Chemical activation of MgH2; a new route to superior hydrogen storage materials†
Simon R. Johnsonab, Paul A. Anderson*a, Peter P. Edwards*b, Ian Gamesona, James W. Prendergasta, Malek Al-Mamouric, David Book*c, I. Rex Harrisc, John D. Speightc and Allan Waltonc
aSchool of Chemistry, The University of Birmingham, Edgbaston, Birmingham, UK B15 2TT. E-mail: p.a.anderson@bham.ac.uk
bInorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK OX3 3QR. E-mail: peter.edwards@chemistry.oxford.ac.uk
cDepartment of Metallurgy and Materials, School of Engineering, The University of Birmingham, Edgbaston, Birmingham, UK B15 2TT. E-mail: d.book@bham.ac.uk
First published on 26th April 2005
Abstract
We report the discovery of a new, chemical route for ‘activating’ the hydrogen store MgH2, that results in highly effective hydrogen uptake/release characteristics, comparable to those obtained from mechanically-milled material.
Hydrogen is widely regarded as a promising alternative to carbon-based fuels; it can be produced from a variety of renewable resources and, when coupled with fuel cells, offers the prospect of near-zero emission of pollutants and greenhouse gases.1 However, the development of hydrogen as a major energy carrier will require solutions to many scientific and technological challenges.2 Foremost among these is the issue of hydrogen storage; the lightest of all the chemical elements has an excellent energy content per unit weight, but a low energy content per unit volume.3
The current generation of solid state hydrogen stores employ metal hydrides (e.g. LaNi5H6) that have excellent volumetric storage densities—higher than for both compressed gas and liquid hydrogen—but which have poor gravimetric storage densities (1.37 wt% for LaNi5H6), thereby precluding their use for mobile storage applications in hydrogen fuel-cell vehicles, for which a capacity of 6–7 wt% is regarded as a minimum requirement.2 Magnesium hydride offers the highest energy density of all reversible hydrides applicable for hydrogen storage.4 However, hydrogen adsorption/desorption kinetics are too slow to form the basis of a practical hydrogen store. It has been demonstrated previously by a number of groups that the hydrogen sorption kinetics of Mg and Mg-based alloys can be greatly improved if the materials are prepared with a nanocrystalline microstructure through high velocity ball milling.5 However, this technique is energy intensive, especially for batch-milling large quantities of material.
Recently, a number of new hydrogen storage systems have been proposed6 based on the interaction between light metal hydrides and amides. Luo reported7 that a 1 ∶ 2 mixture of MgH2 and LiNH2 desorbed hydrogen reversibly at around 200 °C. Here we report a dramatic enhancement in the absorption/desorption kinetics of MgH2 through reaction with small amounts of LiBH4.8
MgH2 and LiBH4 (Sigma-Aldrich) were mixed together in a mole ratio of 1 ∶ 0.1 in an argon atmosphere glove box (O2 content < 10 ppm) and placed in a quartz tube. The tube was capped with a Young's tap, via an Ultra Torr fitting, removed from the glove box and placed on a vacuum line, where it was evacuated to 10−6 mbar before being sealed with a gas torch. The sealed evacuated tube was then placed in a muffle furnace and heated at 300 °C for 12 hours.
For comparison, MgH2 (typical size ∼ 100 µm) was milled at 300 rpm (PM400 Retsch planetary ball mill) for 15 hours in an argon atmosphere in a stainless steel milling pot (250 ml) using 8 mm balls with a 10 ∶ 1 ball to powder ratio. All samples were prepared for characterization in the glove box.
The hydrogen desorption kinetics of MgH2/LiBH4 after five hydrogen desorption and reabsorption cycles are shown in Fig. 1, compared to MgH2 and MgH2 milled for 15 h (i, ii and iii respectively).9 It is apparent that MgH2/LiBH4 desorbed hydrogen significantly faster than both the MgH2 and milled MgH2. The average desorption rates, therefore, were 0.28 × 10−2 wt% s−1, 0.01 × 10−2 wt% s−1 and 0.12 × 10−2 wt% s−1 for MgH2/LiBH4, MgH2 and milled MgH2 respectively.10
 | ||
| Fig. 1 Hydrogen desorption kinetics at 300 °C under 10 mbar H2 of i) MgH2/LiBH4, ii) as-received MgH2 and iii) MgH2 milled for 15 h; all samples measured on the sixth hydrogen desorption. | ||
The subsequent sixth hydrogen absorption at 300 °C and 10 bar H2 for the same three materials is shown in Fig. 2. Again the MgH2/LiBH4 material reabsorbed hydrogen faster than MgH2, and at a rate comparable to MgH2 milled for 15 hours. The wt% values of hydrogen absorbed at 60 min for MgH2/LiBH4, MgH2 and ball milled MgH2 were 5.86, 4.86 and 6.01 respectively, and the absorption rates within this time scale were 1.03 × 10−2 wt% s−1, 0.16 × 10−2 wt% s−1 and 1.24 × 10−2 wt% s−1 respectively.11
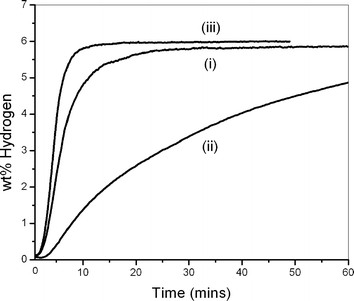 | ||
| Fig. 2 Hydrogen absorption kinetics at 300 °C under 10 bar H2 of i) MgH2/LiBH4, ii) MgH2 and iii) MgH2 milled for 15 h; all samples measured on the sixth hydrogen absorption. | ||
Clearly, the reaction of MgH2 with quite modest amounts of LiBH4 resulted in a spectacular increase in the kinetics of hydrogen desorption/absorption. This improvement, however, was not apparent in the ‘as-prepared’ material: importantly, the rate of hydrogen desorption/absorption increased with each successive cycle for the first 4–5 cycles before stabilizing. Both the heat treatment and the initial cycling were necessary to activate the material: cycling a simple mixture of MgH2 and LiBH4 did not result in such an ‘active’ material with fast kinetics.
There are a number of possible factors that could contribute to the observed enhancement in hydrogen absorption/desorption properties. Powder XRD studies revealed that MgH2/LiBH4 contained only phases with the tetragonal α-MgH2 structure (rutile).12 Evidently a change in the crystal structure of MgH2 is not responsible. The XRD pattern of the as-prepared material exhibited narrow, split diffraction lines, indicating that the material contained two different phases with the same structure, present in roughly equal amounts. After cycling, the active material was apparently single phase but with noticeably broader lines, and a unit cell slightly expanded (∼0.8%) from that of as-received MgH2.13 This expansion could contribute to enhanced diffusion of H atoms in Mg/MgH2.
Previously observed enhancements in the hydrogen absorption/desorption kinetics of Mg and Mg-based alloys after ball milling have been attributed to a nanocrystalline microstructure with an increased volume of grain boundary material, which acts as a preferred pathway for hydrogen diffusion.14 Changes in sample morphology and particle size may also be important. SEM images of MgH2/LiBH4, before and after six hydrogen desorption/absorption cycles, are shown in Fig. 3, along with those of as-received and milled MgH2.15 Before cycling, MgH2/LiBH4 (Fig. 3i(c)) had a very similar appearance to MgH2 (Fig. 3i(a)) consisting of large particles of up to 100 µm with a smooth surface. After six cycles of repeated crystallization of Mg and MgH2, some finer particles were visible in MgH2, but a significant proportion of larger lumps remained (Fig. 3ii(a)). MgH2/LiBH4 (Fig. 3ii(c)) in contrast consisted of a fine, apparently porous material of coral-like appearance with characteristic length scale ca. 2 µm. This is roughly the same as the particle size of MgH2 milled for 15 hours (Fig. 3i(b) & ii(b)), although the morphologies were noticeably different.
 | ||
| Fig. 3 Scanning electron microscope images, i) before hydrogen desorption of a) MgH2, b) MgH2 milled for 15 h and c) MgH2/LiBH4 and ii) taken after the sixth hydrogen reabsorption of a) MgH2, b) MgH2 milled for 15 h and c) MgH2/LiBH4. | ||
The fine powder structure observed may be a factor in the fast kinetics of both MgH2/LiBH4 and milled MgH2 materials, but this may not be the most important factor in the former, as it differs chemically, owing to the presence of LiBH4. Analysis of MgH2/LiBH4 after the sixth hydrogen absorption by XPS16 indicated that lithium and boron were still present in the sample. The FTIR spectrum17 of MgH2/LiBH4 before cycling exhibited peaks at 1126 cm−1, which corresponds to a BH2 deformation, and four peaks at 2225 cm−1, 2238 cm−1, 2291 cm−1 and 2386 cm−1, corresponding to B–Ht (terminal) stretching.18 This may be compared to LiBH4, which showed peaks at 1120 cm−1 (BH2 deformation) and 2220 cm−1, 2231 cm−1, 2282 cm−1 and 2381 cm−1 (B–Ht stretches). The spectrum of the same material after six hydrogen desorption/absorption cycles contained no B–H vibrations, suggesting that BH4− anions did not persist at this stage. This conclusion was supported by proton-decoupled solid state 11B NMR spectra19 of the MgH2/LiBH4 before and after cycling. Before hydrogen desorption the spectrum was very similar to that of LiBH4 with a single central transition at ca. −41 ppm. After the first hydrogen desorption the spectrum was transformed with peaks at 12.7 ppm, 0.9 ppm, −16.0 ppm and −43.3 ppm, and retained multiple peaks on subsequent absorption/desorption cycles.
The decomposition of BH4− after the first hydrogen desorption at 300 °C is to be expected as compounds such as LiBH4 and NaBH4 are well known to lose hydrogen at temperatures lower than this.20 The fact that the anion did not reform on subsequent hydrogen adsorption, however, is significant, indicating that LiBH4 served as a convenient method for introducing both lithium and boron into the system, but that BH4− compounds were not present in the active material. In fact, it was possible to make a material with identical properties starting from a mixture of magnesium powder and LiBH4. Although the presence of both Li and B in MgH2/LiBH4 after cycling is not in doubt, their exact chemical role in the fast hydrogen absorption/desorption has not yet been identified. One suggestion is that these elements might have a catalytic effect at the surface of the magnesium or magnesium hydride particles, but preliminary XPS data suggest that boron enters the subsurface layers of the material and is not just confined to the surface.
Doping of boron and/or lithium into MgH2 could also be consistent with the small increase observed in lattice parameters and could change the thermodynamic properties of the compound. So far, however, there is little evidence for significant thermodynamic differences from MgH2, as there was no observed shift in the hydrogen plateau pressure at 300–350 °C. The observation that use of NaBH4 in the reaction instead of LiBH4 resulted in no improvement in the hydrogen absorption/desorption kinetics, however, suggests that lithium doping of MgH2 could be significant. The presence of Li+ in the MgH2 lattice might lead to hydride anion vacancies which could facilitate diffusion of hydrogen in the hydride phase. More detailed characterization including measurements of thermodynamic parameters and microstructural studies of MgH2/LiBH4 is in progress.
We gratefully acknowledge The European Framework V (FUCHSIA) and EPSRC (SUPERGEN) for funding and we thank colleagues at the Johnson Matthey Technology Centre, UK for technical support.16
Notes and references
- P. Hoffman, Tomorrow's Energy: Hydrogen, Fuell Cells and The Proposals for a Cleaner Planet, MIT Press, Cambridge, MA, 2001 Search PubMed.
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004,(December), 39 CrossRef CAS.
- (a) L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef; (b) W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1315 CrossRef CAS; (c) I. R. Harris, D. Book, P. A. Anderson and P. P. Edwards, Fuel Cell Rev., 2004,(June/July), 17–23 Search PubMed.
- B. Bogdanovic, K. Bohmhammel, B. Christ, A. Reiser, K. Schlichte, R. Vehlen and U. Wolf, J. Alloys Compd., 1999, 282, 84–92 CrossRef CAS.
- (a) W. Oelerich, T. Klassen and R. Bormann, J. Alloys Compd., 2001, 315, 237–242 CrossRef CAS; (b) O. Gutfleisch, N. Schlorke-de Boer, N. Ismail, M. Herrich, A. Walton, J. Speight, I. R. Harris, A. S. Pratt and A. Zuttel, J. Alloys Compd., 2003, 356, 598–602 CrossRef.
- (a) P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS; (b) Y. Nakamori, G. Kitahara and S. Orimo, J. Power Sources, 2004, 138, 309–312 CrossRef CAS; (c) F. E. Pinkerton, G. P. Meisner, M. S. Meyer, M. P. Balogh and M. D. Kundrat, Phys. Chem. B Lett., 2005, 109, 6–8 Search PubMed.
- W. Luo, J. Alloys Compd., 2004, 381, 284–287 CrossRef CAS.
- UK patent application no GB 0415561.0.
- Hydrogen absorption/desorption properties were measured on a constant pressure thermogravimetric balance (Hiden IGA). The desorption kinetics were monitored at 10 mbar of hydrogen and 300 °C, absorption at 10 bar and 300 °C. Samples were transferred quickly from the glove box to the IGA to minimize contact with the atmosphere.
- The desorption rates were calculated as the ratio between the amount desorbed and time taken to go from 80 to 20% of full capacity. The error in the wt% was ±0.05, while the error in the desorption rate was ±0.01 × 10−2 wt% s−1.
- The absorption rates were calculated as the ratio between the amount absorbed and time taken to go from 20 to 80% of capacity measured at 60 min. The error in the wt% was ±0.05, while the error in the absorption rate was ±0.01 × 10−2 wt% s−1.
- In addition to α-MgH2, a number of other crystal structures, observed and predicted, have been reported for MgH2; see M. Bortz, B. Bertheville, G. Bøttger and K. Yvon, J. Alloys Compd., 1999, 287 Search PubMed L4. α-MgH2 is sometimes referred to in the literature as the ‘β-MgH2 phase’, which is not to be confused with the proposed high temperature, high pressure β-MgH2 structure.
- Structural characterisation of the samples was carried out on a Siemens D5000 X-ray diffractometer, using a CuKα source. The structure of α-MgH2 is tetragonal (space group P42mnm) with a = 4.508 ± 0.001 Å, c = 3.017 ± 0.001 Å; MgH2/LiBH4 as-prepared contained two phases with a = 4.510 ± 0.001 Å, c = 3.017 ± 0.001 Å and a = 4.502 ± 0.001 Å, c = 3.011 ± 0.001 Å; MgH2/LiBH4 after cycling, a = 4.522 ± 0.003 Å, c = 3.025 ± 0.003 Å.
- G. Liang, J. Huot, S. Boily and R. Schulz, J. Alloys Compd., 2000, 305, 239–245 CrossRef CAS.
- SEM images were recorded on a JEOL 6300 electron microscope.
- XPS measurements were carried out at the Johnson Matthey Technology Centre, UK on a VG ESACLAB 250 instrument. A monochromatic Al Kα X-ray source at 1464.6 eV was used.
- FTIR spectra were acquired at a resolution of 2 cm−1, from KBr disks containing the samples, on a Nicolet Magna FTIR spectrometer, equipped with a liquid nitrogen cooled MCTB detector.
- (a) T. J. Marks, W. J. Kennelly, J. R. Kolb and L. A. Shimp, Inorg. Chem., 1972, 11, 2540 CrossRef CAS; (b) J. A. A. Ketelaar and C. J. H. Schutte, Spectrochim. Acta, 1961, 17, 1240 CrossRef CAS; (c) T. C. Waddington, J. Chem. Soc., 1958, 4783 Search PubMed.
- Solid state 11B NMR spectra were acquired on a 400 MHz Varian Inova spectrometer. Samples were packed into 3.2 mm o.d. rotors and spun at the magic angle at rates of 10 to 15 KHz. A pulse width of 0.4 to 1.4 µs was employed for data acquisition.
- A. Züttel, S. Rentsch, P. Fischer, P. Wenger, P. Sudan, Ph. Mauron and Ch. Emmenegger, J. Alloys Compd., 2003, 356, 515–520 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: XPS and 11B NMR spectra. See http://www.rsc.org/suppdata/cc/b5/b503085d/ |
| This journal is © The Royal Society of Chemistry 2005 |