Construction of functional porphyrin polystyrene nano-architectures by ATRP†
Femke
de Loos
,
Irene C.
Reynhout
,
Jeroen J. L. M.
Cornelissen
*,
Alan E.
Rowan
* and
Roeland J. M.
Nolte
Department of Organic Chemistry, Institute for Molecules and Materials (IMM), Radboud University Nijmegen, Toernooiveld 1, 6525 ED Nijmegen, The Netherlands. E-mail: j.cornelissen@science.ru.nl; a.rowan@science.ru.nl; Fax: (+31) 24 365 2929; Tel: (+31) 24 365 2323
First published on 1st December 2004
Abstract
A series of mono-functionalized metallo-porphyrin polystyrenes have been synthesized using atom transfer radical polymerization and their self-assembling behavior was studied by electron microscopy showing that the polystyrene tail-length influences the aggregate architecture.
The construction of well-defined functional nano-objects is nowadays one of the main topics in supramolecular chemistry. The use of porphyrins as components of the nano-system is of interest since they may introduce functional properties such as energy transfer and catalysis. To construct such porphyrin-functionalized nano-structures we decided to prepare amphiphilic porphyrins, since these can be expected to self-assemble into a large variety of structures. Small molecular weight amphiphilic porphyrins have already been shown to assemble into interesting architectures.1 The need to increase the stability of the aggregates prompted us to develop polystyrene-based porphyrin amphiphiles. Polymeric amphiphiles, for example block copolymers, are known to assemble into various (kinetically) stable structures.2 Our goal is to control the aggregate morphology by adjusting the ratio between the polar and apolar part of the amphiphile and thereby influence the functionality of the aggregated system. To this end, we have utilized atom transfer radical polymerization (ATRP) to construct porphyrin-functionalized polystyrenes. ATRP is a rather well established polymerization process, allowing good control over the degree of polymerization and chain length distribution.3,4 Recently, Beil and Zimmerman have used ATRP in combination with a porphyrin complex to perform polymer imprinting.5
We prepared a series of ATRP initiators, all based on a mono initiator-functionalized tritolylporphyrin, which differ in porphyrin inner-core functionalization. Scheme 1 depicts the synthesis of the free-base porphyrin initiator 2a, the copper porphyrin initiator 2b, the manganese porphyrin initiator 2c and the zinc porphyrin initiator 2d. With these initiators we performed bulk-polymerization reactions of styrene under ATRP conditions (Scheme 1).‡
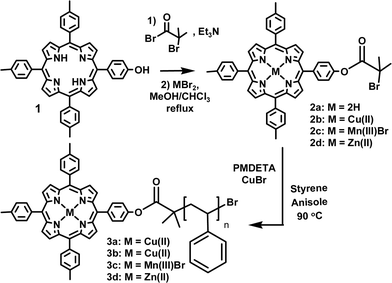 | ||
| Scheme 1 Synthesis of porphyrin-functionalized polystyrene. | ||
Polymerization of styrene with free-base initiator 2a did not take place with one equivalent of copper, due to preferential insertion of copper(II) into the porphyrin core. UV-vis spectroscopy showed that this insertion was already complete after the first 15 min of the reaction.6 As a result, no Cu(II) was left to be converted into Cu(I) to enter the catalytic cycle. However, both with two and ten equivalents of copper, polymerization did take place (Fig. 1). Using 2 equivalents of copper bromide the polymerization progressed in a controlled fashion resulting in polystyrene-functionalized Cu(II)–porphyrin with low polydispersity, as was confirmed by GPC (Table 1 and Fig. 2a) and MALDI-TOF mass spectrometry. When 10 equivalents of copper bromide were used GPC showed that the formed polystyrene had a much higher molecular weight than expected. Also, a low molecular weight peak was observed, which had a strong absorption at 420 nm pointing to the presence of a high concentration of free porphyrin. Probably, the large amount of copper leads to the cleavage of the ester bond between the porphyrin and the growing polymer chain. This alters the initiator ∶ monomer ratio in favour of high molecular weight polymer. Another possible explanation is that the increase in Cu(I) concentration results in an increased number of radicals at the start of the polymerisation and hence to an increase in termination reactions. When Cu–porphyrin 2b was applied as the initiator, 1 equivalent of CuBr proved to be sufficient to perform a controlled polymerization, as expected.
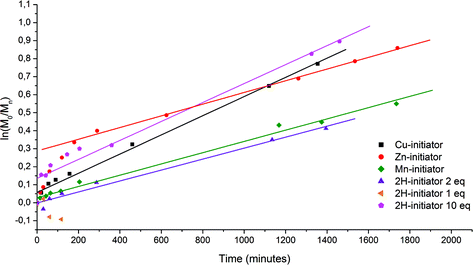 | ||
| Fig. 1 First order log plots of the conversion of styrene as a function of time for the polymerization of styrene with porphyrin initiators 2a–2d. | ||
 | ||
| Fig. 2 (A) GPC-traces of Cu–porphyrin polystyrene measured at two different wavelengths, 254 nm (polystyrene), 420 nm (Cu–porphyrin). (B) GPC-traces (254 nm) of Mn–porphyrin polystyrene fractions collected after size exclusion column. | ||
| Initiator | Polystyrene product | Polymerisation conditions | PDIa | M n/kg mol−1a | M w/kg mol−1a |
|---|---|---|---|---|---|
| a Measured by GPC. b Pentamethyl diethylene triamine. | |||||
| 2a | 3a (2 equiv.) | 2 equiv. CuBr, 2 equiv. PMDETAb | 1.39 | 5.8 | 8.0 |
| 2a | 3a (10 equiv.) | 10 equiv. CuBr, 20 equiv. PMDETAb | 1.28 | 16.0 | 20.0 |
| 2b | 3b | 1 equiv. CuBr, 2 equiv. PMDETAb | 1.22 | 4.2 | 5.2 |
| 2c | 3c | 1 equiv. CuBr, 2 equiv. PMDETAb | 1.57 | 3.9 | 6.2 |
| 2d | 3d | 1 equiv. CuBr, 2 equiv. PMDETAb | 1.14 | 3.6 | 4.2 |
In order to construct amphiphilic polystyrenes with catalytic properties polymer 3c was prepared from initiator 2c having a positively charged manganese headgroup. The Mn(III)–porphyrin–polystyrene 3c showed a relatively high polydispersity of 1.5.
Only a limited number of metals inside the porphyrin catalyst can be used, since these metals may interact with the copper catalyst. The use of free-base initiator followed by insertion of the metal after polymerization is not an option due to insertion of copper during the polymerization of styrene. In addition, the copper is difficult to remove from the porphyrin core and will result in decomposition of the macromolecule. We decided therefore to synthesize Zn–porphyrin–polystyrene 3d. Zinc is easily removed from the porphyrin in this compound under mild acidic conditions. This will give the possibility to insert any metal after the polymerization reaction. The polymerization of styrene from the Zn–porphyrin initiator 2d was performed in a similar controlled fashion yielding a Zn–porphyrin–polystyrene with low polydispersity (Table 1).
For the free-base porphyrin initiator with 2 equivalents of CuBr as well as for all three metallo-porphyrin initiators the conversion of styrene in time showed first-order kinetics (Fig. 1). The rate constants varied from 5.1 × 10−6 s−1 to 8.9 × 10−6 s−1, depending on the initiator that was used.
The aggregation behavior of the Cu–porphyrin–polystyrene prepared with 2 equivalents of CuBr (3a) was studied in water. When the porphyrin-functionalized polystyrene was injected from a THF-solution into water while sonicating at 60 °C, highly monodisperse (DI = 1.02) micellar structures, 20–35 nm in size, were formed (Fig. 3a). In subsequent experiments, the aggregation behavior of the Mn–porphyrin–polystyrene was investigated. 3c was fractionated in different samples (f1–f5) with Mn's ranging from 1495 to 9973 and polydispersities ranging from 1.09 to 1.27 (Fig. 2b). With two of these fractions, f1 (Mn = 9973, PDI = 1.24) and f5 (Mn = 1495, PDI = 1.11), aggregation studies were performed. For the more hydrophilic Mn(III)–porphyrin–polystyrene, sonication was not required to obtain well defined architectures. The polymer was dissolved in THF and water was slowly added until the solution turned cloudy. TEM studies showed the formation of small spherical micellar aggregates for f1 (Fig. 3b). The Mn(III)–porphyrin–polystyrene with the shortest apolar tail (f5), showed the formation of large spherical architectures 0.5–2 µm in size, which are vesicular-like in nature. SEM studies showed that these structures are porous, suggesting they are hollow spheres (Fig. 3c and 3d). In the case of f5 the average length of the apolar tail is comparable to the diameter of the porphyrin head (estimated from calculations). However, for f1, the length of the polystyrene tail is ten times larger. The difference in the ratio head ∶ tail results in a different aggregation behavior. The structures proved to be stable over time for at least one month.
 | ||
| Fig. 3 (A) and (B) Transmission electron micrographs of the aggregation behaviour of amphiphilic Cu–porphyrin–polystyrene (A) and Mn–porphyrin–polystyrene Mn = 9973 (B). (C) and (D) Scanning electron micrographs of the aggregation behaviour of Mn–porphyrin–polystyrene with Mn = 1495. | ||
In summary, it is shown that the polymerization of styrene by ATRP using a series of metallo-porphyrin initiators proceeds in a controlled fashion and yields well-functionalized metallo-porphyrin polystyrenes with low polydispersities. The Cu–porphyrin–polystyrene (3a) formed well defined highly monodispersed spheres in water. Aggregation studies of the Mn–porphyrin–polystyrene (3c) showed that the morphology of the architectures formed in water was controlled by the polystyrene tail length. Currently we are investigating the catalytic properties of the different Mn(III)–porphyrin–polystyrene architectures.
The Council for the Chemical Sciences of the Netherlands Organization for Scientific Research is acknowledged for financial support to R. J. M. Nolte (TOP grant), J. J. L. M. Cornelissen (Veni grant) and A. E. Rowan (Vidi grant).
Notes and references
- A. P. H. J. Schenning, M. C. Feiters and R. J. M. Nolte, Tetrahedron Lett., 1993, 34, 7077 CrossRef CAS; J.-H. Fuhrhop, U. Bindig and U. Siggel, J. Am. Chem. Soc., 1993, 115, 11036 CrossRef CAS.
- (a) N. S. Cameron, M. K. Corbierre and A. Eisenberg, Can. J. Chem., 1999, 77, 1311 CrossRef; (b) M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323 CrossRef CAS.
- (a) V. Coesens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337 CrossRef CAS; (b) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- (a) G. Deng and Y. Chen, Macromolecules, 2004, 37, 18 CrossRef CAS; (b) R. Narain and S. P. Armes, Macromolecules, 2003, 36, 4675 CrossRef CAS; (c) D. M. Haddleton and K. Ohno, Biomacromolecules, 2000, 1, 152 CrossRef CAS; (d) L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967 CrossRef CAS.
- J. B. Beil and S. C. Zimmerman, Macromolecules, 2004, 37, 778 CAS . It should be noted that in the work of Beil and Zimmerman, 10 equiv. of Cu(I)Br are used for 8 initiator sites. Even if one copper is inserted into the porphyrin (a possibility not studied in the paper), there would still be an excess of copper per initiator (1.1 equiv.) and hence catalysis occurs. In addition, the basicity of the ligand may also play a role in the efficiency of copper insertion, when a stronger ligand is used the copper may not be extracted by the porphyrin.
- The radical formed upon homolytic cleavage of the carbon–bromine bond must be immediately trapped since no conversion of styrene is observed. From entry 3, Table 1 it appears that a copper–porphyrin does not act as the radical trap; its precise nature is still under investigation.
Footnotes |
| † Electronic supplementary information (ESI) available: ATRP procedure and characterization data. See http://www.rsc.org/suppdata/cc/b4/b412067a/ |
| ‡ Mono hydroxy-functionalized tritolylporphyrin 1 was functionalized with the initiator group by stirring it for 30 min in dry dichloromethane with a small excess of 2-bromoisobutyryl bromide in the presence of an equimolar amount of triethylamine. Insertion of the desired metal was achieved by refluxing this compound in a mixture of methanol–chloroform (1 ∶ 1, v ∶ v) with the corresponding metal(II) bromide. The conversion of styrene during the polymerization was followed by GC. Anisole was used as a standard. |
| This journal is © The Royal Society of Chemistry 2005 |