesis, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 2000 Search PubMed.
- D. Voet, J. G. Voet and C. W. Pratt, Fundamentals of Biochemistry, Wiley, New York, 2002 Search PubMed.
- (a) A. Chuchuryukin, H. P. Dijkstra, B. M. J. M. Suijkerbuijk, R. J. M. Klein Gebbink, G. P. M. van Klink, A. M. Mills, A. L. Spek and G. van Koten, Angew. Chem. Int. Ed., 2003, 42, 228 CrossRef CAS; (b) S. Anderson, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2255–2267 RSC.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017 CrossRef CAS.
- (a) S. Höger and A.-D. Meckenstock, Chem. Eur. J., 1999, 5, 1686–1691 CrossRef CAS; (b) B. C. Gibb, Chem. Eur. J., 2003, 9, 5180 CrossRef.
- (a) Molecular Catenanes, Rotaxanes and Knots, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999 Search PubMed; (b) O. Lukin and F. Vögtle, Angew. Chem. Int. Ed., 2005, 44, 1456–1477 CrossRef CAS; (c) S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1–9 CrossRef CAS; (d) L. Wang, M. O. Vysotsky, A. Bogdan, M. Bolte and V. Böhmer, Science, 2004, 304, 1312–1314 CrossRef CAS.
- S. C. Zimmerman, M. S. Wendland, N. A. Rakow, I. Zharov and K. S. Suslick, Nature, 2002, 418, 399–403 CrossRef CAS.
- Y. Kim, F. M. Mayer and S. C. Zimmerman, Angew. Chem. Int. Ed., 2003, 42, 1121–1126 CrossRef CAS.
- F. Hof, L. C. Craig, C. Nuckolls and J. Rebek, Jr., Angew. Chem. Int. Ed., 2002, 41, 1488–1508 CrossRef CAS.
- M. O. Vysotsky, A. Bogdan, L. Wang and V. Böhmer, Chem. Commun., 2004, 1268–1269 RSC.
- R. K. Castellano, B. H. Kim and J. Rebek, Jr., J. Am. Chem. Soc., 1997, 119, 12671–12672 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS.
- See for example: A. Pop, M. O. Vysotsky, M. Saadioui and V. Böhmer, Chem. Commun., 2003, 1124–1125 Search PubMed.
- P. Wang, S. F. Moorefield and J. Newkome, Angew. Chem. Int. Ed., 2005, 44, 1579–1683.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details for the synthesis of macrocycles 7 and selected examples for 6 and 10; X-ray crystal structure of 6 (m = 10, Y = CH3) in CIF format. See http://www.rsc.org/suppdata/cc/b5/b505223h/index.sht |
‡ Single crystals of 6
(m
= 10, Y = CH3) could be obtained by slow crystallisation from THF–methanol (2∶1). Crystal data for 6: C100H128N8O16·8 CH4O·1.5 H2O, M
= 1981.46, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a
= 18.8106(15)
Å, b
= 23.1537(18)
Å, c
= 28.968(2)
Å, α
= 98.409(6)°, β
= 104.443(6)°, γ
= 102.706(6)°, V
= 11645.5(15)
Å3T
= 173 K, Z
= 4, Dc
= 1.130 g cm−3, λ
(Mo Kα)
= 0.71073 Å, 93167 reflections measured, 38675 unique (Rint
= 0.198) which were used in all calculations. The structure was solved by direct methods (SHELXL-9715) and refined by full-matrix least-squares methods on F2 with 2548 parameters. R1
= 0.1560 [I > 2σ(I)] and wR2
= 0.3756, GOF = 1.252; max/min residual density 1.379/−0.590 e Å−3. CCDC reference number 265476. See http://www.rsc.org/suppdata/cc/b5/b505223h/index.sht for crystallographic data in CIF or other electronic format. , a
= 18.8106(15)
Å, b
= 23.1537(18)
Å, c
= 28.968(2)
Å, α
= 98.409(6)°, β
= 104.443(6)°, γ
= 102.706(6)°, V
= 11645.5(15)
Å3T
= 173 K, Z
= 4, Dc
= 1.130 g cm−3, λ
(Mo Kα)
= 0.71073 Å, 93167 reflections measured, 38675 unique (Rint
= 0.198) which were used in all calculations. The structure was solved by direct methods (SHELXL-9715) and refined by full-matrix least-squares methods on F2 with 2548 parameters. R1
= 0.1560 [I > 2σ(I)] and wR2
= 0.3756, GOF = 1.252; max/min residual density 1.379/−0.590 e Å−3. CCDC reference number 265476. See http://www.rsc.org/suppdata/cc/b5/b505223h/index.sht for crystallographic data in CIF or other electronic format. |
| § General procedure for the synthesis of macrocycles7: the compound 6 (m = 8, 10, 14, 20) (0.025 mmol) was refluxed in acetic acid (20 mL) for 24 hours. After cooling, acetic acid was removed in vacuo. The residue was separated and purified by column chromatography on silica gel (eluent CH2Cl2–MeOH = 20∶1 ) giving 7 as the first eluted compound and 8. Macrocycle 11 was prepared in a similar way.7 (m = 8): yield: 50%; m.p. > 300 °C, phase transition 136–140 °C; 1H NMR (dmso-d6): δ = 9.79 (s, 4H, NH), 6.77 (br d, 8H, ArH), 6.14 (br t, 4H, ArH), 3.87 (t, 3J = 6.5 Hz, 16H, OCH2), 1.99 (s, 12H, C[O]CH3), 1.66 (m, 16H, OCH2CH2), 1.38–1.24 (m, 32H, OCH2CH2CH2CH2); ESI-MS m/z: calcd for C64H92N4O12Na (M + Na) 1132.4, found 1132.7.7 (m = 10): yield: 66%. m.p. > 300 °C, phase transition 130–135 °C; 1H NMR (dmso-d6): δ = 9.78 (s, 4H, NH), 6.77 (d, 4J = 2.0 Hz, 8H, ArH), 6.14 (br t, 4H, ArH), 3.87 (t, 3J = 6.5 Hz, 16H, OCH2), 1.99 (s, 12H, C[O]CH3), 1.67–1.63 (m, 16H, OCH2CH2), 1.36–1.23 (m, 48H, OCH2CH2CH2CH2); ESI-MS m/z: calcd for C72H108N4O12Na (M + Na) 1244.7, found 1243.6.11 (n = 1): yield: 65%. m.p. > 300 °C, phase transition 65–70 °C; 1H NMR (dmso-d6): δ = 9.80 (s, 4H, –NH), 6.79 (br d, 8H, ArH), 6.20 (br t, 4H, ArH), 3.99 (br t, 16H, OCH2CH2OCH2), 3.63 (br t, 16H, OCH2CH2O), 3.43 (br t, 16H, OCH2CH2O), 1.99 (s, 12H, C[O]CH3), 1.53 (m, 16H, OCH2CH2CH2); ESI-MS m/z: calcd for C64H92N4O20Na (M + Na) 1260.4, found 1259.6. |
| ¶ In a very recent publication (3,5-dialkenyloxy)benzyl ether residues were attached to a bis(terpyridine) ligand, which forms a cyclic hexameric complex with six Ru(III) cations. A metathesis reaction leads to a connection of six units similar to 1 or 2, but the yield of the resulting macrocycle is not reported; see ref. 14. While the preorganisation of six “monomeric units” is on principle advantageous, if wrong connections can be avoided, an extension to the regular incorporation of different monomeric units A, B, C is clearly not possible in this case. |
| This journal is © The Royal Society of Chemistry 2005 |