Inversion of enantioselectivity of asymmetric biocatalytic decarboxylation by site-directed mutagenesis based on the reaction mechanism†
Yoichiro
Ijima
,
Kaori
Matoishi
,
Yosuke
Terao
,
Nobuhide
Doi
,
Hiroshi
Yanagawa
and
Hiromichi
Ohta
*
Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: hohta@bio.keio.ac.jp; Fax: +81-(0)45-566-1551; Tel: +81-(0)45-566-1703
First published on 10th January 2005
Abstract
The introduction of two mutations (G74C/C188S) based on the estimated reaction mechanism resulted in the inversion of enantioselectivity of arylmalonate decarboxylase, which catalyses the asymmetric decarboxylation of arylmethylmalonate to give optically active arylpropionate.
We have been investigating a novel enzymatic (arylmalonate decarboxylase = AMDase, EC. 4.1.1.76) asymmetric decarboxylation of α-aryl-α-methylmalonate to give optically active α-arylpropionate [eqn. (1)].1,2
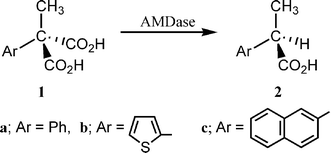 | (1) |
The chemical and optical yields of the reaction are generally high, the configuration of the products being R (S for Ar = thienyl, because of the priority rule).3,4 It was demonstrated that a free cysteine residue plays an essential role in this reaction5,6 and its location was revealed to be 188 on the basis of point mutation.7 Recently we proposed that the Cys188 is working as a proton donor to the intermediate enolate (Scheme 1).8
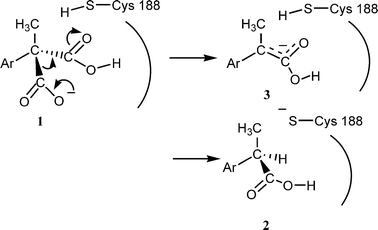 | ||
| Scheme 1 | ||
The inversion of the enantioselectivity of the reaction might be possible by changing the binding mode of the substrate or by shifting the key cysteine residue from the si-face to the re-face of the enolate intermediate. In the former case, to exchange the binding sites of the aromatic ring and methyl group, the volume of the binding sites as well as the location of some amino acid residues should be changed to realise a strong binding between the substrate and the enzyme. The latter strategy seems more practical, because replacing the Cys188 residue with another amino acid that has little or no proton-donating ability is not difficult. Then, the introduction of a new proton donor at a suitable position would bring about the expected inversion of enantioselectivity. Herein, we report that the introduction of only two mutations, i.e. G74C and C188S, led to the formation of the opposite enantiomer, although the activity of the mutant was lower than that of the native enzyme.
As the tertiary structure of AMDase is not yet known, the most serious problem is to predict the position at which the new proton donor should be introduced. The only clue that might be effective is a homology search as well as the comparison of the amino acid sequence and the function of the enzymes. The characteristic features of AMDase are (1) the reaction proceeds via an enolate-type transition state,5 (2) the cysteine residue plays an essential role,5,7 and (3) the reaction involves an inversion of configuration on the α-carbon of the carboxyl group.9
Some enzymes were found that had about 30% homology and some common functions via multiple alignment using the PSI-BLAST program (position-specific iterative basic local alignment search tool). These were glutamate racemase from Lactobacillus fermenti,10 aspartate racemase from Streptococcus thermophilus,11 hydantoine racemase from Pseudomanas sp. strain NS671,12 and maleate isomerase from Alcaligenes faecalis.13 The important feature that is consistent for all of these enzymes is Cys188. On the other hand, while all of the isomerases have another cysteine residue at around 74, AMDase has no corresponding cysteine residue around this region as shown in Fig. 1.
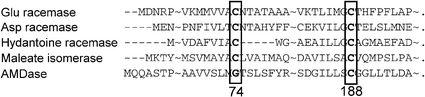 | ||
| Fig. 1 Amino acid homology between some racemases and AMDase. | ||
The reaction mechanism for glutamate racemase has been studied extensively.14–16 It has been proposed that the key for the racemisation activity is that the two cysteine residues of the enzyme are located in both sides of the substrate bound to the active site. Thus one cysteine residue abstracts the α-proton from the substrate, while the other delivers a proton from the opposite side of the intermediate enolate of the amino acid. In this way the racemase catalyses the racemisation of glutamic acid via a so-called two-base mechanism (Scheme 2).
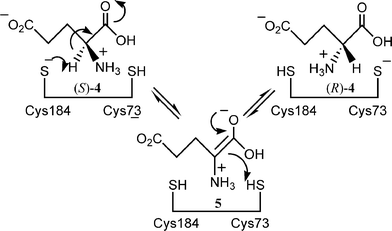 | ||
| Scheme 2 | ||
The tertiary structure of glutamate racemase has already been resolved, and it has also been clarified that a substrate analogue glutamine binds between two cysteine residues.17 These data enabled us to predict that the new proton-donating amino acid residue should be introduced at position 74 replacing Gly for the inversion of enantioselectivity of the decarboxylation reaction.
First, we examined the enantioselectivity of a C188S mutant enzyme (purified from E. coli JM 109, pAMD 101, IPOD 12908).7 Although the proton-donating ability of Ser is weaker than Cys, the location of the proton-donor does not change in this case. Thus, we presumed that the configuration of the product would be the same as in the case of the reaction by the wild-type enzyme. To our surprise, however, the reality was entirely different. α-Methyl-α-thienyl- (1b) and α-methyl-α–naphthylmalonate (1c) gave the corresponding monobasic acids with configurations opposite to that given by the native enzyme (Table 1). This fact suggests that there are some other proton donors on the opposite side of the enantiomeric face of the intermediate enolate, although their effect is far smaller compared with Cys188. In the case of the C188S mutant, as the proton-donating ability of serine is weaker than that of cysteine, the hidden effect of the other proton donors might be reflected in the product. Then, higher enantiomeric excess will be attained if the Cys188 of the native enzyme is changed by an amino acid that has no acidic proton. Thus we prepared the C188A mutant. However, this mutant had no enzyme activity and in addition, was very unstable. This means that a Cys or Ser residue is inevitable at position 188, probably to construct a hydrogen bonding network. Thus we decided to introduce a Cys residue as a proton donor instead of the Gly74 of the native enzyme.
| Enzyme | Sub | Km/mM | kcat/s1 | Relative activity | Enzyme/mg mL−1 | Reaction time/h | Yield (%) | Ee (%) |
|---|---|---|---|---|---|---|---|---|
| a The substrate concentration is 0.1 mmol mL−1. b The non-enzymatic decarboxylation of the substrates under the same reaction conditions was less than 3%. | ||||||||
| WT | 1b | 12.5 | 200 | 100 | 0.87 | 1 | quant | 99, S |
| C188S | 1b | 14.5 | 1.63 | 0.70 | 0.77 | 3 | 17 | 50, R |
| G74C | 1b | 6.24 | 0.79 | 0.79 | 1.07 | 1 | 37 | 0 |
| G74C/C188S | 1b | 5.03 | 0.34 | 0.42 | 0.52 | 72 | 60 | 94, R |
| WT | 1c | 0.43 | 31 | 100 | 0.87 | 1 | 96 | 97, R |
| C188S | 1c | 1.26 | 2.54 | 2.80 | 0.77 | 3 | 6 | 70, S |
| G74C | 1c | 1.56 | 0.50 | 0.44 | 1.07 | 1 | 13 | 6, R |
| G74C/C188S | 1c | 1.25 | 0.12 | 0.13 | 0.52 | 72 | 17 | 96, S |
The G74C mutant was prepared via a PCR (polymerase chain reaction) using the plasmid that contains the gene coding native AMDase (pAMD 101).7 The amplified gene was digested by Hind III and Pst I followed by ligation with pUC 19. The mutant enzyme was purified from the transformed E. coli cells. Although the change in amino acid is drastic, the mutant still exhibited some activity. As expected, the products were entirely or nearly racemic in the case of both 1b and 1c (Table 1). These results demonstrate that this position is effective to give a proton to the intermediate of the reaction.
If the proton-donating ability of the amino acid at 188 is weaker, then the enantioselectivity of the reaction is expected to become the opposite. Thus we prepared a double mutant G74C/C188S gene starting from the one that already contains the codon for C188S mutation. As shown in Table 1, the absolute configuration of the products are opposite to those of the products obtained by the native enzyme, and the ee of the products dramatically increased to 94 and 96% for 1b and 1c, respectively (Table 1). This inversion of the enantioselectivity of the reaction supports the reaction mechanism where the Cys188 of the native enzyme is working as the proton donor.8
This work was partly supported by the “Research for the Future Program (JSPS-RFTF 97100302)” and the “Science and Technology Program on Molecules, Supra-Molecules and Supra-Structured Materials” of the Academic Frontier Promotional Project of the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Notes and references
- H. Ohta, Bull. Chem. Soc. Jpn., 1997, 70, 2895–2911 CAS.
- H. Ohta, Advances in Biochemical Engineering/Biotechnology, ed. K. Faber, Springer, Berlin, 1998, vol. 63, pp. 1–30 Search PubMed.
- K. Miyamoto and H. Ohta, J. Am. Chem. Soc., 1990, 112, 4077–4078 CrossRef CAS.
- K. Miyamoto and H. Ohta, Biocatalysis, 1991, 5, 49–60 Search PubMed.
- K. Miyamoto and H. Ohta, Eur. J. Biochem., 1992, 210, 475–481 CAS.
- K. Miyamoto and H. Ohta, Appl. Microbiol. Biotechnol., 1992, 38, 234–238 CrossRef CAS.
- M. Miyazaki, H. Kakidani, S. Hanzawa and H. Ohta, Bull. Chem. Soc. Jpn., 1997, 70, 2765–2769 CAS.
- K. Matoishi, M. Ueda, K. Miyamoto and H. Ohta, J. Mol. Catal. B: Enzym., 2004, 27, 161–168 CrossRef CAS.
- K. Miyamoto, S. Tsuchiya and H. Ohta, J. Am. Chem. Soc., 1992, 114, 6256–6257 CrossRef CAS.
- K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3981–3990 CrossRef CAS.
- M. Yohda, H. Okada and H. Kumagai, Biochim. Biophys. Acta, 1991, 1089, 234–240 CAS.
- K. Watanabe, T. Ishikawa, Y. Mukohara and H. Nakamura, J. Bacteriol., 1992, 174, 3461–3466.
- K. Hatakeyama, Y. Asai, Y. Uchida, M. Kobayashi, M. Terasawa and H. Yukawa, Biochem. Biophys. Res. Commun., 1997, 239, 74–79 CrossRef CAS.
- S. Glavas and M. E. Tanner, Biochemistry, 1999, 38, 4106–4113 CrossRef CAS.
- K. A. Gallo, M. E. Tanner and J. R. Knowles, Biochemistry, 1993, 32, 3991–3997 CrossRef CAS.
- M. E. Tanner, K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3998–4006 CrossRef CAS.
- K. Y. Hwang, C.-S. Cho, S. S. Kim, H.-C. Sung, Y. G. Yu and Y. Cho, Nat. Struct. Biol., 1999, 6, 422–425 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: preparation of the substrates, site-directed mutagenesis of AMDase, purification of AMDase and enzymatic decarboxylation of α-methyl-α-arylmalonic acid (1). See http://www.rsc.org/suppdata/cc/b4/b416398b/ |
| This journal is © The Royal Society of Chemistry 2005 |